Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Kaposi’s Sarcoma-Associated Herpesvirus K8

Is Derived from
a Spliced Intermediate of K8 Pre-mRNA and Antagonizes
K8
␣
(K-bZIP) To Induce p21 and p53 and Blocks
K8
␣
-CDK2 Interaction
Koji Yamanegi, Shuang Tang, and Zhi-Ming Zheng*
HIV and AIDS Malignancy Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892
Received 12 March 2005/Accepted 22 August 2005
Kaposi’s sarcoma-associated herpesvirus (KSHV) is a lymphotropic DNA tumor virus that induces Kaposi’s sarcoma and AIDS-related primary effusion lymphoma. KSHV open reading frame 50 and K8 genes in early viral lytic infection express, respectively, a tricistronic and a bicistronic pre-mRNA, which undergo alternative splicing to create two major spliced mRNA isoforms,␣and, by inclusion () or exclusion (␣) of an intron at nucleotides 75563 to 75645. This intron contains some suboptimal features, which cause the intron 5ⴕ splice site (ss) to interact weakly with U1 snRNA and the 3ⴕ ss to bind a U2 auxiliary factor, U2AF, with low affinity. Optimization of this intron in K8 (K8 intron 2) promoted the interaction of the 5ⴕ ss with U1 and the 3ⴕ ss with U2AF, resulting in a substantial increase in intron splicing. Splicing of K8 intron 2 has also been shown to be stimulated by the splicing of a downstream intron. This was confirmed by the insertion of a human-globin intron into the K8exon 3-exon 4 splice junction, which promoted splicing of K8intron 2 and conversion of the K8mRNA to the K8␣mRNA that encodes a K-bZIP protein. Intron 2 contains a premature termination codon, yet the K8mRNA is insensitive to nonsense-mediated mRNA decay, suggesting that the truncated K8 protein may have a biological function. Indeed, although the truncated K8 protein is missing only a C-terminal leucine zipper domain from the K-bZIP, its expression antagonizes the ability of the K-bZIP to induce p53 and p21 and blocks K-bZIP-CDK2 interaction through interfering K8␣ mRNA production.
Kaposi’s sarcoma-associated herpesvirus (KSHV), or human herpesvirus 8, is a human gammaherpesvirus that closely re-sembles human Epstein-Barr virus (7). As with other gamma-herpesviruses, infection with KSHV is known to be strongly associated with lymphoproliferative disorders and malignan-cies. It is present in all forms of Kaposi’s sarcoma (16, 47) and in two other diseases, body cavity-based B-cell lymphoma (also known as primary effusion lymphoma [PEL]) and multicentric Castleman’s disease (40, 41).
Like infection with other herpesviruses, KSHV infection dis-plays two viral life cycles. Latent KSHV infection in Kaposi’s sarcoma tissues and B-cell lines features the highly restricted expression of only five viral genes (9, 62). The lytic KSHV life cycle is characterized by the production of progeny virus from infected cells and can be induced by chemicals such as tetra-decanoyl phorbol acetate (TPA) and n-butyrate in PEL-de-rived B cells with latent KSHV infection (26, 27, 34). Chemical induction in PEL-derived B cells initiates the expression of a KSHV transactivator, open reading frame 50 (ORF50), which is absolutely required for the lytic switch from KSHV latency (24, 43).
KSHV ORF50 is an immediate-early gene transcribed as a polycistronic RNA with five exons and four introns. It encodes
a pivotal transactivator that mediates transcription of all early genes (24, 43). ORF50 exon 1 and exon 2 contain the coding region of ORF50, whereas exons 3, 4, and 5 of ORF50 are noncoding exons that overlap with the K8 and K8.1 coding regions (43, 46). This overlap enables ORF50 to use a common poly(A) site at nucleotide (nt) 76714, downstream of the K8.1 coding region. This unique gene structure means that KSHV ORF50, K8, and K8.1 are positioned side by side in the virus genome, and the transcripts of the three genes overlap each other (56). However, each gene initiates its transcription at different stages of the viral life cycle (23, 36, 45) and undergoes extensive RNA splicing during gene expression (56). KSHV K8 is an early gene transcribed as a bicistronic RNA with four exons and three introns that expresses a K-bZIP protein (K8␣) involved in viral DNA replication (1, 22, 51) and transcription (19, 21, 28, 49) as well as induction of cell cycle arrest (20, 52, 53). KSHV K8.1 is a late gene transcribed as a monocistronic RNA with two exons and one intron that encodes an envelope glycoprotein (6, 32, 45).
We have profiled the splicing patterns of KSHV ORF50, K8, and K8.1 through multiple approaches and have reported that splicing of these pre-mRNAs is extremely complex and often results in inclusion of an intron (intron 3 of ORF50, intron 2 of K8) at nt 75563 to 75645 (Fig. 1A), presumably due to subop-timal features of the intron splice sites (46, 56). mRNAs with this intron inclusion are referred to as the isoform; those without the intron are referred to as the␣isoform. We have also shown by using KSHV⫹cell lines that exon 3 of the K8
* Corresponding author. Mailing address: HIV and AIDS Malignancy Branch, Center for Cancer Research, NCI/NIH, 10 Center Dr., Rm. 10 S255, MSC-1868, Bethesda, MD 20892-1868. Phone: (301) 594-1382. Fax: (301) 480-8250. E-mail: zhengt@exchange.nih.gov.
14207
on November 8, 2019 by guest
http://jvi.asm.org/
pre-mRNA (exon 4 of ORF50) includes most of a previously defined intron 3 and has three 5⬘splice sites (ss), at nt 75838, 76155, and 76338 (46). Selection of the nt 75838 5⬘ss dictates the production of K8 mRNA and overwhelms the RNA pro-cessing. The alternative selection of the other two 5⬘ ss is feasible and leads to the production of two additional bicis-tronic mRNAs: K8/K8.1␣and -. However, only a little K8 and no detectable K8.1 protein are translated from the novel bi-cistronic K8/K8.1 mRNAs in 293 cells, suggesting that
[image:2.585.83.499.71.479.2]produc-tion of the K8/K8.1 mRNAs may be an essential way to limit K8 mRNAs, especially K8␣, to a threshold at the RNA pro-cessing level (46). Altogether, we have concluded that a sub-stantial proportion of pre-mRNAs transcribed from the ORF50, K8, and K8.1 genes undergo alternative RNA splicing, leading to the production of at least 19 species of spliced and unspliced transcripts (56). In this regard, the inclusion or ex-clusion of the intron from nt 75563 to 75645 plays an important role in the alternative RNA splicing of both ORF50 and K8
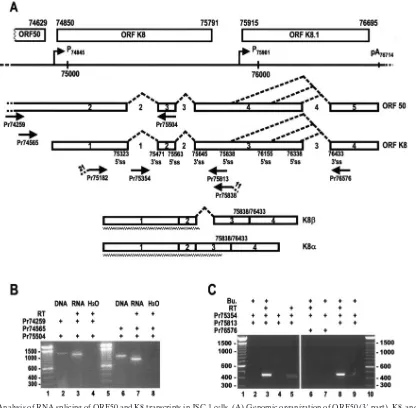
FIG. 1. Analysis of RNA splicing of ORF50 and K8 transcripts in JSC-1 cells. (A) Genomic organization of ORF50 (3⬘part), K8, and K8.1 and the primary structure of the partial ORF50 and K8 RNAs. The numbers above each ORF are the nucleotide positions of the start and termination codons in the KSHV genome (GenBank accession number U75698 [35]). The continuous line under the ORFs represents a linear form of the virus genome for better presentation of relative promoters (arrows) and the poly(A) site. The promoters are named with the transcription start site for each gene, and the poly(A) site was named with the first nucleotide position of the polyadenylation signal AAUAAA for ORF50, K8, and K8.1 in the virus genome. Below the virus genome are primary transcripts of ORF50 (partial) and K8 (full-length) RNA with the numbered exons (boxes) and introns (spaces between exons). Splicing directions to remove introns are depicted by dashed lines between exons. A 5⬘ss and a 3⬘ss are indicated below each intron. The arrows underneath the pre-mRNAs are the primers used for RT-PCR or PCR analysis and are named by the locations of their 5⬘ends. Below the arrows are two major spliced products, K8and K8␣, and their corresponding protein products (jagged lines). Total cell RNA extracted from JSC-1 cells with (B and C, lanes 2, 3, and 6 to 9) or without (C, lanes 4 and 5) butyrate (Bu.) stimulation were analyzed by RT-PCR using different combinations of the indicated primer pairs in each panel for the presence of a specific RNA isoform derived from alternative RNA splicing. JSC-1 DNA was also included in panel B as a KSHV viral DNA control for the detection of ORF50 intron 2 splicing.
on November 8, 2019 by guest
http://jvi.asm.org/
pre-mRNA. However, what controls this intron splicing and how it affects the biology of ORF50 and K8 remains to be understood.
In this report, we have characterized K8 intron 2 (intron 3 of ORF50) and demonstrated that the suboptimal features of intron 2 determine its inefficient interaction with the cellular splicing machinery. Further experiments showed that the in-clusion of this intron in K8mRNA results in the production of a truncated protein that plays a dominant negative role in fully spliced K8␣mRNA expression, K8␣-mediated induction of p21 and p53, and K8␣-CDK2 interaction.
