0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
Multiple Regions within EBNA1 Can Link DNAs
DAVID MACKEY, TIM MIDDLETON,ANDBILL SUGDEN*
McArdle Laboratory for Cancer Research, Department of Oncology, Medical School, University of Wisconsin-Madison, Madison, Wisconsin
Received 24 April 1995/Accepted 14 July 1995
Epstein-Barr virus nuclear antigen 1 (EBNA1) can bind specifically to two clusters of sites within the Epstein-Barr virus plasmid origin of DNA replication (oriP). EBNA1 activates DNA replication mediated by oriPand can also activate transcription and retain DNA in cells when bound site specifically. EBNA1 bound tooriPphysically links the two clusters of EBNA1-binding sites, resulting in loop formation by the intervening DNA. To elucidate the contribution of DNA linking by EBNA1 to its biological activities, we identified regions within it that can independently link DNAs to which they are bound. An electrophoretic mobility shift assay was used to detect this activity. Proteins which link DNA aggregate that DNA into large lattices. Proteins which cannot link DNA but still bind to DNA retard the mobility of that DNA but do not cause it to form lattices. Amino-terminal truncations were used to map the amino-terminal limit of a minimal DNA-linking domain approximately to amino acid 372 of EBNA1. To map the carboxy-terminal limit of this minimal domain, fusion proteins containing the DNA-binding domain of GAL4 and fragments of EBNA1 were generated and studied. This approach identified the carboxy-terminal limit of this minimal domain to be approximately amino acid 391 and verified its amino-terminal limit. Internal deletions within a truncated EBNA1 derivative verified the importance of this region. Two additional fragments of EBNA1, each of which independently conferred DNA-linking activity on the domain of GAL4 which binds DNA, were identified within amino acids 54 to 89 and amino acids 331 to 361. Therefore, EBNA1 contains at least three regions that can act independently to link DNAs and that may act in concert within intact EBNA1.
Epstein-Barr virus (EBV) infects primary human B lympho-cytes and, in vitro, efficiently induces and maintains them in a proliferating state. Infections in vivo are associated with sev-eral lymphoproliferative diseases, including Burkitt’s lym-phoma. In some of the lymphomas, EBNA1 is the only viral protein expressed detectably (30). These and other findings indicate that EBNA1 and cis-acting elements within the viral DNA are sufficient viral contributions to replicate and main-tain the viral DNA within the nucleus and to regulate the expression of the EBNA1 protein.
All known activities of EBNA1 require that EBNA1 main-tain its ability to bind to DNA and are mediated through EBNA1-binding sites on the EBV DNA (22, 25, 26, 38). Most of these binding sites are found in the origin of latent DNA replication, oriP (27, 37). oriP is composed of two clusters of EBNA1-binding sites, the family of repeats (FR) and the dyad symmetry element (DS) (29). FR is composed of twenty high-affinity binding sites (12). FR, in cis, is sufficient for two activ-ities dependent upon EBNA1. First, incorporation of FR into plasmids lowers the rate at which they are lost from cells expressing EBNA1 (22). This retention may occur because FR is part of a DNA domain which, in cells expressing EBNA1, associates with the nuclear matrix (11). Second, FR activates transcription at EBV’s BamC and LMP promoters (7, 36) and from heterologous promoters (28) in an EBNA1-dependent manner. The other element of oriP, DS, is about 1 kbp away from FR and is composed of four binding sites with lower affinity for EBNA1 than those of FR (12). Replication initiates at or near DS and is activated by FR in a manner largely independent of the spacing or orientation of the two elements (6, 29). Replication initiates once per cell cycle during the S
phase, and the replicated molecules are efficiently segregated to daughter cells (1, 13, 39). EBNA1 and oriP are the only viral contributions needed to mediate viral DNA replication in la-tently infected cells (19, 29, 37, 40).
Little information is available to explain EBNA1’s activation of replication, transcription, and retention of DNA within cells. EBNA1 from the prototypical B95-8 strain of EBV is a 641-amino-acid nuclear protein. The amino- and carboxy-ter-minal domains of the protein are separated by 235 amino acids of Gly-Gly-Ala repeats (see Fig. 2A). The portion of EBNA1 required to dimerize and bind site specifically to DNA lies between amino acids 460 and 607 (2, 10). EBNA1 has been shown to bind to RNA through Arg-Gly-Gly motifs, although no specificity of binding is apparent (31). Enzymatic activities of EBNA1 have not been identified; in particular, EBNA1 purified from insect and monkey cells lacks detectable helicase and ATPase activities (5, 21). EBNA1 is phosphorylated on serine(s), but effects of phosphorylation on any of EBNA1’s activities have not been reported (5, 9). None of these findings reveals the mechanisms by which EBNA1 activates replication, transcription, and retention of DNA within cells.
One insight to EBNA1’s mode of action may come from the finding that EBNA1 can link, or physically associate, regions of DNA to which it binds specifically (4, 21, 34). EBNA1 has been shown by electron microscopy to link FR and DS, looping out the intervening sequence (4, 34). The ability of EBNA1 to link DNA seems likely to play a role in EBV replication for several reasons. First, linking by a prokaryotic protein that binds to its origin of replication has been demonstrated to contribute to its function (14, 23). Second, proteins that bind origins of repli-cation of other animal viruses have been shown to link DNA (15, 16). Finally, the bipartite nature of oriP in EBV, which is conserved in the related herpesvirus papio, is consistent with a functional role for the separation of FR and DS (18). A do-main of EBNA1 which mediates DNA linking has been iden-tified. The amino-terminal limit of this domain has been
* Corresponding author. Mailing address: McArdle Laboratory for Cancer Research, 1400 University Ave., Madison, WI 53706. Phone: (608) 262-6697. Fax: (608) 262-2824.
6199
on November 9, 2019 by guest
http://jvi.asm.org/
mapped to amino acid 350 (3). It has also been demonstrated that EBNA1’s carboxy-terminal 22 amino acids are not re-quired for this activity (8).
We have used three assays which measure the ability of EBNA1 to link DNA. An electrophoretic mobility shift assay (EMSA) in which all reactants are in solution has been devel-oped as one means to assess DNA linking. EMSA provides the advantages of being rapid, reproducible, and amenable to the testing of unpurified proteins. These advantages allowed the screening of a large collection of proteins. The EMSA takes advantage of the observation that DNA-binding proteins which can link DNA incorporate that DNA into large lattices which fail to or barely enter gels composed of 4% polyacrylamide. Those proteins which cannot link DNA but still bind to DNA reduce the mobility of that DNA but do not incorporate it into lattices. This assay has been complemented with two additional assays, one that measures an enhancement of ligation of DNAs (8, 23) and one that measures retention of radiolabeled DNA by DNA covalently bound to a matrix (21), used previously to demonstrate EBNA1-mediated DNA linking.
We have identified three regions of EBNA1 which can in-dependently link DNAs. Within wild-type EBNA1, two of these regions are nearly contiguous and are separated from the third by Gly-Gly-Ala repeats. We have identified a minimal linking domain, consisting of amino acids 372 to 391, within EBNA1. This minimal domain linked DNA independently of the rest of the EBNA1 protein when fused to the dimerization and DNA-binding domain of GAL4. Also, deletion of this domain ablated the ability of truncated EBNA1 to link DNA. The study of additional fragments of EBNA1 fused to the dimerization and DNA-binding domain of GAL4 identified two more regions, amino acids 331 to 361 and 54 to 89, suffi-cient to link DNAs. The ability of EBNA1 to link DNAs with various numbers of binding sites was determined, and the de-pendence of linking upon specific DNA binding was estab-lished. These results identify multiple regions within EBNA1 that encode independently acting DNA-linking activity.