MATERIALS AND METHODS
Cells.The KSHV-positive B-cell line JSC-1 was a generous gift from Richard Ambinder of the Johns Hopkins University. BCBL-1 cells were obtained from Michael McGrath and Don Ganem of the University of California at San Fran-cisco through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health. Both PEL-derived B-cell lines were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA). Human 293 cells and HeLa cells were obtained from the American Type Culture Collection (Manassas, VA) and were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen). Both culture media were supplemented with 10% fetal bovine serum (HyClone, Logan, UT) and with 2 mML-glutamine, 100 U penicillin/ml, and 100g streptomycin/ml. The cells were incubated in the complete culture medium at 37°C in the presence of 5% CO2.
KSHV activation by chemical induction.To activate KSHV lytic gene expres-sion in JSC-1 cells, 1⫻107
cells were treated with sodium butyrate (Aldrich, Milwaukee, WI) at a final concentration of 3 mM for 24 h. BCBL-1 cells (1⫻ 107
) were induced with 20 ng/ml TPA (Sigma, St. Louis, MO) for 48 h. Total cell RNA and total protein samples were then prepared for further analysis of K8 expression. To analyze nonsense-mediated RNA decay (NMD) of KSHV K8 RNA, a protein synthesis inhibitor, cycloheximide (CHX; Sigma, St. Louis, MO), was added at a concentration of 50g/ml to the JSC-1 cells after 4 h of butyrate induction (63). The cells were further cultured for 24 h in the presence of both CHX and butyrate, after which total cell RNA was prepared for RNase protec-tion assays (RPA).
Cell transfection.For transfection of 293 cells and HeLa cells, plasmid DNA was transfected into 1⫻107cells using Lipofectamine 2000 (Invitrogen)
accord-ing to the manufacturer’s instructions. Total cell RNA or protein was prepared 24 or 48 h after transfection, as indicated in the figures. The RNA was used for reverse transcription (RT)-PCR and RPA, and the protein was used for Western blot analysis. To study the NMD of the KSHV K8, 293 cells were transfected with plasmid pST1, a K8 gene expression vector (46); 8 h later, the cells were cultured in the presence of CHX (28g/ml) (5) for 24 h prior to total cell RNA preparation.
Electroporation was used for transfection of KSHV⫹BCBL-1 cells with plas-mid pORF50, pST1, pST3, and pcDNA 3.0 vector. Briefly, BCBL-1 cells were washed twice with cold phosphate-buffered saline (PBS) (calcium and magne-sium free) and resuspended at 1⫻107
cells/ml in RPMI 1640 without any supplements (GIBCO). Approximate 400l of the cell suspension was mixed with 20g of each plasmid and transfected in a prechilled 0.4-cm cuvette (Bio-Rad, Hercules, CA) by electroporation with an Electro Cell Manipulator BTX ECM630 (Genetronics, Inc., San Diego, CA) at the following conditions: voltage, 350 V; capacitance, 960F; and resistance, 50⍀. Under such electro-poration conditions, over 50% of the cells remained viable, and half of those viable cells could be transfected with a green fluorescent protein fusion plasmid as a transfection control. The cell suspensions after electroporation were then incubated at 37°C in 20 ml of complete RPMI 1640 medium for 72 h using regular cell culture conditions prior to total cell RNA extraction and protein sample preparation.
Plasmid construction.The plasmids pKY1 and pKY2 are KSHV K8 expres-sion plasmids based on the wild-type K8 gene expresexpres-sion vector pST1 (46), but with an optimized intron 2. pKY1 has an optimized (mutated) nt 75645 3⬘ss, and pKY2 contains an optimized nt 75563 5⬘ss in K8 intron 2. These plasmids were constructed using an overlapping PCR technique (60). Basically, two primer sets were used to introduce mutations into each splice site. To amplify PCR frag-ments for the construction of plasmid pKY1, the primer set Pr74850 (oST41, 5⬘-CACC/ATGCCCAGAATGAAGGAC-3⬘) and Pr75644 (oZMZ320, 5⬘-CTG
AAAAGAAAGAAAGGTGGCGCCATTGTTCCCATTTGAG-3⬘) and the
primer set Pr75625 (oZMZ321, 5⬘-CCACCTTTCTTTCTTTTCAGGCATTAG
AAGAAAAGGATGC-3⬘) and Pr76687 (oST42, 5⬘-TCC/AGGGTTTCTTAC GCCG-3⬘) were used. The amplified PCR products from each set of primers were mixed, heated at 95°C for 5 min, annealed at room temperature for 30 min, and reamplified using the primer Pr74850 in combination with the primer Pr76687. Following gel purification, the amplified DNA fragment was inserted into an expression vector, pcDNA3.1/V5-His–TOPO (Invitrogen). The same strategy was used to construct plasmid pKY2 with two different primer sets, Pr74850 in combination with Pr75589 (oZMZ 332, 5⬘-GATAGACACCTGTTT
CCCGTACTTACCTGCTGCAGCTGTCT-3⬘) and Pr75549 (oZMZ 331,
5⬘-AGACAGCTGCAGCAGGTAAGTACGGGAAACAGGTGTCTATC-3⬘)
in combination with Pr76687. The plasmid pKY3 is optimized at both the 75563 5⬘ss and the 75645 3⬘ss and was derived from pKY1 using the same primer sets as for the pKY2 construction. All optimization mutations described for pKY1, pKY2, and pKY3 were verified by sequencing.
The plasmid pKY4 was derived from plasmid pST3 (46), a full-length KSHV K8cDNA expression vector, and has a human-globin intron inserted at the K8exon 3-exon 4 junction. To introduce human-globin into the exon 3-exon 4 junction of K8, a two-step overlapping PCR approach was taken. Briefly, the first step of the overlapping PCR was carried out with two separate PCR prod-ucts: the K8 exon 4 product and a human-globin intron product obtained from two separate PCRs. K8 exon 4 was amplified from plasmid pST3 using a sense primer, Pr76433 (oZMZ 364, 5⬘-GATCATATTCATCTGGGGAACC-3⬘), in combination with an antisense primer, Pr76687. The human-globin intron was amplified from plasmid pSP64-H⌬6 (Promega, Madison, WI) using a K8 exon 3–-globin chimeric sense primer, Pr146 (oZMZ 365, 5⬘-ACCATGCCAGACT TTGTGTG/GTTGGTATCAAGGTTACAAG-3⬘), in combination with a K8 exon 4–-globin chimeric antisense primer, Pr275 (oZMZ 366, 5⬘-GGTTCCC
CAGATGAATATGATC/CTAAGGGTGGGAAAATAGACC-3⬘). The two
separate PCR products were then mixed for overlapping PCR using the sense primer Pr146 combined with the antisense primer Pr76687; the resulting PCR product had part of exon 3, a-globin intron, and exon 4. The first-step overlapping PCR was then mixed with another PCR product obtained from plasmid pST3 using the sense primer Pr74850 in combination with an anti-sense primer, Pr75838 (oZMZ 367, 5⬘ -CACACAAAGTCTGGCATGGTTC-3⬘), which contained all K8sequences except exon 4. The second step of the overlapping PCR was performed on the mixture using the primer pair Pr74850 and Pr76687, followed by gel purification. The amplified product was then cloned into the expression vector pcDNA3.1/V5-His–TOPO. The plas-mid inserts were verified by sequencing.
RT-PCR.Total cell RNA was extracted from cells by using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Following DNase I digestion, reverse transcription of 1g of total RNA was performed at 42°C for 1 h using random hexamers and then amplified by PCR with AmpliTaqDNA polymerase (Roche, Indianapolis, IN) using different primer sets as described in each figure. The following list includes the primer sequences, which are named by the locations of their 5⬘ ends. Pr74259 (oZMZ317, 5⬘-GACCATCGG AGGCACAAC-3⬘), Pr74565 (oST43, 5⬘-ACGAGGTACAGGAGTCCG-3⬘),
Pr75182-T7 (oST1, 5⬘-TAATACGACTCACTATAGGG/ACCACCAAGA
GGACCACACATTTC-3⬘), and Pr75354 (oST23, 5⬘-GAACTGACAAGGGGT GACGC-3⬘) are forward primers; Pr75504 (oST4, 5⬘-GCGTGGGAGAATGTG ACT-3⬘), Pr75813 (oST32, 5⬘-CGGACGACGGCACAACCA-3⬘), Pr75838
(oST3, 5⬘-GTACTCACCCC/CACACAAAGTCTGGCATGGTTCTCCC-3⬘),
and Pr 76576 (oST31, 5⬘-TGTAGTGCGCGTCTCTTCCTC-3⬘) are reverse primers. The final PCR products were detected using agarose gel electrophoresis and ethidium bromide staining and analyzed with a ChemiDoc system (Bio-Rad, Hercules, CA). All RT-PCR products were gel purified and confirmed by se-quencing through the splicing junction.
Human GAPDH RNA in each RNA sample was amplified by RT-PCR using a sense primer, Pr6732 (oZMZ269, 5⬘-GTCATCAATGGAAATCCCATCACC-3⬘), in combination with an antisense primer, Pr7207 (oZMZ257, 5⬘-TAATACGAC
TCACTATAGGGA/CCGTTCAGCTCAGGGATG-3⬘).