MATERIALS AND METHODS
DNA.The DNA fragments used as probes in this study were derived from cloned DNAs as follows: zero binding sites, 106 bp, SacI to KpnI from pBKS (Stratagene); 1 EBNA1-binding site, 108 bp, SalI to XbaI from p896 (21); 2 EBNA1-binding sites, 131 bp, SalI to PstI from p880 (22); 4 EBNA1-binding sites (DS), 156 bp, BamHI fragment of 141EH2 (composed of the EcoRV to HpaI fragment of EBV containing DS inserted via BamHI linkers into the BamHI site of pHyg [35]); 10 EBNA1-binding sites (half of FR), 516 bp, BstXI fragment from p304 (21); 5 GAL4-binding sites, 109 bp, HindIII to XbaI from G5BCAT (17). DNA fragments were purified from agarose gels. They were quantified by making serial dilutions in the presence of ethidium bromide (0.5mg/ml) and comparing their intensities, when exposed to UV light, to that of similar dilutions of a standard DNA. This method can distinguish less-than-twofold differences in DNA concentrations. DNAs were labeled with the Klenow fragment of DNA polymerase 1 in the presence of dATP, dGTP, dTTP, and [a-32P]dCTP (Amer-sham) and precipitated three times to remove unincorporated label. The DNA used for enhanced ligation (p1361) contained two sets of five GAL4-binding sites cloned into the HindIII and BstEII sites of pKAN2 (37). 1361 was cut with SgrAI generating a linear DNA of 5,065 bp with the clusters of five GAL4-binding sites 380 and 393 bp from its ends. The linear DNA was purified from agarose gels prior to use.
Protein expression vectors.bEBNA1 was produced by a baculovirus expres-sion vector (5) (kindly provided by Mike O’Donnell). Truncated EBNA1 deriv-atives were cloned into the pET3A vector (Novagen). The PCR was used to amplify DNA encoding EBNA1 from p220.2, a derivative of p201 (35). One primer used in the construction of each mutant was downstream of the EBNA1 stop codon and contained a BamHI site at its 59end. The other primers con-tained an NdeI site at the 59end and a sequence of nucleotides from within the EBNA1 open reading frame. The amplified DNA fragment was digested with NdeI and BamHI and cloned into these sites in pET3A. The ATG within the NdeI site encodes the initiating methionine, and only EBNA1 amino acids follow. The protein designations used include the number of amino acids deleted from the amino terminus of EBNA1; for example, ND360 has a methionine followed
by amino acid 361 to the COOH terminus (residue 641 of EBNA1). The internal deletions were introduced into the ND360 expression vector by using a site-directed mutagenesis kit (Clontech).
The GAL4 fusion proteins were all cloned into TP-3 (T7-based expression vector for GAL4 amino acids 1 to 94; kindly provided by Greg Peterson, Tularik, San Francisco, Calif.). Fragments of EBNA1 were amplified by PCR with prim-ers containing EcoRI and SacI sites at their 59ends. Cloning of these fragments into the EcoRI and SacI sites of TP-3 resulted in a fusion protein composed of GAL4 amino acids 1 to 94, Arg, Tyr, Pro, Gly, Glu, Phe, EBNA1 amino acids, Glu, and Leu. GAL4(1-94) is composed of the same amino acids without any EBNA1 residues. One exception is GAL4(1-94)&1-43, which is composed of GAL4 amino acids 1 to 94, Arg, Tyr, Pro, Gly, Glu, Phe, EBNA1 amino acids 1 to 43, Asp, Arg, Ala, Glu, and Leu. DNAs from all EBNA1 expression vectors were sequenced. Expression vectors for the following amino acid fragments of EBNA1 fused to GAL4 were sequenced: 390 to 457, 361 to 391, 372 to 391, 361 to 378, 331 to 378, 331 to 361, 1 to 330, 1 to 361, 1 to 43, 41 to 330, 54 to 330, 41 to 89, and 54 to 89. For all other fusions, multiple clones were tested. All sequencing was done by the Sanger method with the Sequenase kit.
Protein production.bEBNA1 was produced in SF21 cells as previously de-scribed (5). All other proteins were produced in BL21 bacteria carrying pLysS and containing a DE3 lysogen (33). Expression vectors were introduced into bacteria, and transformants carrying both plasmids were selected in the presence of 50mg each of chloramphenicol and ampicillin per ml. Bacteria were grown in Terrific Broth (Sigma) and induced with 1 mM isopropyl-b-D -thiogalactopyrano-side (IPTG) at an optical density at 600 nm of 1.0. Shaking at 378C was continued for 4 h. Bacteria were pelleted and washed with 50 mM Tris (pH 7.5). Pellets were resuspended in 1/30 of the original culture volume of 50 mM Tris (pH 7.5)–2.5 mM EDTA–1 mM phenylmethylsulfonyl fluoride–protease inhibitor cocktail (24). Chicken egg lysozyme was added to 250mg/ml, and the solution was incubated at room temperature for 10 min. One-sixteenth of a volume of 5 M NaCl was added, and the mixture was sonicated. These bacterial extracts were the starting material for protein purification. Preparation of crude extracts for use in gel shift assays was done as follows. Four milliliters of bacteria was grown at 378C until turbid. IPTG was then added to a 1 mM final concentration, and bacteria were agitated at 378C for an additional 4 h. Bacteria were pelleted and resuspended in 140ml of buffer containing 50 mM Tris (pH 7.5), 2.5 mM EDTA, and 200mg of chicken egg lysozyme per ml. After 10 min at room temperature, 10ml of 5 M NaCl was added and the mixture was sonicated. The lysate was then clarified by microcentrifugation at 12,000 rpm for 10 min. This supernatant was then used in EMSAs.
Protein purification.All purification was done in protein buffer (TEP) con-taining 50 mM Tris (pH 7.5), 2.5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and the NaCl concentrations indicated elsewhere in this report. bEBNA1 was purified as previously described (5), with minor modifications. EBNA1 derivatives produced in bacteria were purified in three steps. First, bacterial extracts, typically from 3 liters of culture, were clarified by centrifuga-tion at 10,000 rpm for 10 min in an SS-34 rotor (Sorvall) and passed over a 15-ml DEAE Sepharose Fast Flow (Pharmacia) column equilibrated in TEP at 0.3 M NaCl. Second, the flowthrough was loaded directly onto a 10-ml heparin-agarose (Sigma) column, also equilibrated in TEP at 0.3 M NaCl, eluted with a 200-ml gradient of 0.3 to 2.0 M NaCl, and collected in 6-ml fractions. Peak fractions, detected by UV absorbance, dot blot enzyme-linked immunosorbent assay (de-scribed below), or DNA filter binding (21), eluted at approximately 0.65 M NaCl. Dot blot enzyme-linked immunosorbent assay analysis was conducted as follows: fractions were serially diluted in TEP at 0.3 M NaCl, dilutions were spotted onto nitrocellulose with a dot blot apparatus (Schleicher & Schuell, Inc.), and EBNA1 was detected with rabbit polyclonal anti-EBNA1 antibodies by standard immu-noblotting techniques (32). Third, the peak fractions were dialyzed and/or di-luted to 0.3 M NaCl and loaded onto a 10-ml EBNA1 DNA affinity column equilibrated in TEP at 0.3 M NaCl. This resin was constructed by coupling HindIII-linearized p778, which contains two and one-half copies of FR, to Sepha-rose CL-4B (Pharmacia) in accordance with the manufacturer’s instructions. The resin contained 200 pmol of p778 per ml. After loading, the affinity resin was washed with 7 column volumes of buffer at 0.5 M NaCl. The EBNA1 was then eluted with buffer at 3.0 M NaCl. EBNA1 was concentrated and equilibrated to 0.3 M NaCl by desiccation in carboxymethyl cellulose (Aquacide IV; Calbio-chem) and dialysis, respectively. All EBNA1 derivatives were.90% pure as judged by Coomassie blue staining of polyacrylamide gels. The concentrations and DNA-binding activities of EBNA1 proteins were determined by Coomassie blue staining of polyacrylamide gels, bicinchoninic acid protein assay (Pierce), and DNA filter binding.
GAL4 and GAL4 fusions were purified by two methods. GAL4(1-94), GAL4(1-94)&361-378, and GAL4(1-94)&372-391 were purified as follows. First, bacterial extracts, typically from 3 liters of culture, were clarified by centrifuga-tion at 10,000 rpm for 10 min in an SS-34 rotor and passed over a 15-ml DEAE Sepharose Fast Flow column equilibrated in TEP at 0.3 M NaCl. Second, the flowthrough was diluted to 0.1 M NaCl in TEP and loaded onto a 10-ml SP Sepharose (Pharmacia) column equilibrated in TEP at 0.1 M NaCl. This resin was washed with four column volumes of TEP at 0.3 M NaCl. Protein was then eluted with TEP at 0.5 M NaCl and collected in 2-ml fractions. Peak fractions were detected by DNA filter binding. Third, the peak fractions were dialyzed and/or diluted to 0.1 M NaCl in TEP and loaded onto 1 ml of GAL4 DNA affinity
on November 9, 2019 by guest
http://jvi.asm.org/
resin equilibrated in TEP at 0.1 M NaCl. Protein was then eluted with 2 column volumes of TEP at 0.3 M NaCl. This resin was constructed by using PCR to amplify a DNA fragment with five GAL4-binding sites. One of the PCR primers had a biotin at the 59end, and the products of the PCR were coupled to streptavidin-agarose (Sigma) for 24 h at 48C in TEP at 1.0 M NaCl. The resin contained 500 pmol of DNA per ml. The concentrations and DNA-binding activities of GAL4 and GAL4 fusion proteins were estimated by using Coomassie blue-stained polyacrylamide gels and DNA filter binding.
GAL4(1-94)&331-361 and GAL4(1-94)&54-89 were purified as described above, through loading of SP Sepharose resin. Protein was then eluted from the SP Sepharose in a 200-ml gradient from 0.1 to 1.0 M NaCl in TEP and collected in 4-ml fractions. The fractions were assayed by DNA filter binding and by Coomassie blue staining of polyacrylamide gels. A fraction selected for maximum purity and binding activity was dialyzed to 0.1 M NaCl in TEP. This fraction was loaded onto 1 ml of heparin-agarose equilibrated to 0.1 M NaCl in TEP. The column was washed with 2 ml of TEP at 0.1 M NaCl and step eluted with 2 ml of TEP at 0.3, 0.5, 0.75, and 1.0 M NaCl. Fractions (400ml) were collected and assayed by DNA filter binding activity and Coomassie blue staining of polyacryl-amide gels. Gal4(1-94)&54-89 eluted with 0.5 M NaCl, and GAL4(1-94)&331-361 eluted with 0.75 M NaCl. Peak fractions were pooled and dialyzed to a final NaCl concentration of 0.3 M in TEP.
Linking assays.Assays for the retention of labeled DS by immobilized FR were done as previously described (21). The enhanced ligation assay was con-ducted essentially as previously described (8), with the following changes: the final NaCl concentration was 0.05 rather than 0.15 M, and reactions were incu-bated with 0.05 Weiss unit of T4 DNA ligase (Boehringer Mannheim) for 4 min at room temperature prior to termination. Gels were imaged with the IS-1000 digital imaging system (Alpha Innotech Corporation), and the data were quan-tified with IPLab Gel software (Signal Analytics). Linking EMSAs were done in a buffer containing 20 mM N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid (HEPES; pH 7.6), 2 mM EDTA, 10% glycerol, 0.1% Nonidet P-40, and 0.3 M NaCl. The probe DNA and 5mg of poly(dI)zpoly(dC) (Pharmacia) were simul-taneously added to the protein. The reactions were incubated at room temper-ature for 30 min in a volume of 23ml (29ml for Fig. 7). Reactions were then separated by electrophoresis at 15 to 25 mA through a 4% polyacrylamide gel (or at 10 mA through 1% agarose). Gels were dried on 3MM chromatography paper (Whatman) at 808C for 1 h and then exposed to film or PhosphorImager screens. The percentage of probe DNA linked was determined by dividing the amount of linked probe (indicated by the bracket in each figure) by the total amount of probe in that lane. Background values of linked and nonlinked DNAs from control lanes were subtracted before this calculation was made. For example, the amount of DNA in the well in a no-protein lane was subtracted from the amount of DNA in the well for all of the other lanes in which that DNA was used as a probe.
RESULTS
Establishment of the EMSA for measurement of DNA link-ing.Electrophoresis of purified bEBNA1 bound to a 131-bp DNA with two EBNA1-binding sites through a 4% polyacryl-amide gel resulted in retention of DNA in the wells. We pos-tulated that this retention resulted from incorporation of the DNA into a lattice. EBNA1 has been shown via electron mi-croscopy to incorporate DNA into lattices (3, 8, 34). The size of the complex retained in the wells was investigated by con-ducting EMSAs with 1% agarose slab gels. When bound by bEBNA1, 5% of a DNA with two binding sites migrated more slowly than did a 20-kbp linear DNA marker in a parallel lane of the agarose gel (data not shown).
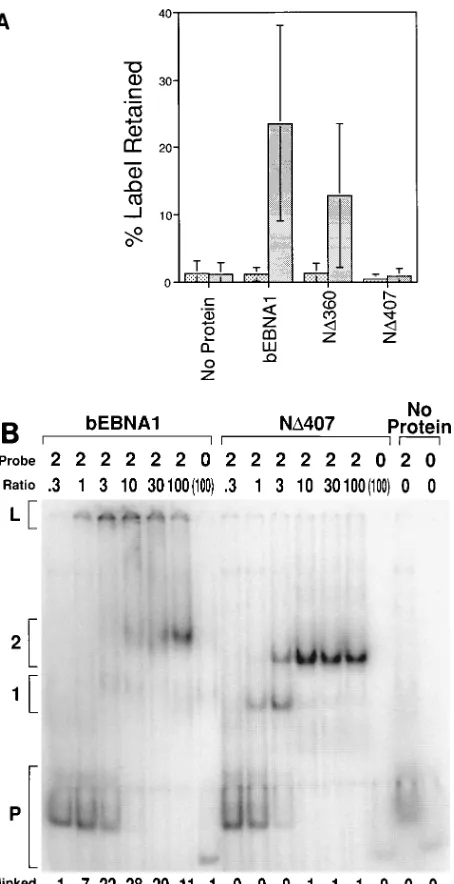
To test whether retention of DNA in the wells was a result of the previously identified linking activity of EBNA1, we com-pared derivatives of EBNA1 for the ability to link DNAs in an EMSA and in a previously characterized linking assay (21). The latter assay measures the ability of EBNA1 to associate a labeled DNA containing the DS with a DNA containing the FR which is covalently bound to a matrix. bEBNA1 linked DNA in both assays (Fig. 1A and B). An EBNA1 derivative with amino acids 1 to 360 deleted (ND360; Fig. 2A) linked DNA in both assays (Fig. 1A and 2A), but an EBNA1 deriv-ative with amino acids 1 to 407 deleted (ND407; Fig. 2A) failed to link DNA in either assay (Fig. 1A and B). These two assays identified the same 47 amino acids of EBNA1 as being neces-sary to link DNAs to which it binds. The EMSA also indicated that high concentrations of bEBNA1 can compete for the
formation of linked complexes (Fig. 1B, lane 6). This observa-tion is more apparent in Fig. 6.
Mapping of a minimal linking domain within EBNA1.To confirm previous mapping and to allow further mapping of the amino-terminal limit of a minimal linking domain within EBNA1, a series of truncated EBNA1 derivatives was gener-ated (Fig. 2A). These proteins were produced in bacteria and, after purification, were estimated to be greater than 90% ho-mogeneous. All of these proteins retain the ability to dimerize and bind site specifically to DNA. bEBNA1 and the series of truncated EBNA1s were tested for the ability to link DNA molecules with two EBNA1-binding sites (Fig. 2A). ND360 and larger derivatives of EBNA1 linked DNA (.13% of probe retained in the well, except ND330, for which we isolated an insufficient amount of protein to measure accurately). ND371 reproducibly linked;50% of the DNA that ND360 did. ND389 and smaller derivatives of EBNA1 consistently linked less than 3% of this DNA.
To determine whether the apparent intermediate linking activity of ND371 was reproducible, we conducted EMSAs with a DNA containing 10 (one-half of FR) EBNA1-binding sites. The increased signal achieved with this DNA allowed interme-diate linking activity to be observed clearly. The ability of ND360, ND371, and ND389 to link a DNA with 10 binding sites was measured (Fig. 2B). ND360 linked .83% of this DNA over a 10-fold range of concentrations (ratio of 3 to 30 dimers per binding site) and maximally linked 93% of the DNA (Fig. 2C). The percentage of DNA with 10 binding sites linked by ND371 peaked at 47%, approximately half of that linked by ND360, confirming that the ability of ND371 to link DNA is reduced relative to that of ND360 (Fig. 2C). Retention of this DNA in the wells by ND389 increased to 18% at the highest concentration tested (Fig. 2C) and may have represented in-creased nonspecific retention of protein, and bound DNA, in the wells. The decreased linking activity of ND371 relative to that of ND360 was also apparent when a DNA with four EBNA1-binding sites was tested (data not shown). The inter-mediate activity of ND371 also was observed with an indepen-dent preparation of ND371 purified in a different manner (data not shown).