In vitro RNA splicing assay.DNA templates were obtained separately from plasmids pST1, pKY1, pKY2, and pKY3 by PCR using a sense chimeric SP6/
KSHV primer, Pr75484 (oZMZ335, 5⬘-ATTTAGGTGACACTATAG/ATC
AGTCACATTCTCCCACGC-3⬘), combined with the antisense primer Pr75813. In vitro transcription using SP6 RNA polymerase was performed on each tem-plate DNA in the presence of a cap analog (m7GpppG) and [␣-32P] GTP. An in
vitro RNA splicing assay was then conducted on each transcript using a HeLa cell nuclear extract in an optimized splicing condition. The spliced products were resolved in an 8% denaturing polyacrylamide gel electrophoresis (PAGE) gel and analyzed as described previously (58).
RPA.Antisense RNA probes were prepared as described above by in vitro transcription from individual DNA templates obtained from plasmids pST1,
on November 8, 2019 by guest
http://jvi.asm.org/
pST3, pKY1, pKY2, pKY3, and pKY4 by PCR using a sense primer, Pr75605 (oST36, 5⬘-CTCAAATGGGAACAATGGCG-3⬘), combined with an antisense chimeric T7/KSHV primer, Pr75935 (oST33, 5⬘-TAATACGACTCACTATAG GG/CGAATCTGTGTGGAACTCA-3⬘). A human cyclophilin probe was tran-scribed from a commercially available DNA template (Ambion, Austin, TX) and used as an internal control. For RPA, 100g of total cell RNA was hybridized with 4 ng of an antisense RNA probe in hybridization buffer at 42°C overnight, followed by RNase (RNase A and T1) digestion as instructed by the manufac-turer of the RPA III kit (Ambion). Protected RNA fragments were separated in an 8% denaturing PAGE gel. A gel image was captured using a Molecular Dynamic PhosphorImager Storm 860 and analyzed with ImageQuant software.
RNase H cleavage protection assay and U1 and U2 depletion.Individual single-strand DNA oligonucleotides complementary to the exon-intron junctions of K8 intron 2 were used for oligonucleotide-directed RNase H digestion assays. The following antisense DNA oligonucleotides were used: oligonucleotide oKY2 (5⬘-TCTATACCTGCTG-3⬘), complementary to the exon 2-intron 2 junction of wild-type (wt) K8 RNA transcribed from plasmid pST1, and oKY4 (5⬘-TAC TTACCTGCTG-3⬘), complementary to the same region of mutant (mt) K8 RNA transcribed from plasmid pKY3; and oKY3 (5⬘-AAGACAGCAAGGT-3⬘), complementary to the intron 2-exon 3 junction of wt K8 RNA transcribed from plasmid pST1, and oKY5 (5⬘-AAGAAAGAAAGGT-3⬘), complementary to the same region of mt K8 RNA transcribed from plasmid pKY3. A RNase H digestion reaction was carried out in 20l of reaction mixture containing 40% HeLa nuclear extract, 2.5 mM MgCl2, 1 U of RNasin, and 4 ng of32P-labeled
pre-mRNA as described previously (61). Each reaction mixture was incubated for 10 min at 30°C in the absence of a DNA oligonucleotide and then incubated for another 10 min in the presence of an oligonucleotide (2M) before diges-tion for 10 min with 2 or 4 U of RNase H (Promega). The digesdiges-tion was terminated by adding 3 volumes of proteinase K solution (5 mg/ml proteinase K, 50 mM EDTA, 0.5% sodium dodecyl sulfate [SDS]) (10), incubated at 75°C for 30 min, and extracted with phenol-chloroform. The RNA cleavage products were then resolved in an 8% polyacrylamide gel containing 8 M urea. The RNase H digestion efficiency for each pre-mRNA was calculated as the total amount of digestion products divided by the sum of the total digestion products plus the remaining pre-mRNA.
The oligonucleotide-directed RNase H digestion described above was also used to deplete small nuclear RNA U1 and U2 from the HeLa cell nuclear extract. Briefly, a human U1 (oKY6, 5⬘-TGCCAGGTAAGTA-3⬘) or U2 (oKY7, 5⬘-AGGCCGAGAAGCGA-3⬘) oligonucleotide was incubated at 30°C for 10 min with the RNase H reaction mixture described above in the absence of a
32
P-labeled pre-mRNA. After the addition of RNase H (2 U) to the reaction, the incubation was extended for another 10 min. A32
P-labeled pre-mRNA and a specific K8 oligonucleotide (oKY2 or oKY3 for pST1; oKY4 or oKY5 for pKY3) were added, and the mixture was incubated for 10 min at 30°C before proteinase K digestion.
Western blot analysis.293 cells or HeLa cells with or without transfection and JSC-1 or BCBL-1 cells with or without butyrate or TPA induction were washed twice with PBS and lysed in 2⫻ SDS protein gel loading solution (Quality Biological, Inc., Gaithersburg, MD) containing 10%-mercaptoethanol or lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) (Boston Bio-Products, Ashland, MA). If RIPA buffer was used, the cell lysates were first incubated on ice for 30 min and then sonicated several times (5 s each) before centrifugation for 10 min at 10,000 rpm. The supernatant was mixed with 2⫻ SDS sample buffer containing 10%-mercaptoethanol. Protein samples were separated on a 4% to 12% Bis-Tris gel (Invitrogen) and transferred onto a nitrocellulose membrane. The membrane was blocked for 2 h at room temper-ature in a blocking buffer containing 5% skim milk in Tris-buffered saline (TBS) (10 mM Tris, 150 mM NaCl [pH 7.4]) and then incubated for 90 min with a 1:2,000 dilution of rabbit polyclonal anti-K-bZIP antibody (a gift from Don Ganem at the University of California, San Francisco), a 1:500 dilution of mouse monoclonal anti-human p21 antibody (BD Pharmingen, San Diego, CA), or a 1:100 dilution of mouse monoclonal anti-human p53 antibody in TTBS buffer (10 mM Tris, 150 mM NaCl, 1% Tween 20) containing 5% skim milk, after which the membrane was washed with TTBS three times (10 min each time). The membrane was then incubated for 60 min at room temperature with a 1:20,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G antibody for K-bZIP detection or with a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G antibody for p21 and p53 detection and visualized with a SuperSignal West Pico chemiluminescence detection system (Pierce, Rockford, IL). The same membrane was stripped in a stripping buffer (Pierce) and reprobed using a polyclonal goat CDK2
anti-body (BD PharMingen) for immunoprecipitated CDK2 detection or using a monoclonal anti--tubulin antibody (BD PharMingen) for sample loading.
p53 reporter assays.HeLa cells (5⫻105) were transfected with 2g of pST1
(mainly expressing K8␣) or pST3 (K8) in the presence of vector pcDNA3 (2g) or cotransfected with both pST1 (2g) and pST3 (2g) using Lipofectamine 2000 (Invitrogen). After 48 h of incubation, the transfected HeLa cells were transfected again with 2g of a p53 reporter plasmid (PG13-Luc, a gift from John Brady of the National Cancer Institute) (31) and incubated for additional 24 h at 37°C and 5% CO2. The cells were then collected and washed twice with
calcium- and magnesium-free PBS. Part of the cell pellets were resuspended in 0.2 ml of Promega reporter lysis buffer at room temperature for 15 min. After centrifugation, 100l of the supernatant was collected for luciferase assay (Promega). Relative promoter activity was calculated by the light unit readings obtained from each plasmid-transfected cell lysate divided by the light unit readings obtained from pcDNA3-transfected cell lysate. The other part of the cell pellet was then lysed in RIPA buffer on ice for 30 min with sonication (several times, for 5 s each time). After centrifugation for 10 min at 10,000 rpm, the supernatant was mixed with 2⫻SDS sample buffer containing 10% -mer-captoethanol for the p53 Western blot assay.
CDK2 immunoprecipitation.One hundred microliters of a total of 700l of cell lysate in RIPA buffer obtained from HeLa cells (1⫻107cells) transfected
with pST1 or pST3 or pST1 and pST3 in combination was precleaned with 20l of a 50% slurry of protein A-Sepharose beads (Upstate, Waltham, MA) for 3 h at 4°C and washed twice with 1⫻immunoprecipitation (IP) buffer (59). After centrifugation at 13,000 rpm for 3 min, 20l of the supernatant was collected and incubated with 10l of anti-CDK2-coated beads (BD PharMingen) in 500l of 1⫻IP buffer overnight at 4°C. The beads were then gently washed four times with 1⫻IP buffer, and the proteins bound on the beads were eluted with 2⫻SDS sample buffer containing 10%-mercaptoethanol. Protein samples were sepa-rated on a 4 to 12% Bis-Tris gel (Invitrogen) and transferred onto a nitrocellu-lose membrane for Western blotting as described above.
RESULTS
Alternative splicing of ORF50 and K8 pre-mRNAs produces
␣and, but not␥, mRNAs during KSHV lytic gene expres-sion.In our previous study, we demonstrated that alternative splicing of K8 pre-mRNA produces two major forms of the spliced products (␣and). The␣form differs from theby the exclusion of intron 2. That is, the K8␣ mRNA is a fully spliced mRNA, whereas the K8is a partially spliced mRNA that retains intron 2. Since the 3⬘half of the ORF50 transcript overlaps the K8 transcript and has the same sequence compo-sition as the entire K8 transcript (Fig. 1A), the part of the ␣andisoforms of the K8 transcripts detected from KSHV⫹ cells must be spliced products of ORF50. However, exon 2 of ORF50 pre-mRNA has a size of 2,751 nt, which is too large to be recognized as an internal exon by the cellular splicing ma-chinery (42). Whether such an exon structure of ORF50 could permit cross talking between intron 1 and intron 2 over exon 2 and therefore restrain ORF50 pre-mRNA from splicing has been questioned. In this study, RT-PCR was performed with total cell RNA purified from butyrate-stimulated JSC-1 cells with a primer from ORF50 exon 2 in combination with the primer Pr75504 (positioned in ORF50 exon 3 or K8 exon 2). The RT-PCR analysis demonstrated that splicing of ORF50 pre-mRNAs was efficient and did not retain intron 2 (K8 intron 1) (Fig. 1B), suggesting that a unknown function resid-ing in ORF50 exon 2 could promote recognition of an over-sized internal exon by cellular splicing machinery.
Previous reports from other laboratories have suggested that there is a ␥isoform of the ORF50 and K8 message due to alternative RNA splicing in lytic KSHV gene expression (23, 63). In these reports, the␥RNA is depicted as retaining two introns, one from nt 75323 to 75471 and another from nt 75563
on November 8, 2019 by guest
http://jvi.asm.org/
to 75645, but lacking the intron from nt 75838 to 76433 due to RNA splicing. A computer analysis of all three of these introns in our laboratory suggested that both the intron from nt 75323 to 75471 and the intron from nt 75838 to 76433 contain con-sensus splice sites and should be efficiently recognized by the cellular splicing machinery; it would be unusual for the cellular splicing machinery to miss recognizing an intron with consen-sus splice site sequences. In fact, we found that splicing of the
[image:5.585.128.459.68.504.2]intron from nt 75323 to 75471 is efficient. To further test this observation, we carried out an RT-PCR strategy to detect a ␥RNA in butyrate-stimulated JSC-1 cells and TPA-stimulated BCBL-1 cells using an intron primer, Pr75354, from the intron region of nt 75323 to 75471, in combination with an antisense exon primer, Pr75813 or Pr76576 (Fig. 1C). The predicted ␥ RNA would be 636 nt in size when primer Pr76576 was paired with primer Pr75354 for the amplification. However,
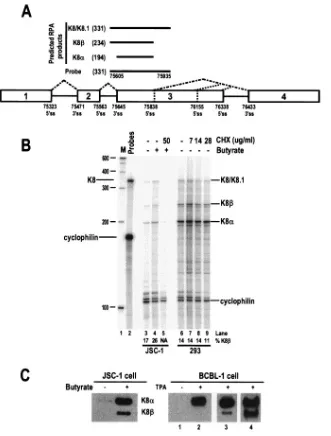
FIG. 2. Detection of nonsense-mediated RNA decay of KSHV K8RNA by RPA. (A) A K8 pre-mRNA with the position of an antisense RNA probe (331 nt) and the protected RPA products (nucleotides in parentheses) that are predicted. (B) CHX at the concentration indicated in each lane was added 4 h after butyrate induction of JSC-1 cells or 8 h after plasmid pST1 transfection of 293 cells. Total cell RNA was prepared 24 h after butyrate stimulation or plasmid transfection and analyzed by RPA using the antisense RNA probe depicted in panel A. A cellular cyclophilin antisense RNA probe was also used in each RPA to control for RNA sampling. The identities of the protected RPA products are indicated on the right of the 8% denaturing PAGE gel. The ratio of KSHV K8to K8␣was calculated based on individual band density. Lane M, size marker (100-bp DNA ladder). (C) Expression of the K8␣and K8proteins during viral lytic replication in PEL cells. JSC-1 cells (1⫻107) with or without 3 mM butyrate treatment for 24 h (left panel) and BCBL-1 cells (1⫻107) with or without 20 ng/ml TPA for 48 h (right panel) were compared for expression of K8␣and K8. Lane 3 in the right panel shows the K8expression in TPA-treated BCBL-1 cells after a relatively long exposure time compared to lane 2, and lane 4 shows K8␣and K8expression from the same sample when double the amount of sample was loaded and exposed as in lane 3.
on November 8, 2019 by guest
http://jvi.asm.org/
total cell RNA prepared from butyrate-stimulated JSC-1 cells did not have a band of this size (Fig. 1C, lane 6), despite the existence of RNAs containing the intron from nt 75323 to 75471 and the intron from nt 75563 to 75645 (Fig. 1C, lanes 3, 5, and 8) as amplified with a primer pair of Pr75813 and Pr75354. The same was true for BCBL-1 cells (data not shown). The RNA containing these two introns but lacking a 3⬘ terminal exon sequences may more likely be a partially transcribed, unspliced pre-mRNA of ORF50 and/or K8.
KSHV K8RNA is not subject to NMD and encodes a K8 protein during lytic infection.K8 intron 2 (positioned from nt 75563 to 75645) contains a premature termination codon (PTC), and its retention in the K8mRNA introduces a PTC into the mRNA. K8should therefore be subject to NMD. As currently understood, NMD serves as a posttranscriptional
[image:6.585.81.499.65.376.2]control system by which cells recognize and degrade mRNAs carrying a PTC positioned⬎50 nt upstream of a splice junction to prevent the possible toxic effects of truncated peptides (25). It has been shown that translation or a translation-like process is a prerequisite for NMD, and inhibition of translation by CHX results in the abolishment of NMD and, subsequently, the accumulation of the PTC⫹mRNA (5, 33). We therefore compared the quality and quantity of the K8RNA by RPA in 293 cells transfected with a K8 expression vector and in bu-tyrate-activated KSHV⫹ JSC-1 cells with or without CHX treatment (Fig. 2). The expected RPA products are shown in Fig. 2A. We postulated that cells treated with CHX would have more K8mRNA due to a minimized NMD response. How-ever, K8␣and K8were present in very similar amounts in the presence or absence of CHX in 293 cells transfected with the
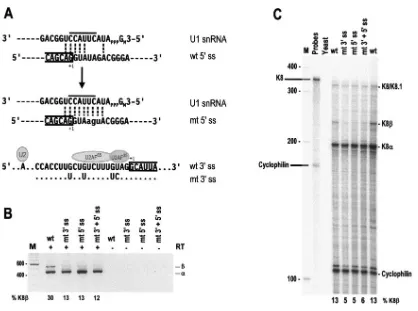
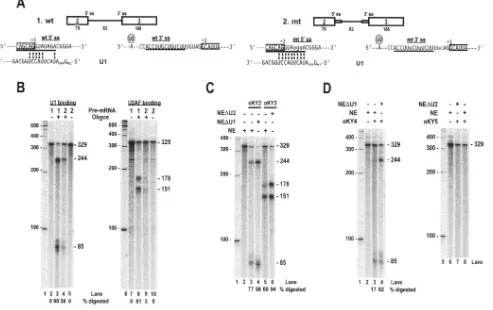
FIG. 3. Optimization of K8 intron 2 splice sites promotes splicing of K8 pre-mRNA. (A) Suboptimal features of the 5⬘ss and 3⬘ss of K8 intron 2 and their optimizing mutations. The sequences of both the wt and the optimized (mt) 5⬘ss with the mt nucleotides in lowercase letters are shown base pairing with the 5⬘end of U1 snRNA. The line above the U1 sequences (top and middle sequences) indicates the most important U1 5⬘end nucleotides for base pairing with each 5⬘ss during recognition. Exon 2-intron 2 junctions are divided by a half box to the exon 2 side, and⫹1 indicates the last nucleotide of exon 2 immediately upstream of the 5⬘end of intron 2. (Bottom sequences) The sequences of the 3⬘ss of intron 2 are depicted with the locations of the U2 snRNA–branch-point (symbolized as an A) interaction and U2 axillary factor (a dimer of U2AF65and U2AF35) binding. The intron 2-exon 3 junctions are divided by a half box to exon 3, and⫹1 indicates the first nucleotide of exon 3. Interposed purines in the polypyrimidine tract are mutated to uridine, and UAG is mutated to CAG, as shown immediately below the sequence of the wt 3⬘ss. Dots indicate nucleotides unchanged in the mt 3⬘ss. (B) RT-PCR and (C) RPA of the constructs with or without optimization of K8 intron 2. Total cell RNA was isolated from 293 cells transfected with plasmid pST1 (wt K8), pKY1 (K8 with a mt intron 2 3⬘ss), pKY2 (K8 with a mt intron 2 5⬘ss), or pKY3 (K8 with mutations in both intron 2 splice sites) and was analyzed by RT-PCR using a primer set of Pr75182 and Pr75838 (see Fig. 1A) and by RPA using the probe described in Fig. 2A. See legend to Fig. 2A for other details. (D) In vitro splicing of K8 intron 2 with or without optimization. Pre-mRNAs with two exons (boxes) and one intron (lines) shown at the top of the panel were used for the assay. Mutated splice sites are diagrammed as a small box on the end of the intron. The numbers above the pre-mRNAs are nucleotide positions in the KSHV genome, and the numbers below the pre-mRNAs are the sizes of the exons or introns (nt). Splicing efficiency (% spliced) was calculated from the splicing gel as described previously (54). The splicing products were resolved on an 8% denaturing PAGE gel, and their identities are indicated. Lanes M, size marker (100-bp DNA ladder).