To determine whether amino acids within the dimerization and DNA-binding domain of EBNA1 also contribute to DNA linking by EBNA1, we constructed a fusion protein [GAL4(1-94)&331-488] with the dimerization and DNA-binding domain of yeast transactivator GAL4 (20) and amino acids 331 to 488 of EBNA1 (Fig. 3A). This fusion protein, in a crude bacterial extract, linked DNAs with five GAL4-binding sites. The GAL4(1-94) protein extract did not link DNA, although it bound DNA, as expected. This observation demonstrated that a linking domain within EBNA1 can act independently of the dimerization and DNA-binding domain of EBNA1.
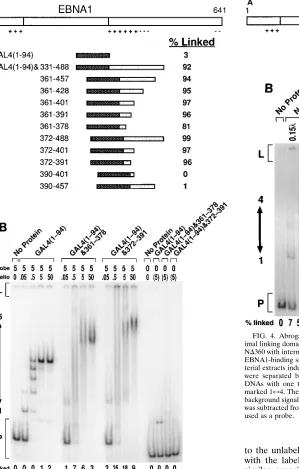
A series of GAL4-EBNA1 fusion proteins was constructed (Fig. 3A). Each protein in the entire series was tested, in crude bacterial extracts, for the ability to link a DNA with five GAL4-binding sites (Fig. 3A). The amino-terminal limit of a minimal linking domain identified with the fusion proteins in Fig. 3A agreed with the results obtained with truncations of EBNA1 as shown in Fig. 2. EBNA1 amino acids 331 to 371 were dispens-able for linking by this set of fusion proteins. When amino acids 372 to 389 were deleted from these fusion proteins, linking activity was lost (Fig. 3A). Importantly, this failure to link was observed even when a large fragment of EBNA1 (amino acids 390 to 457) was fused to GAL4. These fusions allowed mapping of the carboxy-terminal limit of this minimal linking domain. The carboxy-terminal limit of the fragment of EBNA1 fused to GAL4 was reduced from amino acids 488 to
on November 9, 2019 by guest
http://jvi.asm.org/
391 with no apparent loss of linking activity (Fig. 3A). Further carboxy-terminal deletion to amino acid 378 [GAL4(1-94)&361-378] resulted in a protein with reduced activity rela-tive to the other acrela-tive fusions. EBNA1 amino acids 372 to 391 are sufficient to link DNA when fused to the dimerization and DNA-binding domain of GAL4.
To determine whether other factors in the bacterial extract affected the linking activity of the GAL4 fusion proteins and to verify that the linking activity of GAL4(1-94)&361-378 was intermediate, three of the proteins were purified. GAL4(1-94) and GAL4(1-94)&372-391 were purified to.90% homogene-ity, and GAL4(1-94)&361-378 was purified to;50% homoge-neity. The ability of these purified proteins to link DNA was then tested (Fig. 3B). The maximum percentage of DNA linked by GAL4(1-94)&372-391 and GAL4(1-94)&361-378 was reduced with each step of the purification, but the relative linking activity of the two remained constant. Purified GAL4(1-94)&372-391 was able to link 25% of the DNA, com-pared with .90% in a crude extract. Purified GAL4(1-94)&361-378 linked 7% of the DNA, compared with 81% in a crude extract. GAL4(1-94) linked 2% of the DNA. Either factors in the extract affected the magnitude of DNA linked by the fusion proteins or the DNA-linking activity was unstable during purification. However, purification did not qualitatively affect the activities of these proteins relative to one another.
Deletion of residues within the minimal linking domain.To test whether deletion of this minimal linking domain within EBNA1 would abrogate DNA linking by EBNA1, deletions were generated within ND360 (Fig. 4A). These proteins, in crude bacterial extracts, were tested for the ability to link DNA with four EBNA1-binding sites (Fig. 4B). Deletion of amino acids 372 to 378 (ND360&372D378) reduced the amount of DNA linked by ND360 from 72 to 40%. Deletion of amino acids 372 to 391 (ND360&372D391) reduced linking by ND360 nearly to background levels (6%). The behavior of these de-letions does not bear on the existence of other regions with linking activity within amino acids 1 to 360 of EBNA1. It does show that amino acids within this minimal domain are neces-sary for linking by ND360.
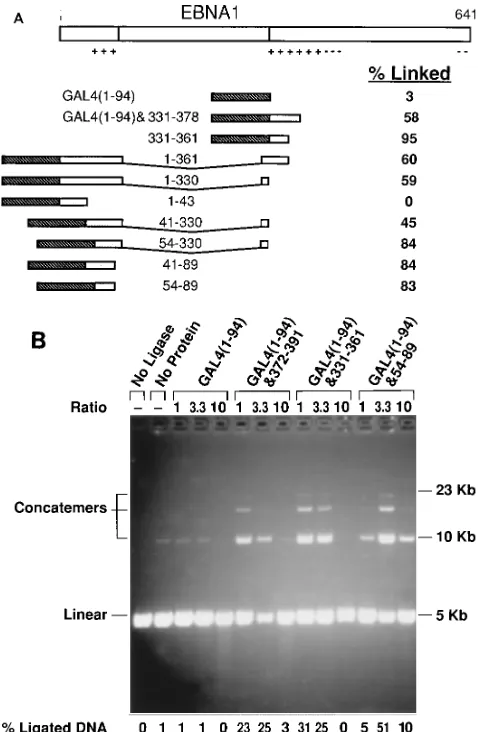
Mapping of other regions of EBNA1 which have linking activity. To test whether amino acids 1 to 360 of EBNA1 contain additional regions with linking activity, another set of GAL4 fusion proteins was generated (Fig. 5A). For fusions in which the fragment of EBNA1 spans the Gly-Gly-Ala repeats, that fragment of EBNA1 was cloned from p205 (40). p205 encodes an EBNA1 molecule of which all but 15 amino acids of the Gly-Gly-Ala repeats have been deleted but which retains its replicational and transcriptional activity. These proteins, in crude extracts, were tested for the ability to link DNAs con-taining five GAL4-binding sites. Two additional regions of EBNA1 which have linking activity were identified, amino ac-ids 331 to 361 and 54 to 89. Fusion proteins containing amino acids 331 to 361 and 54 to 89 had linking activity comparable to that obtained with amino acids 372 to 391 when each was tested in purified form (.90% homogeneity) (data not shown). Examination of the three regions of EBNA1 with linking ac-tivity (amino acids 372 to 391, 331 to 361, and 54 to 89) showed that the amino acid sequences are not similar. Each region is, however, rich in glycines and basic residues. Our experimental findings do not preclude amino acids 41 to 53 or 90 to 330 of EBNA1 from also having linking activity.
[image:4.612.69.296.73.515.2]An enhanced ligation assay was used to demonstrate that, in an independent assay, each of these three regions of EBNA1, when fused to GAL4, can link DNA. This assay measures the ability of a protein to link DNAs as an increase in ligation of those DNAs. A GAL4 fusion protein which can link DNA FIG. 1. Correlation of EMSA with a previously described linking assay. The
ability of purified bEBNA1 and purified EBNA1 derivatives produced in bacteria to link DNA in two independent assays was tested. (A) DNA linking by EBNA1 derivatives. A 20-fmol sample of radiolabeled DNA containing either four (DS) or zero EBNA1-binding sites was incubated with 1,000 fmol of the indicated EBNA1 derivative (described in Fig. 2A). Agarose beads with DNA containing FR covalently bound were then added to the reaction and incubated. The beads were washed, and the radioactivity in both the beads and the supernatant was counted. The percentage of the label retained represents the percentage of counts linked to the beads of the total counts in the reaction. The average and standard deviation from three different experiments are shown. (B) A 20-fmol sample of DNA containing either two or zero EBNA1-binding sites was incubated with bEBNA1 or ND407. The amount of protein included in each reaction is indicated as a ratio and is the number of protein dimers per binding site on the DNA. In lanes where the ratio is in parentheses, the amount of protein included in the reaction was 4,000 fmol of dimers. Reactions were separated by electrophoresis through a 4% polyacrylamide gel. The positions of the unbound probe (P), the probe with one and two binding sites occupied (1 and 2, respectively), and the linked probe (L) are indicated. PhosphorImager analysis was used to determine the percentage of linked probe of the total probe in each reaction, and the result is shown beneath each lane. The background signal measured with no protein added in the region marked linked was subtracted from the signal for that region in all lanes in which that DNA was used as a probe. The difference in mobility between 1 and 2 with bEBNA1 and ND407 is a result of the difference between the sizes of the proteins.1, zero binding sites;u, four binding sites (DS).
on November 9, 2019 by guest
http://jvi.asm.org/
increases the local concentration of DNA ends, thus increasing the efficiency with which they are ligated. The linear DNA used in these experiments contains five GAL4-binding sites near each end of the molecule. The GAL4 dimerization and DNA-binding domain alone [GAL4(1-94)] did not enhance ligation of the DNA to which it binds (Fig. 5B). When GAL4 was fused to any of the three regions of EBNA1 determined to have linking activity, the ligation of the DNA was enhanced signif-icantly (Fig. 5B). Thus, amino acids 54 to 89, 331 to 361, and 372 to 391 of EBNA1 displayed linking activity in two inde-pendent assays (EMSA and enhanced ligation).