on November 8, 2019 by guest
http://jvi.asm.org/
K8 expression vector (pST1, lanes 6 to 9), suggesting no accu-mulation of K8 and therefore no NMD-mediated K8 mRNA degradation. In the butyrate-activated JSC-1 cells, CHX treatment inhibited both K8␣and K8gene expression (Fig. 2B, lanes 3 to 5). This was not surprising, since several studies have reported that expression of K8 in KSHV⫹ BCBL-1, BC-1, and JSC-1 cells is sensitive to CHX treatment (44, 63). Thus, it was impossible to draw any conclusion on NMD-mediated K8degradation at the RNA level in JSC-1 cells. However, we were able to detect K8protein from both JSC-1 and BCBL-1 cells after butyrate and TPA stimulation (Fig. 2C), respectively, suggesting that K8RNA resists NMD-mediated RNA degradation in those cells and is capable of being translated.
Optimization of K8 intron 2 splice sites promotes splicing of K8 pre-mRNA.As proposed in our previous report (46), the inclusion of intron 2 in K8 mRNA could result from its suboptimal nature, since this intron contains a nonconsensus
5⬘ ss and a weak 3⬘ ss. The sequence of the 5⬘ ss of intron 2 misses pairing a few nucleotides from the U1 5⬘ end. Most importantly, the 5⬘ ss misses two nucleotides, at intron posi-tions 4 and 5, for base pairing to the six core nucleotides of the U1 5⬘ end (Fig. 3A, top). A missing base pairing of a U1 snRNA 5⬘core nucleotide to the RNA 5⬘ss at intron position 5 has been reported to inhibit U1 snRNA binding and splicing (15, 61). The sequences of the intron 3⬘ss contain a few purine Gs interspersed in its putative polypyrimidine tract (PPT), which lies between the branch point sequence (BPS) and the AG dinucleotide (Fig. 3A, bottom). Recognition of the BPS by U2 snRNA initiates the first step of RNA splicing. The PPT is a run of 15 to 40 pyrimidines (usually U’s) and contains binding sites for U2AF, a heterodimer of U2AF65and U2AF35.
Bind-ing of U2AF to the PPT strengthens U2 recognition of the BPS (57). It has been reported that any purines interspersed in the PPT will weaken the binding of U2AF and affect U2 interac-tion with the BPS. Moreover, the nature of the nucleotide preceding the AG dinucleotide has a profound effect on splic-ing (39), and CAG is twice as common as UAG in mammalian 3⬘ ss (55). Together, these features of K8 intron 2 would be expected to decrease the inherent strength of its recognition by U1 and U2 snRNAs.
To test our hypothesis, we constructed three K8 expression vectors. pKY1 has an optimized 3⬘ss (Fig. 3A, bottom, mt 3⬘ss), pKY2 has an optimized 5⬘ss (Fig. 3A, middle), and pKY3 has optimized 3⬘ss and 5⬘ss. Following transient transfection of 293 cells with these expression vectors, we used RT-PCR and RPA to examine the expression of K8␣and K8in parallel with the expression of a wild-type K8 expression vector, pST1. As shown in Fig. 3B and C, the optimization of K8 intron 2 increased the splicing efficiency of the K8 pre-mRNA and resulted in at least twofold reduction of K8 production in both assays. The data suggest that strengthening either the 3⬘ ss or 5⬘ss in K8 intron 2 improved the recognition of the intron by the cellular splicing machinery and led to the splicing of intron 2.
In vitro splicing of pre-mRNAs transcribed from the opti-mized and wt plasmids showed similar results. The pre-mRNA transcribed from wt K8 had a poor splicing efficiency (only 2%) (Fig. 3D, pre-mRNA 1), while the optimized pre-mRNAs ex-hibited an increased (⬎9-fold) splicing efficiency (Fig. 3D, pre-mRNAs 2 to 4). Notably, the pre-mRNA with both the 5⬘ss and 3⬘ ss optimized showed the highest splicing efficiency (13-fold greater than the wild type) (Fig. 3D).
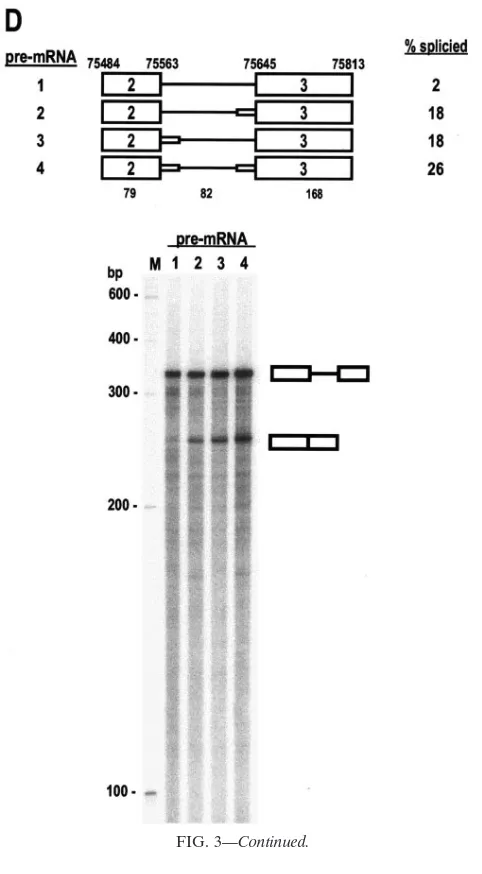
The escape of K8 intron 2 from splicing is due to its ineffi-cient recognition by U1 snRNA and U2AF. To understand further how intron 2 escapes RNA splicing, the RNAs tran-scribed from pST1 (wt K8) and pKY3 (K8 with both of the intron 2 splice sites optimized) were also used for a DNA oligonucleotide-directed RNase H digestion assay. As men-tioned above, we would expect that the suboptimal features of intron 2 do not favor efficient interactions between U1 and the 5⬘ss or between U2AF and the 3⬘ss. If this prediction is right, the optimization of both splice sites should lead to increased U1-5⬘ss interactions and U2AF-3⬘ss interactions and, there-fore, to increased splicing efficiency of intron 2.
[image:7.585.43.284.67.504.2]RNase H digests only RNA-DNA hybrids in which a DNA oligonucleotide hybridizes to a specific RNA sequence through base pairing (8, 14). An RNA-RNA hybrid in the same region
FIG. 3—Continued.
on November 8, 2019 by guest
http://jvi.asm.org/
or a region of RNA with bound protein will prevent a DNA oligonucleotide from base pairing and will consequently pre-vent RNase H digestion. Based on this theory, we synthesized antisense DNA oligonucleotides (12 or 13 nt in length) specific to the 5⬘ss or the 3⬘ss of wt or mt K8 intron 2, as diagrammed in Fig. 4A. Using this approach, we were able to quantify a U1-5⬘ss interaction and a U2AF-3⬘ss interaction in the presence of HeLa cell nuclear extracts containing enriched U snRNAs and splicing factors. As shown in Fig. 4B, the wt 5⬘ss and 3⬘ss of the K8 intron 2 have a weak binding affinity for U1 and U2AF, allowing more DNA oligonucleotides to bind to their corresponding splice sites; as a result, approximately 90% and 61% of the RNA was digested by RNase H directed by a 5⬘ ss-specific DNA oligonucleotide and a 3⬘ ss-specific DNA oligonucleotide, respectively. In contrast, RNase H digestion
was much less effective when both the intron 5⬘ ss and 3⬘ ss were optimized: only about 38% of the RNA was subjected to the oligonucleotide-directed digestion of the RNA 5⬘ ss, and approximately 3% of the RNA was subjected to the oligonu-cleotide-directed digestion of the RNA 3⬘ss.
To further confirm that the reduction in digestion of the RNA intron 2 splice sites by RNase H was specifically due to the improvement in the U1-5⬘ss and U2AF-3⬘ss interactions, we depleted either U1 or U2 snRNAs from HeLa nuclear extracts using RNase H digestion directed by an antisense U1 or U2 oligonucleotide. The U1- or U2-depleted nuclear ex-tracts were then used in DNA oligonucleotide-directed diges-tion of wt or mt K8 intron 2. Since U2AF is abundant in HeLa nuclear extracts and complete depletion of U2AF would be difficult by antibody affinity or poly(U) approaches, we
de-FIG. 4. Optimization of K8 intron 2 splice sites promotes recognition of K8 intron 2 by the RNA splicing machinery. (A) Maps of K8 pre-mRNAs containing part of exon 2, all of intron 2 (line), and part of exon 3 that were used for in vitro RNase H digestion. Wild-type (wt) K8 pre-mRNA (pre-mRNA 1) transcribed from plasmid pST1 contains a native intron 2 with suboptimal splice sites; the splice site sequences and the possible base-pairing of the 5⬘ss with the U1 5⬘end are shown below the map. The mutant (mt) K8 pre-mRNA (pre-mRNA 2) transcribed from pKY3 has optimized intron 2 splice sites (as indicated by a small, horizontal box on each end of the intron). The mutated nucleotides are indicated in lowercase letters, and possible base pairing with U1 snRNA is shown. The horizontal lines between the U1 5⬘end base pairing at the exon 2-intron 2 junction and underneath the intron 2 3⬘ss indicate the positions of the antisense DNA oligonucleotides used for the oligonu-cleotide-directed RNase H digestion. See other details in the legend to Fig. 3A. (B) DNA oligonuoligonu-cleotide-directed RNase H digestion assays. K8 pre-mRNA 1 (wt intron) and 2 (mt intron) were incubated with a HeLa nuclear extract containing nuclear small RNAs and digested with RNase H (4 units) in the presence of oligonucleotide oKY2 (for wt 5⬘ss), oKY4 (for mt 5⬘ss), oKY3 (for wt 3⬘ss), or oKY5 (for mt 3⬘ss). (C and D) Depletion of U1 or U2 from the HeLa nuclear extract promotes DNA oligonucleotide-directed RNase H digestion. U1 or U2 depletion from the HeLa nuclear extract was carried out using antisense U1 DNA oligonucleotide-directed or antisense U2 DNA oligonucleotide-directed RNase H digestion. The nuclear extracts with U1 or U2 depletions were then used in DNA oligonucleotide (oKY2 or oKY3)-directed RNase H (2 units) digestion assays of wt K8 intron 2 (C) or used in DNA oligonucleotide (oKY4 or oKY5)-directed RNase H (2 units) digestion assays of mt K8 intron 2 (D). The digested products were resolved in 8% denaturing PAGE gels, and their sizes are indicated to the right of each gel. Size markers (100-bp DNA ladder) are indicated on the left of each gel.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.47.540.76.386.2]pleted U2 instead of U2AF from HeLa nuclear extracts on the assumption, based on the U2 recruitment model, that the RNA-U2AF interaction is dynamic and less U2AF would be fixed on the RNA in the absence of U2 snRNA (11, 12). Depletion of U1 from HeLa nuclear extracts did increase the oligonucleotide-directed RNase H digestion of both the wt (Fig. 4C, compare lanes 3 and 4) and the mt (Fig. 4D, compare lanes 3 and 4) K8 intron 2, indicating a specific U1-5⬘ ss interaction. In addition, an increase in oligonucleotide-di-rected RNase digestion of the wt K8 intron 2 was observed when U2-depleted HeLa nuclear extracts were used (Fig. 4C, compare lanes 5 and 6). However, we were unable to see a similar result from the oligonucleotide-directed RNase H di-gestion of the 3⬘ss of mt K8 intron 2 (Fig. 4D, compare lanes 7 and 8), suggesting that optimization of the 3⬘ ss might also create an additional binding site (UCUU) for another protein, most likely a PPT-binding protein (PTB) (29), which is unre-lated to U2 recruitment.
Insertion of a human -globin intron into the K8 exon 3-exon 4 splice junction stimulates splicing of K8 intron 2 and converts K8to K8␣.We have reported that K8 exon 3 con-tains three alternative 5⬘ ss, at nt 75838, 76155, and 76338. Selection of the nt 75838 5⬘ss is subject to exon definition and predominates over the other two 5⬘ ss during RNA splicing, even though all three 5⬘ss are optimal splice sites with con-sensus U1 binding sequences. In vivo experiments also suggest
that utilization of the nt 75838 5⬘ss stimulates splicing of an upstream nt 75645 3⬘ss, crossing over exon 3 (46). Disruption of the nt 75838 5⬘ss promotes either the inclusion of intron 2 or the exclusion of exon 3 due to alternative splicing of exon 2 to exon 4 (data not shown). This implies that intron 3 5⬘ss recognition may play an important role in the inclusion and exclusion of K8 intron 2 and the production of K8mRNA.
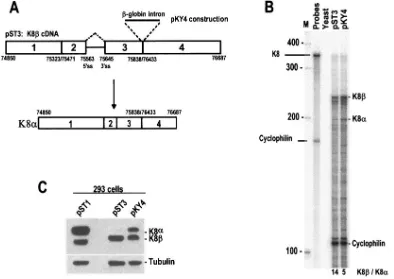
To test this role of downstream intron recognition in up-stream intron splicing, we inserted a human-globin intron into the exon 3-exon 4 splice junction of a K8 cDNA con-struct, pST3, to create plasmid pKY4 (Fig. 5A). Since the human-globin intron has optimal splice sites, the splicing of this inserted intron was expected to stimulate removal of the K8 intron upstream. When pKY4 was used to transfect 293 cells, the splicing of the-globin intron in the pKY4-derived pre-mRNA did stimulate splicing of the upstream K8intron, and much of the K8RNA was converted into K8␣ mRNA (Fig. 5B).
[image:9.585.93.487.71.350.2]Western blot analysis was performed with a polyclonal K-bZIP antibody to examine K8␣and K8protein expression simultaneously in the transfected 293 cells. As shown in Fig. 5C, a K8 genomic DNA construct, pST1, expressed mainly K8␣ protein in 293 cells, accompanied by a lower amount of K8 expression. In contrast, the K8 cDNA construct pST3 expressed K8protein but very little K8␣protein. The pKY4 construct, which had the-globin intron inserted into the pST3
FIG. 5. Recapitulation of enhanced removal of K8 intron 2 by splicing of K8 intron 3 downstream. (A) Structure of K8(pST3) and diagram of K8cDNA containing an inserted human-globin intron at its exon 3-exon 4 splice junction (pKY4). (B) Quantitative analysis of K8and K8␣ mRNAs was performed by RPA on total cell RNA isolated from individual plasmid-transfected 293 cells. Cellular cyclophilin RNA was used as an internal control for sampling. Lane M, size marker (100-bp DNA ladder). (C) Removal of the K8intron in pST3 converts the K8mRNA into K8␣mRNA (pKY4) and, consequently, K8␣(K-bZIP) protein is produced, as shown by a Western blot. Plasmid pST1 contains a full-length
K8 gene (four exons and three introns).
on November 8, 2019 by guest
http://jvi.asm.org/
exon 3-exon 4 splice junction, expressed almost equal amounts of K8␣and K8protein, as predicted (Fig. 5C).
K8regulates K8␣function.As described above, the inclu-sion of K8 intron 2 also creates a PTC in the K8mRNA and results in the production of the truncated protein K8 in KSHV⫹JSC-1 cells and BCBL-1 cells as well as in transient transfected epithelial cells. Thus, both K8␣protein and K8 protein have the same N-terminal structure, but they differ at their C-terminal ends, where a leucine zipper domain in K8␣is missing in K8. Recent reports indicate that one of the func-tions of KSHV K8␣during viral lytic replication in PEL cells is to induce expression of the universal CDK inhibitor p21 by stabilizing p21 from proteasome-mediated degradation (20, 52, 53). This observation prompted us to wonder whether the expression of K8might antagonize the activity of K8␣if this truncated protein cannot similarly induce p21.
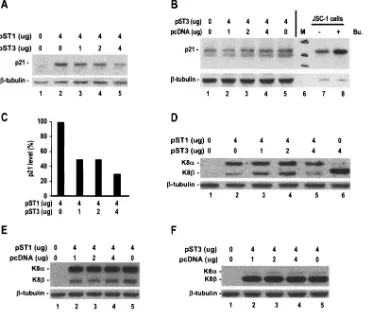
To examine the biological functions of K8, we investigated the effect of KSHV K8␣and K8by transient transfection on the expression of p21 initially in HeLa cells. We transiently transfected HeLa cells with a K8 expression vector that pre-dominantly expresses K8␣(pST1) or with the K8expression
vector (pST3) and found that only the pST1-transfected HeLa cells strongly induced p21 (Fig. 6A, lane 2). Although pST3 itself had no effect on p21 induction in HeLa cells (Fig. 6B), pST3 did suppress the pST1-induced p21 expression in a dose-dependent manner (Fig. 6A, compare lanes 3 to 5 and lane 2; Fig. 6C). At a 1:1 ratio of pST1 and pST3, the expression of both K8␣and K8was reduced (Fig. 6D, lane 5). Interestingly, this reciprocal suppression of K8␣and K8appeared only in the HeLa cells cotransfected with pST1 and pST3, not in cells cotransfected with pST1 or with pST3 and an empty vector, pcDNA3 (Fig. 6E and F, lanes 4). Thus, the expression of K8 antagonizes the function of K8␣.