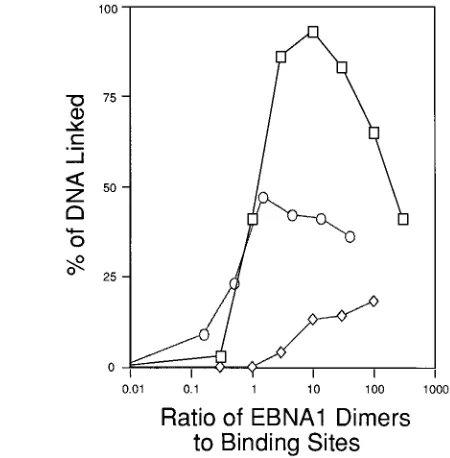
Linking of DNAs with different numbers of EBNA1-binding sites.DNAs with 1, 2, 4 (DS), and 10 (one-half of FR) binding sites were used to test the relationship between the number of EBNA1-binding sites on a DNA and the ability of EBNA1 to link that DNA. These DNAs were incubated with both bEBNA1 (Fig. 6A) and ND360 (Fig. 6B) over a wide range of protein concentrations. Products of these reactions were re-solved by EMSA, and PhosphorImager analysis was used to determine the percentage of DNA linked in each reaction. The compiled results of these experiments are shown in Fig. 6. Both bEBNA1 and ND360 compete with formation of linked com-plexes when their concentrations are increased beyond that needed to occupy their specific binding sites on the DNA (Fig.
6 and data not shown). The profiles observed are similar for bEBNA1 and for ND360, with the exception that bEBNA1 more efficiently linked DNAs with two binding sites. bEBNA1 maximally linked an average of 21% of a DNA with two bind-ing sites. ND360 maximally linked an average of 13% of a DNA with two binding sites. This difference may result from the contributions of the sum of the regions with DNA-linking activity that are present in bEBNA1, only one of which is present in ND360.
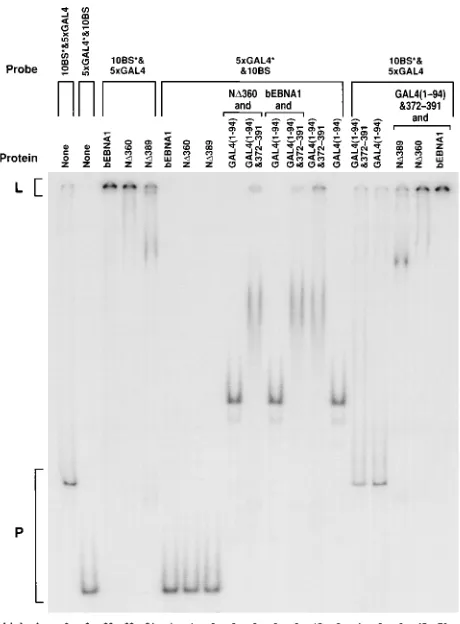
[image:5.612.331.556.71.300.2]Determination of the specificity of DNA linking.We exam-ined the specificity of DNA linking by EBNA1 and GAL4-EBNA1 fusions. The abilities of linked complexes to incorpo-rate naked DNA, DNA bound by nonlinking proteins, and DNA bound by unlike linking proteins were tested (Fig. 7). Proteins were combined, and equimolar amounts of two DNAs were added, one with 10 EBNA1-binding sites and one with 5 GAL4-binding sites. In each reaction, one of the DNAs was labeled and the other was not. bEBNA1 and ND360 bound to and efficiently linked the DNA with EBNA1-binding sites. ND389 bound to this DNA but did not link it, although the amount of probe in the well in this experiment was aberrantly high (contrast with Fig. 2B; the dimer/binding site ratio in this experiment was 15). None of these three proteins, when bound FIG. 2. Mapping of the amino-terminal limit of a minimal linking domain within EBNA1. (A) Schematic representation of the EBNA1 derivatives purified from insect cells (bEBNA1) and bacteria (ND330 through ND407). Full-length EBNA1 is depicted at the top. The plus and minus signs represent clustered basic and acidic residues, respectively. Amino acids previously shown to be important for dimerization and DNA binding by EBNA1 are represented by the striped box. The residues present in the derivatives are represented by shaded boxes. bEBNA1 lacks the first 7 amino acids and all but 15 amino acids of the Gly-Gly-Ala repeats. Each percent linked value is the percentage of a 20-fmol sample of DNA with two binding sites linked by purified EBNA1 at a ratio of three dimers per binding site. (B) A 15-fmol sample of DNA containing 10 binding sites for EBNA1 was incubated with ND360, ND371, or ND389 at the dimer/binding-site ratio indicated. Reactions were separated by electrophoresis through a 4% poly-acrylamide gel. Probe DNAs with 1 through 10 binding sites occupied migrated to the region marked 1710. The percentage of linked probe is indicated below each lane. The background signal measured with no protein added in the region marked linked was subtracted from the signal for that region in all lanes in which that DNA was used as a probe. (C) Linking of a DNA with 10 binding sites. A graphic representation of the data in panel B is shown. The percentage of linked DNA was plotted against the protein dimer/binding-site ratio. Symbols: h, ND360;E, ND371;{, ND389.
on November 9, 2019 by guest
http://jvi.asm.org/
to the unlabeled DNA with EBNA1-binding sites, interacted with the labeled DNA with GAL4-binding sites. Also, in a similar experiment in which no poly(dI)zpoly(dC) was in-cluded, ND360 did not incorporate DNA lacking EBNA1-binding sites into the linked complex (data not shown). Neither ND360 nor bEBNA1, when in linked complexes with the un-labeled DNA with EBNA1-binding sites, interacted with GAL4(1-94) bound to the DNA with GAL4-binding sites. Also, neither ND360 nor bEBNA1, when in linked complexes with the unlabeled DNA with EBNA1-binding sites, incorpo-rated GAL4(1-94)&372-391 bound to the labeled DNA with GAL4-binding sites into those linked complexes. Rather, ND360 and, to a greater extent, bEBNA1 inhibited linking of the labeled DNA with GAL4-binding sites by GAL4(1-94)&372-391. The ability of bEBNA1 and ND360 to inhibit linking by GAL4 fusion proteins was dose dependent, and ND389 failed to inhibit this linking (data not shown). GAL4(1-94) bound to, and GAL4(1-GAL4(1-94)&372-391 bound to and linked, the labeled DNA with GAL4-binding sites. Neither of these proteins, when bound to the unlabeled DNA with GAL4-bind-ing sites, interacted with the labeled DNA with EBNA1-bind-FIG. 3. A minimal linking domain within EBNA1 acts independently of
[image:6.612.105.548.68.459.2]other regions of EBNA1. (A) Schematic representation of GAL4-EBNA1 fusion proteins constructed. The fragments of EBNA1 align with the full-length EBNA1 structure shown at the top and are represented by shaded boxes. The striped boxes represent the dimerization and DNA-binding domain of GAL4. Each percent linked value is the percentage of a 20-fmol sample of a DNA with five GAL4-binding sites linked by 15ml of a crude extract of E. coli induced to express the fusion protein indicated. Serial dilutions of each extract were tested by EMSA (the same experiments in which the percentage of DNA linked was determined), and the disappearance of unbound DNA indicates that all extracts contained comparable amounts of binding activity (data not shown). (B) A 20-fmol sample of a DNA containing five GAL4-binding sites was incubated with purified GAL4(1-94), GAL4(1-94)&361-378, or GAL4(1-94)&372-391. The amount of protein included in each reaction is indicated in the row marked ratio and is the number of protein dimers per binding site on the DNA. In lanes where the ratio is in parentheses, the amount of protein included in the reaction was 500 fmol of dimers. Reactions were separated by electrophoresis through a 4% polyacrylamide gel. Probe DNAs with one through five binding sites occupied migrated to the region marked 175. The percentage of linked probe is indicated below each lane. The background signal measured with no protein added in the region marked linked was subtracted from the signal for that region in all lanes in which that DNA was used as a probe.