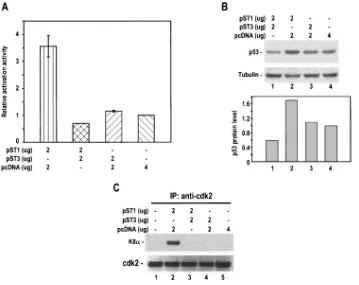
[image:10.585.107.477.71.386.2]Since p21 expression is usually induced by p53, it is conceiv-able that K8␣-mediated p21 accumulation might occur be-cause of an accumulated p53 expression in cells overexpressing K8␣. To address this question, we examined p53 expression by using HeLa cells transfected with pST1 (K8␣), pST3 (K8), or pST1 and pST3 in combination. HeLa cells were selected for the study, since they contain only a residual amount of p53. As shown in Fig. 7A and B, the overexpression of K8␣(pST1) in HeLa cells was found to induce⬎2.5-fold activation activity of
FIG. 6. KSHV K8antagonizes the induction of p21 expression by K8␣. HeLa cells (1⫻107) at 75% confluence in a 10-cm petri dish were transfected with a K8 expression vector, pST1, which predominantly expresses K8␣, or in combination with a K8expression vector, pST3, as indicated (A). The protein samples in 700l of RIPA buffer were prepared 48 h after transfection and analyzed by Western blotting for expression of p21 (A). (B) K8(pST3) does not induce p21 expression in the transient transfection. Cell lysates obtained from JSC-1 cells with or without butyrate (Bu.; 3 mM) treatment for 24 h were used as controls for p21 induction. (C) The p21 expression in panel A was quantified and expressed as a bar graph. (D) Western blot analysis of the samples in panel A for K8␣and K8expression. (E and F) A pcDNA3 empty vector (Invitrogen) without KSHV sequence insertion was used as a vector control in the cotransfection assays, and its effect on expression of K8␣and K8was examined by Western blotting. The same membrane in each panel was examined for tubulin as a control for sample loading.
on November 8, 2019 by guest
http://jvi.asm.org/
p53 in p53 reporter assays, which could be translated into a 71% higher p53 protein accumulation in the cells by Western blotting. Although the overexpression of K8(pST3) had no effect on p53 accumulation, HeLa cells cotransfected with both K8␣(pST1) and K8(pST3) expression vectors showed no p53 accumulation or activation activity but, rather, p53 reduction. Data suggest that the K8␣-mediated p21 accumulation and its suppression by K8may be attributable partially to K8␣ -me-diated p53 expression.
A recent study also suggests that the inhibition of CDK2 activity by K-bZIP through direct CDK2-K-bZIP interaction is partially responsible for the increased p21 protein level (20), since phosphorylation by CDK2 targets p21 for degradation (38). We wondered whether K8could also affect the interac-tion of K8␣and CDK2 and examined this interaction by using HeLa cells transfected with pST1 (K8␣) or pST3 (K8) or pST1 plus pST3 in combination. The cell lysates were immu-noprecipitated with anti-CDK2-coated beads, and the precip-itates were immunoblotted with anti-K-bZIP. As shown in Fig. 7C, K8␣(pST1) did interact with CDK2 (lane 2), but this interaction could be completely eliminated in the presence of K8 (pST3, lane 3) despite the fact that the latter had no detectable interaction with CDK2 (lane 4). These data, to-gether with the p53 results described above, suggest that K8␣
mediates the increase in p21 expression, presumably through several pathways.
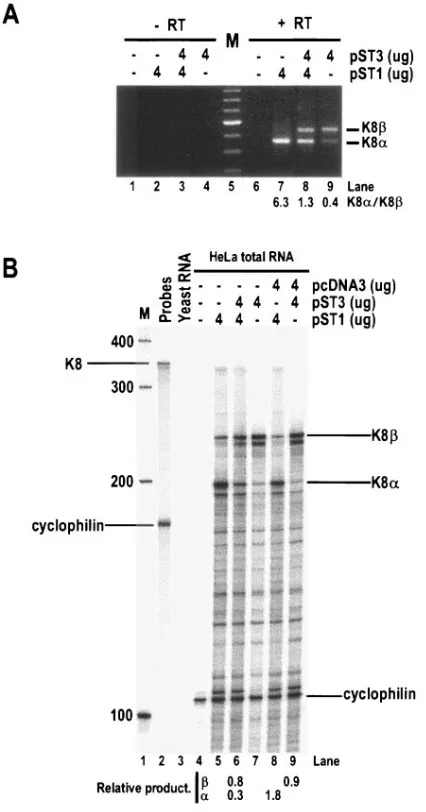
[image:11.585.113.472.69.354.2]K8 affects K8␣ expression at the mRNA level.As stated above, K8differs from K8␣by lacking a C-terminal leucine zipper motif which is required for K8␣dimerization and func-tions. Thus, it is very unlikely that K8could form a dimer with K8␣as a mechanism of the K8-mediated suppression. Con-sidering the fact that K8also reduces K8␣protein expression in HeLa cell cotransfection assays (Fig. 6D, lane 5), we were interested to know if the K8-mediated K8␣protein reduction is a result of K8-mediated suppression of K8␣RNA expres-sion, since K8has the same exon structures in K8␣ mRNA and differs from K8␣only by the inclusion of a K8 intron 2 as an extra piece in its mRNA. As demonstrated in Fig. 8, the cotransfection of HeLa cells with a K8 expression vector (pST3) resulted in an approximately fivefold reduction of K8␣ mRNA (pST1) in RT-PCR assays (Fig. 8A, compare lanes 7 and 8). To further confirm our RT-PCR results, total cell RNA obtained from separate experiments was analyzed by RPA with an antisense K8 probe and showed a sixfold reduction of K8␣ mRNA (pST1) in the presence of K8(pST3) (Fig. 8B, com-pare lanes 6 and 8). Although K8␣ mRNA expression was reduced greatly in the presence of K8, the K8mRNA level remained relatively constant in the presence of K8␣(compare
FIG. 7. KSHV K8antagonizes K8␣-mediated p53 accumulation and affects K8␣-CDK2 interaction. HeLa cells (5⫻105) in a 6-cm petri dish were transfected with a K8␣(pST1) or K8(pST3) expression vector or in combination, as indicated in panels A and B, for 48 h and followed by transfection with a p53 reporter, PG13-Luc, for additional 24 h. The cell lysates were then prepared as described in Materials and Methods and analyzed for p53 expression by p53 reporter assays (A) or by Western blotting (B). Data shown in panel A were obtained from three separate experiments, each performed in triplicate. A bar graph below the gel profile in panel B is the quantified p53 expression (relative density ratio of p53 to tubulin) from the corresponding sample shown above. For CDK2 detection, the cell lysates in 700l of RIPA buffer were prepared 48 h after transfection, and 20l of the lysates was immunoprecipitated with anti-CDK2-coated beads and then immunoblotted with anti-K-bZIP followed with anti-CDK2 antibody for sample loading (C).
on November 8, 2019 by guest
http://jvi.asm.org/
lanes 6 and 9). Together, the results from two different assays lead us to conclude that K8-mediated suppression of K8␣ function is most likely a consequence of K8-mediated inhibi-tion of K8␣RNA expression.
Since KSHV⫹B cells express both K8␣and K8via alter-native RNA splicing during lytic infection, it is difficult to dissect the function of K8␣from K8during lytic infection in those cells. More importantly, the K8 gene overlaps the ORF50 gene in the virus genome despite the fact that each gene has its own ORF, as shown in Fig. 1A. Accordingly, it is
not feasible to study K8 function in its native context without disruption of the overlapped ORF50. To duplicate our obser-vation with HeLa cells, we analyzed K8on K8␣function with BCBL-1 cells by transient transfection. As shown in Fig. 9A, TPA activation of BCBL-1 cells (lane 2) or expression of KSHV ORF50 (lane 4) or K8␣(lane 5, pST1) alone in BCBL-1 cells promotes p21 expression. The expression of K8 alone (lane 7, pST3) in BCBL-1 cells did not induce p21 expression, as was done by its vector control (pcDNA, lane 3). As ex-pected, we were able to demonstrate that the expression of K8 could antagonize K8␣-mediated induction of p21 (Fig. 9A, compare lanes 5 and 6) in BCBL-1 cells. In parallel, BCBL-1 cotransfection with pST1 (K8␣) and pST3 (K8) had much less production of K8␣mRNAs (Fig. 9B, lane 7) than the cells transfected with pST1 alone (Fig. 9B, lanes 6), implying that K8diminishes K8␣-mediated function most likely at the RNA level.
DISCUSSION
In this report, we have analyzed alternative splicing of two overlapping primary transcripts expressed during the KSHV lytic cycle, the ORF50 and K8 pre-mRNAs. As we (46) and others (13, 23, 63) previously reported, this alternative RNA splicing leads to the production of two major spliced isoforms (␣and) of mRNAs due to the inclusion () or exclusion (␣) of an intron from nt 75563 to 75645 (intron 3 of the ORF50 pre-mRNA and intron 2 of the K8 pre-mRNA). However, we were unable to detect a previously reported␥isoform message (23, 63).