FIG. 4. Abrogation of ND360-mediated DNA linking by deletion of the min-imal linking domain. (A) Schematic representation of ND360 and derivatives of ND360 with internal deletions. (B) Samples (20 fmol) of a DNA containing four EBNA1-binding sites (DS) were incubated with various amounts of crude bac-terial extracts induced to express the derivatives of ND360 indicated. Reactions were separated by electrophoresis through a 4% polyacrylamide gel. Probe DNAs with one through four binding sites occupied migrated to the region marked 174. The percentage of linked probe is indicated below each lane. The background signal measured with no protein added in the region marked linked was subtracted from the signal for that region in all lanes in which that DNA was used as a probe.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.71.370.74.543.2]ing sites. Also, GAL4(1-94)&372-391, when bound to and link-ing unlabeled DNA with GAL4-bindlink-ing sites, did not affect the interactions between the labeled DNA with EBNA1-binding sites and ND389, ND360, or bEBNA1. GAL4(1-94)&54-89 and GAL4(1-94)&331-361 can each inhibit linking by ND360 in a dose-dependent manner, approximately 10 times more effi-ciently than GAL4(1-94) (data not shown). Proteins which link DNA, when linking DNA to which they bind, do not interact
with DNA to which they do not bind. We have been unable to detect linking between derivatives of EBNA1 which can link DNAs with EBNA1-binding sites and GAL4-EBNA1 fusions which can link DNAs with GAL4-binding sites; however, each has been shown to compete with linking mediated by the other (Fig. 7 and data not shown).
DISCUSSION
We have used an EMSA to measure the ability of EBNA1 and its derivatives to link DNA into large lattices. This assay measures the same activity as do two previously characterized DNA-linking assays and is rapid and versatile (Fig. 1 and 5). Amino-terminal truncations of EBNA1 and fusions of frag-ments of EBNA1 with the GAL4 dimerization and DNA-binding domain were generated and used to map a minimal, independently acting DNA-linking domain within EBNA1. Deletion of amino acids 1 to 360 in EBNA1 did not abrogate its ability to link DNA. Deletion of amino acids 1 to 371 in EBNA1 reduced its linking activity, while deletion of amino acids 1 to 389 abolished its linking activity (Fig. 2). A fusion protein containing EBNA1 amino acids 372 to 391 and the dimerization and DNA-binding domain of GAL4 was able to link DNA with binding sites for GAL4 (Fig. 3). These results identified a minimal, independently acting linking domain within amino acids 372 to 391 of EBNA1.
To demonstrate the necessity of this minimal domain, we generated deletions in ND360. Deletion of amino acids 372 to 378 (ND360&372D378) slightly reduced linking activity, while deletion of amino acids 372 to 391 (ND360&372D391) almost entirely eliminated linking activity (Fig. 4). These results es-tablished that a DNA-linking domain within ND360 requires amino acids 372 to 391.
bEBNA1 and ND360 differed in the ability to link a DNA with two binding sites (Fig. 6). This observation indicated that amino acids 1 to 360 of EBNA1 may contain additional regions with linking activity. GAL4 fusion proteins showed that at least three regions within EBNA1 contain linking activity. Amino acids 54 to 89, amino acids 331 to 361, and amino acids 372 to 391 of EBNA1 contain independently acting DNA-linking ac-tivity when fused to the DNA-binding domain of GAL4 (Fig. 3 and 5). These three regions, and others, may act in concert when wild-type EBNA1 links DNAs. These results explain previous electron microscopic observations which showed that sequential amino-terminal truncations of EBNA1 incremen-tally decrease the ability of these EBNA1 derivatives to form loops between FR and DS (8). As amino-terminal deletions removed more of the regions with linking activity, the number of looped structures observed declined. The fact that only slight differences were observed between bEBNA1 and ND360 in this study probably reflects differences in the assays. DNA linking by intact EBNA1 may result from the combinatorial activity of the three regions identified.
The ability of EBNA1 to bind to RNA has recently been noted, and some regions important for RNA binding overlap with some, but not all, of the regions containing linking activity (31). Because EBNA1 amino acids 54 to 89 do not overlap with regions shown to have RNA-binding activity, RNA binding does not contribute to DNA linking by this region of EBNA1. Therefore, since the three regions of EBNA1 are similar in DNA-linking activity, RNA binding likely does not contribute to DNA linking by the other two regions of EBNA1. Experi-ments demonstrated that RNA is not necessary for DNA link-ing (data not shown).
[image:7.612.59.298.70.436.2]The efficiency with which EBNA1 links DNAs as a function of the number of sites to which it binds was measured. We FIG. 5. Two independent assays demonstrating that multiple regions with
independently acting DNA-linking activity exist within EBNA1. (A) Schematic representation of GAL4-EBNA1 fusion proteins constructed. Symbols are the same as in Fig. 3. Each percent linked value is the percentage of a 20-fmol sample of a DNA with five GAL4-binding sites linked by 15ml of a crude extract of E. coli induced to express the fusion protein indicated. Serial dilutions of each extract were tested by EMSA (the same experiments in which the percentage of linked DNA was determined), and the disappearance of unbound DNA indicates that all extracts contained comparable amounts of binding activity (data not shown). (B) Fusions between Gal4 and EBNA1 which can link DNA also en-hance the ligation of DNAs to which they bind. A 60-fmol sample of a linear DNA with five GAL4-binding sites 380 bp from one end and five GAL4-binding sites 393 bp from the other end was incubated with the purified GAL4 dimer/ binding-site ratio indicated (0.6, 2, or 6 pmol of each protein). The reaction was then incubated at room temperature for 4 min with 0.05 Weiss unit of T4 DNA ligase. Reaction products were separated with a 0.7% agarose gel in the presence of 0.4 mg of ethidium bromide per ml. The reaction with no ligase also contained no protein and shows the initial state of the DNA. The reaction with no protein shows the background level of ligation. Positions to which concatemers of two, three, and four unit lengths migrated are indicated. The percentage of ligated DNA is the percentage of DNA in concatemeric form of the total DNA in the lane.
on November 9, 2019 by guest
http://jvi.asm.org/
tested DNAs with 1, 2, 4 (DS), and 10 (one-half of FR) binding sites for EBNA1. The ability of bEBNA1 and ND360 to link these DNAs increased with increasing numbers of binding sites. As the concentration of bEBNA1 or ND360 increased, the amount of DNA linked first increased and then decreased (Fig. 6). The decrease is dependent upon EBNA1, which is not bound to DNA site specifically; it occurs as the concentration of EBNA1 increases beyond that needed to occupy its specific binding sites. This pool of excess EBNA1, either free or bound to poly(dI)zpoly(dC), competes for formation of the linked complex. The percentage of DNA linked by GAL4 fusion pro-teins also decreased with addition of excess protein (Fig. 3B
and 5 and data not shown). Similarly, excess EBNA1 competed with linking by GAL4 fusions and vice versa (data not shown). Both bEBNA1 and ND360 linked DNAs containing one EBNA1-binding site (Fig. 6). DNAs with one binding site linked by ND360 were unable to enter a 1% agarose gel (data not shown), indicating that these structures were in the form of a large lattice. For EBNA1 to incorporate DNAs with one binding site into a lattice, it must have a divalent linking ac-tivity. A divalent linking activity could be achieved in either of two ways. Each half of the EBNA1 dimer could contain an independent linking domain, or the two halves of the dimer together could form a domain. If the two halves of the dimer were to act together, two independent domains would still have to be formed. The small region with linking activity within ND360 makes it unlikely that the halves of the dimer can interact to form two independent domains. Therefore, we think it likely that ND360 dimers bound to DNA present two independent linking domains, each contained within one monomer. A similar mechanism has been suggested for the E2 protein of bovine papillomavirus (15).
Each of the EBNA1 fragments found to contain linking activity is rich in glycines and basic residues. If linking occurs via protein-protein interactions between regions rich in basic residues, these interactions create an accumulation of positive charges. The ability of EBNA1 dimers to associate detectably with one another requires that they first be bound to DNA. EBNA1 not bound to DNA exists in solution as a dimer (5). Detectable linking between EBNA1 molecules may occur only when they are bound to DNA because the negatively charged backbone of the DNA is a counterion necessary to stabilize such an accumulation of positive charges. Another hypothesis is that the positively charged linking regions interact directly with DNA. The observation that ND360 can link a 20-bp DNA containing an EBNA1-binding site (data not shown) makes the latter idea unlikely; little DNA will be free of EBNA1 and accessible to a second moiety of EBNA1. A third hypothesis is that upon binding to DNA, the conformation of EBNA1 changes in a way that activates linking. The observation that many fragments of EBNA1 are active when fused to a different DNA-binding domain (Fig. 3 and 5) makes such structural requirements unlikely.
We do favor a model for the mechanism by which EBNA1 links DNA which is consistent with our and others’ findings, including observations indicating that the presence of FR can stabilize binding of EBNA1 to DS (3, 5, 34). In this model, we propose that dimers of EBNA1 associate whether or not they are bound to DNA. The affinity between dimers not bound to DNA is low, such that complexes have not been detected in sedimentation analyses (5). Complexes of dimers of EBNA1 bound to DNA have been detected by a variety of assays (4, 8, 21, 34; this study), perhaps because of charge delocalization provided by the DNA. Direct measurements of interactions between one dimer bound to DNA and another dimer not bound to DNA have not been made. This study, however, may measure such interactions indirectly. DNA linking becomes inhibited as the ratio of EBNA1 dimers to binding sites creases (Fig. 6). As the concentration of EBNA1 dimers in-creases, the concentration of dimers bound site specifically to DNA asymptotically approaches a constant (because the amount of DNA is constant), while the concentration of dimers not bound site specifically to DNA increases almost linearly at higher concentrations. As the concentration of EBNA1 increases and the site(s) on the DNA become filled, high-affinity interactions between EBNA1 dimers bound to DNA predominate and lattices are formed. As the concentra-tion of EBNA1 is increased further, the concentraconcentra-tion of FIG. 6. Ability of EBNA1 to link DNAs with various numbers of binding
sites for EBNA1. EMSAs were conducted with bEBNA1 (A) and ND360 (B) and 20-fmol samples of DNAs containing 1, 2, and 4 (DS) binding sites and 15-fmol samples of DNA containing 10 (one-half of FR) binding sites. In the graphs, the percentage of DNA linked is plotted against the EBNA1 dimer/binding-site ratio. One to four experiments were conducted for each curve. Standard devia-tions are shown when two or more experiments were conducted. The same data were also plotted against the protein concentration. The minimal concentrations of protein required to maximally link the DNAs with 1, 2, and 10 binding sites were nearly identical. This concentration for bEBNA1 was between 1.731028 and 1.931028
M, and for ND360 it was between 0.931028
and 1.931028 M. Both proteins required a higher minimal concentration (0.7 31027
M) to maximally link the DNA containing four binding sites (DS). The binding sites in DS, however, are lower affinity than those in the other probe DNAs (12). Symbols:Ç, 10 BS;E, 4 BS;{, 2 BS;h, 1 BS.
on November 9, 2019 by guest
http://jvi.asm.org/
dimers bound site specifically to DNA remains essentially con-stant, while the concentration of dimers not bound site specif-ically to DNA increases. Thus, mass action dictates that inter-actions between dimers not bound to DNA and those bound to DNA will begin to displace the interactions between pairs of dimers both of which are bound to DNA, thus inhibiting link-ing. The concentration of dimers not bound to DNA needed to inhibit linking of a DNA with one binding site reflects the difference in the affinity of the association between two dimers bound to DNA and of the association between one dimer bound to DNA and one dimer not bound to DNA. Figure 6 shows that inhibition of linking of a DNA with one binding site occurs over a 10-fold or greater increase in the concentration of unbound dimers, suggesting that the affinity of interaction between dimers one of which is bound to DNA is less than that between dimers both of which are bound to DNA. This obser-vation would be expected if the negatively charged DNA
served to stabilize interactions between the positively charged linking regions.
This model may explain how EBNA1, upon binding to FR, stabilizes binding to DS (3, 5, 34). EBNA1 binds to the sites in FR with an approximately eightfold higher affinity than to the sites in DS (12). If the affinity with which dimers free in solu-tion associate with EBNA1 dimers bound to the 20 sites in FR is greater than, or approximates, the affinity with which they associate with the 4 sites in DS, the resulting increase in the local concentration will facilitate binding of EBNA1 to DS. Association between EBNA1 bound to FR and EBNA1 bound to DS also could serve to prevent diffusion of EBNA1 dimers which dissociate from their sites in DS, thereby increasing the likelihood that they reassociate. In these ways, FR could serve to facilitate and maintain occupancy of DS by EBNA1.
It is likely that the ability of EBNA1 to link DNA, that is, the activity which allows EBNA1 bound to DNA to associate with an identical protein-DNA complex, contributes to phenotypes dependent upon EBNA1. When bound to oriP in vivo, EBNA1 presumably associates FR and DS. This association appears to increase the apparent affinity of EBNA1 for DS and may con-tribute in other ways to initiation of replication at oriP. Also, EBNA1’s ability to link DNA may contribute to maintenance and segregation of plasmid DNAs with EBNA1-binding sites. Cellular factors which can bind to oriP in vitro have been identified (41). By analogy, EBNA1 may bind in vivo to the endogenous binding sites for these proteins. Through DNA linking, EBNA1 bound to cellular DNA could tether plasmid DNAs containing oriP to chromosomes. Association with chro-mosomes could increase the maintenance or efficiency of seg-regation of these DNAs.
We have studied three regions with linking activity from within EBNA1 fused to two dimeric DNA-binding moieties, EBNA1 and GAL4. We have only demonstrated DNA linking between like proteins. The proteins tested have been demon-strated to compete with linking mediated by unlike proteins. The scope with which we have been able to test DNA linking between EBNA1 and an unlike protein is narrow, three frag-ments of EBNA1 fused to one dimerization and DNA-binding domain. By no means do these experiments rule out the pos-sibility that regions of EBNA1 which link DNA can interact with other proteins. Were this true, such interactions could contribute to EBNA1’s ability to activate transcription, repli-cation, and retention of DNA within cells.
ACKNOWLEDGMENTS
We thank Greg Peterson and Vijay Baichwal for providing the TP-3 vector and Mike O’Donnell for providing the bEBNA1-expressing baculovirus. We thank Mark Sandberg and Curtis Tyree for initial characterization of the EMSA. We appreciate critical comments on the manuscript from Dan Loeb and Paul Lambert.
This work was supported by grants CA07175 and CA22443 from NCI and grant GM07215 from NIGMS.
REFERENCES
1. Adams, A. 1987. Replication of latent Epstein-Barr virus genomes in Raji cells. J. Virol. 61:1743–1746.
2. Ambinder, R. F., M. A. Mullen, Y. N. Chang, G. S. Hayward, and S. D.
Hayward.1991. Functional domains of Epstein-Barr virus nuclear antigen EBNA-1. J. Virol. 65:1466–1478.
3. Frappier, L., K. Goldsmith, and L. Bendell. 1994. Stabilization of the EBNA1 protein on the Epstein-Barr virus latent origin of DNA replication by a DNA looping mechanism. J. Biol. Chem. 269:1057–1062.
4. Frappier, L., and M. O’Donnell. 1991. Epstein-Barr nuclear antigen 1 me-diates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 88:10875–10879.
[image:9.612.63.293.69.381.2]5. Frappier, L., and M. O’Donnell. 1991. Overproduction, purification, and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J. Biol. Chem. 266:7819–7826.
FIG. 7. Derivatives of EBNA1 and GAL4 fusions which can link DNA link only DNAs bound by themselves. Samples (1.5 pmol) of the protein(s) indicated were aliquotted. To these reactions 10 fmol of a DNA with 10 (one-half of FR) EBNA1-binding sites and 10 fmol of a DNA with 5 GAL4-binding sites were added simultaneously. In each reaction only one DNA was labeled. The DNA which was labeled is indicated by the asterisk. Reactions were separated by electrophoresis through a 4% polyacrylamide gel. The percentage of linked probe DNA is indicated below each lane. The background signal measured with no protein added in the region marked linked was subtracted from the signal for that region in all lanes in which that DNA was used as a probe. In the third and fourth lanes from the left, bEBNA1 and ND360 linked 80 and 62% of the labeled DNA with 10 binding sites for EBNA1, respectively. However, in the sixth and seventh lanes from the left, bEBNA1 and ND360, which linked the unlabeled DNA with binding sites for EBNA1, did not affect the mobility of the labeled DNA with five binding sites for GAL4. Similarly, bEBNA1 and ND360 did not affect the mobility of labeled DNA bound by GAL4 with nothing fused to it. bEBNA1 and ND360 each competed with linking of labeled DNA with GAL4-binding sites by GAL4 fused to amino acids 372 to 391 of EBNA1.
on November 9, 2019 by guest
http://jvi.asm.org/
6. Gahn, T. A., and C. L. Schildkraut. 1989. The Epstein-Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell 58:527–535.
7. Gahn, T. A., and B. Sugden. 1995. An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J. Virol. 69:2633–2636.
8. Goldsmith, K., L. Bendell, and L. Frappier. 1993. Identification of EBNA1 amino acid sequences required for the interaction of the functional elements of the Epstein-Barr virus latent origin of DNA replication. J. Virol. 67:3418– 3426.
9. Hearing, J. C., and A. J. Levine. 1985. The Epstein-Barr virus nuclear antigen (BamHI K antigen) is a single-strand DNA binding phosphoprotein. Virology 145:105–116.
10. Inoue, N., S. Harada, T. Honma, T. Kitamura, and K. Yanagi. 1991. The domain of Epstein-Barr virus nuclear antigen 1 essential for binding to oriP region has a sequence fitted for the hypothetical basic-helix-loop-helix struc-ture. Virology 182:84–93.
11. Jankelevich, S., J. L. Kolman, J. W. Bodnar, and G. Miller. 1992. A nuclear matrix attachment region organizes the Epstein-Barr viral plasmid in Raji cells into a single DNA domain. EMBO J. 11:1165–1176.
12. Jones, C. H., S. D. Hayward, and D. R. Rawlins. 1989. Interaction of the lymphocyte-derived Epstein-Barr virus nuclear antigen EBNA-1 with its DNA-binding sites. J. Virol. 63:101–110.
13. Kirchmaier, A. L., and B. Sugden. 1995. Plasmid maintenance of derivatives of oriP of Epstein-Barr virus. J. Virol. 69:1280–1283.
14. Kittell, B. L., and D. R. Helinski. 1993. Plasmid incompatibility and repli-cation control, p. 223–242. In D. Clewell (ed.), Bacterial conjugation. Ple-num Press, New York.
15. Knight, J. D., R. Li, and M. Botchan. 1991. The activation domain of the bovine papillomavirus E2 protein mediates association of DNA-bound dimers to form DNA loops. J. Virol. 88:3204–3208.
16. Koff, A., J. F. Schwedes, and P. Tegtmeyer. 1991. Herpes simplex virus origin-binding protein (UL9) loops and distorts the viral replication origin. J. Virol. 65:3284–3292.
17. Lillie, J. W., and M. R. Green. 1989. Transcription activation by the adeno-virus E1a protein. Nature (London) 338:39–44.
18. Loeb, D. D., N. S. Sung, R. L. Pesano, C. J. Sexton, C. Hutchison III, and
J. S. Pagano.1990. Plasmid origin of replication of herpesvirus papio: DNA sequence and enhancer function. J. Virol. 64:2876–2883.
19. Lupton, S., and A. J. Levine. 1985. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence in Epstein-Epstein-Barr vi-rus-derived plasmids in human cells. Mol. Cell. Biol. 5:2533–2542. 20. Ma, J., and M. Ptashne. 1987. Deletion analysis of GAL4 defines two
transcriptional activating segments. Cell 48:847–854.
21. Middleton, T., and B. Sugden. 1992. EBNA1 can link the enhancer element to the initiator element of the Epstein-Barr virus plasmid origin of DNA replication. J. Virol. 66:489–495.
22. Middleton, T., and B. Sugden. 1994. Retention of plasmid DNA in mam-malian cells is enhanced by binding of the Epstein-Barr virus replication protein EBNA-1. J. Virol. 68:4067–4071.
23. Miron, A., S. Mukherjee, and D. Bastia. 1992. Activation of distant replica-tion origins in vivo by DNA looping as revealed by a novel mutant form of an initiator protein defective in cooperativity at a distance. EMBO J. 11: 1205–1216. [Erratum, 11:2002, 1992.]
24. Nishimura, R., M. J. Raymond, I. Ji, R. V. Rebois, and T. H. Ji. 1986. Photoaffinity labeling of the gonadotropin receptor with native, asialo, and deglycosylated choriogonadotropin. Proc. Natl. Acad. Sci. USA 83:6327– 6331.
25. Polvino-Bodnar, M., J. Kiso, and P. A. Schaffer. 1988. Mutational analysis of Epstein-Barr virus nuclear antigen 1 (EBNA-1). Nucleic Acids Res. 16:3415– 3435.
26. Polvino-Bodnar, M., and P. A. Schaffer. 1992. DNA binding activity is re-quired for EBNA1-dependent transcriptional activation and DNA replica-tion. Virology 187:591–603.
27. Rawlins, D. R., G. Milman, S. D. Hayward, and G. S. Hayward. 1985. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell 42:859– 868.
28. Reisman, D., and B. Sugden. 1986. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol. Cell. Biol. 6:3838–3846.
29. Reisman, D., J. Yates, and B. Sugden. 1985. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 5:1822–1832.
30. Rowe, D. T., M. Rowe, G. I. Evan, L. E. Wallace, P. J. Farrell, and A. B.
Rickinson.1986. Restricted expression of EBV latent genes and T-lympho-cyte-detected membrane antigen in Burkitt’s lymphoma cells. EMBO J.
5:2599–2607.
31. Snudden, D. K., J. Hearing, P. R. Smith, F. A. Gra¨sser, and B. E. Griffin.
1994. EBNA-1, the major nuclear antigen of Epstein-Barr virus, resembles ‘RGG’ RNA binding proteins. EMBO J. 13:4840–4847.
32. Sternas, L., T. Middleton, and B. Sugden. 1990. The average number of molecules of Epstein-Barr nuclear antigen 1 per cell does not correlate with the average number of Epstein-Barr virus (EBV) DNA molecules per cell among different clones of EBV-immortalized cells. J. Virol. 64:2407–2410. 33. Studier, F. W., and B. A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130.
34. Su, W., T. Middleton, B. Sugden, and H. Echols. 1991. DNA looping be-tween the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr nuclear antigen 1. Proc. Natl. Acad. Sci. USA 88:10870–10874.
35. Sugden, B., K. Marsh, and J. Yates. 1985. A vector that replicates as a plasmid and can be efficiently selected in B-lymphoblasts transformed by Epstein-Barr virus. Mol. Cell. Biol. 5:410–413.
36. Sugden, B., and N. Warren. 1989. A promoter of Epstein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J. Virol.
63:2644–2649.
37. Yates, J., N. Warren, D. Reisman, and B. Sugden. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of re-combinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. USA
81:3806–3810.
38. Yates, J. L., and S. M. Camiolo. 1988. Dissection of DNA replication and enhancer activation functions of Epstein-Barr virus nuclear antigen 1. Can-cer Cells 6:197–205.
39. Yates, J. L., and N. Guan. 1991. Epstein-Barr virus-derived plasmids repli-cate only once per cell cycle and are not amplified after entry into cells. J. Virol. 65:483–488.
40. Yates, J. L., N. Warren, and B. Sugden. 1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature (Lon-don) 313:812–815.
41. Zhang, S., and M. Nonoyama. 1994. The cellular proteins that bind specif-ically to the Epstein-Barr virus origin of plasmid DNA replication belong to a gene family. Proc. Natl. Acad. Sci. USA 91:2843–2847.