[image:12.585.56.269.70.476.2]The intron from nt 75563 to 75645 (K8 intron 2 or ORF50 intron 3) has suboptimal features, including a nonconsensus 5⬘ss and a weak 3⬘ss with purines interspersed in its putative polypyrimidine tract. As a result, K8 intron 2 interacts with U1 snRNA and U2AF protein with low affinity. The optimization of these splice sites was found to improve the interactions with U1 snRNA and U2AF and, consequently, to promote the splicing of K8 intron 2 in vitro and in vivo. Our data provide strong experimental evidence that the escape of K8 intron 2 (ORF50 intron 3) from RNA splicing can most likely be at-tributed to its suboptimal features. The mRNA that results from the retention of intron 2, K8, contains a PTC in the intron at a distance of⬎50 nt upstream of an exon-exon junc-tion. Despite having this PTC, the K8 mRNA bypasses an mRNA surveillance mechanism that initiates NMD to rapidly degrade mRNAs carrying PTCs. This was surprising, since a major function of NMD is to block the synthesis of truncated proteins that could have dominant negative effects (25). Al-though there are many other characteristics of mammalian transcripts that determine the efficiency of NMD, including a nonsense codon positioned close to the initiation AUG (4, 18, 30, 54), the absence of an NMD-promoting element (3), and the presence of NMD-mediated up-regulation of RNA splicing (48), we do not know how the K8 mRNA avoids NMD. Nevertheless, in PEL cells and transiently transfected mamma-lian cells, the K8mRNA produces a K8protein that is 48 amino acids smaller than the K-bZIP protein encoded by fully spliced K8␣ mRNA. This led us to speculate that the PTC-containing K8 might avoid the NMD pathway because the encoded protein has a function in viral gene expression.
FIG. 8. KSHV K8 reduces K8␣ mRNA expression. Total cell RNA was prepared after 48 h of HeLa cell transfection with the plasmids as indicated on the top of each panel and was analyzed for K8␣(pST1) and K8(pST3) mRNA expression by RT-PCR (A) using a primer set of Pr75182 and 75838 (see Fig. 1A) or by RPA (B) using an antisense K8 probe (see Fig. 2A). The ratio (fold) of K8␣to K8in panel A was calculated based on individual band density. A cellular cyclophilin antisense RNA probe was also used in each RPA (B) to control for RNA sampling. The relative production (fold) of K8␣and K8mRNAs in each cotransfection was expressed as a ratio of K8␣or K8to cyclophilin based on individual band density. Lanes M, size marker (100-bp DNA ladder).
on November 8, 2019 by guest
http://jvi.asm.org/
Despite the suboptimal features of the intron from nt 75563 to 75645, the majority of the K8 pmRNAs completely re-move this intron to produce a fully spliced K8␣message, which encodes a K-bZIP protein important for viral gene transcrip-tion (19, 21, 28, 49), DNA replicatranscrip-tion (1, 22, 51), and cell cycle arrest (20, 52, 53). To investigate what promotes the splicing of this intron, we demonstrated that a ciselement, a nt 75838 5⬘ ss crossing over exon 3, could trigger the splicing. This finding is consistent with a well-established exon definition proposing that cross talk between a 3⬘ss and a 5⬘ ss over the exon promotes splicing of an upstream intron (2, 17, 42, 57). Based on this principle, we inserted a human-globin intron into the K8exon 3-exon 4 splice junction and demonstrated that this intron insertion stimulated efficient splicing of the K8intron and resulted in production of K8␣mRNA from a K8cDNA. Together, our RNA and protein expression data provide conclusive evidence that the splicing of K8 intron 2 is enhanced by the exon definition machinery crossing over exon 3 and that the K8 RNA is derived from an RNA splicing intermediate during viral RNA processing.
The production of K8and its escape from NMD-mediated degradation in early viral lytic infection suggest that the ex-pression of K8may play a role in viral gene expression and pathogenesis. The K8 protein has exactly the same amino acid sequences as the K-bZIP (K8␣) protein in its N-terminal
region, including a basic domain close to the leucine zipper domain, but is missing a C-terminal leucine zipper domain encoded by the K8 exon 3 region (23). Recent studies have shown that the KSHV K-bZIP protein increases p21 expres-sion and causes p21-mediated G1 cell cycle arrest through
[image:13.585.134.454.67.362.2]CCAAT/enhancer-binding protein-␣(C/EBP␣) (52, 53) and/or inhibition of CDK2 (20). The K-bZIP protein binds very effi-ciently to both C/EBP␣ and p21 and stabilizes them from proteasome-mediated protein degradation. Although the only region of the K-bZIP protein that interacts with and stabilizes C/EBP␣is the basic leucine zipper region in the C terminus, both the N-terminal and C-terminal domains of the K-bZIP protein are required to stimulate C/EBP␣-induced expression from both the C/EBP␣ and p21 promoters in cotransfected cells (53). In contrast, only the basic region (residues 123 to 189), not the leucine zipper region, of K-bZIP was found to play a functional role in CDK2 binding and down-modulation of CDK2 activity (20). Consistent with these reports, we also demonstrated in our transient transfection assays that expres-sion of the K-bZIP protein could cause both p53 and p21 induction. However, coexpression of K-bZIP and K8in the cells reduced the K-bZIP-mediated p53 and p21 induction, suggesting that K8acts as an antagonist. Moreover, we fur-ther demonstrated that the expression of K8also blocks the
FIG. 9. Expression of K8 (pST3) antagonizes K8␣ (pST1)-mediated induction of p21 in BCBL-1 cells. BCBL-1 cells at 1 ⫻107were transfected by electroporation with individual plasmid or in combinations as indicated in each panel. Vector pcDNA3 without any insert was included as a negative control or to validate the transfection at the same plasmid DNA level. Cell lysates from BCBL-1 cells with or without TPA treatment for 72 h were also included as controls for induction of p21 (A) and K8 (B) expression. All transfected cells were collected at 72 h after transfection and were analyzed for p21 expression (A) by Western blotting and for K8␣and K8expression (B) by RT-PCR. The same membrane was also blotted for tubulin as sample loading control (A). Total cell RNA in each RT-PCR assay was normalized with GAPDH RNA detection, and a relative ratio (fold) of K8␣to K8was calculated based on individual band density (B).
on November 8, 2019 by guest
http://jvi.asm.org/
interaction of K-bZIP with CDK2. This is the first report of a biological function for K8.
Although enhancement of p21 expression by K-bZIP can be induced in a p53-independent pathway (53), the finding of K-bZIP interaction with p53 (20, 28) in other laboratories also provides some convincing evidence for interpreting our results, indicating that K-bZIP-p53 interaction might stabilize p53 from degradation and result in accumulation of the p53 that activates transcription of the p21 gene. More interestingly, we found a significant reduction of p21 expression, along with a reciprocal suppression of K-bZIP and K8 expression, when equal amounts of expression vector pST1 (mainly for K-bZIP expression) and pST3 (for K8) were cotransfected. In this case, the expression levels of the K8␣mRNAs in the cotrans-fected cells were greatly reduced along with an unrestricted expression of K8 mRNA, suggesting that overexpression of K8RNA might affect splicing of K8␣pre-mRNA. However, how K-bZIP suppresses K8protein expression remains to be further understood. Altogether, these observations clearly in-dicate that expression of K8␣and K8must remain at a con-stant ratio. Production of K8over a threshold is detrimental to K8␣expression and consequently is subtly regulated by K8␣. Supporting this is the fact that it has been found consistently that K8␣and K8mRNA are expressed at approximately 5-6 to 1 (this study and Tang and Zheng [46]). It will be interesting to know how the virus keeps this ratio of K8␣and K8mRNAs in check during viral gene expression and what controls this regulation, in addition to the exon definition we described in this study.
In summary, we have investigated in detail the features of an intron from KSHV nt 75563 to 75645 and its inclusion in a isoform mRNA during KSHV ORF50 and K8 expression. Be-cause the 3⬘UTR of the ORF50 transcript overlaps the entire K8 transcript, we simplified our focus on understanding alter-native splicing of this intron in processing of the K8 pre-mRNA to exemplify the way the inclusion or exclusion of this intron may take place during the splicing of the ORF50 pre-mRNA. Both the ␣ and  isoforms of the ORF50 mRNA presumably encode the same protein (replication and tran-scription activator; RTA) with the same size, since the intron from nt 75563 to 75645 in the ORF50 transcript is positioned in its 3⬘UTR; however, the inclusion of this intron in the K8 mRNA leads to the production of a truncated protein that plays a dominant negative role in K8␣mRNA expression and K8␣function.
ACKNOWLEDGMENTS
This study was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. K.Y. was supported by a research fellowship from the Japan Society for the Promotion of Science at NIH.
We thank Richard Ambinder for providing JSC-1 cells and the NIAID AIDS Research and Reference Reagent Program, Division of AIDS, for providing BCBL-1 cells originally obtained from Michael McGrath and Don Ganem. We also thank Don Ganem for providing the rabbit polyclonal anti-K-bZIP antibody, John Brady for p53 re-porter plasmid, and Yan Yuan for pORF50 expression vector.
REFERENCES
1.AuCoin, D. P., K. S. Colletti, S. A. Cei, I. Papouskova, M. Tarrant, and G. S. Pari.2004. Amplification of the Kaposi’s sarcoma-associated herpesvirus/ human herpesvirus 8 lytic origin of DNA replication is dependent upon a
cis-acting AT-rich region and an ORF50 response element and thetrans -acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology318:542–555. 2.Berget, S. M.1995. Exon recognition in vertebrate splicing. J. Biol. Chem.
270:2411–2414.
3.Bu¨hler, M., A. Paillusson, and O. Mu¨hlemann.2004. Efficient downregula-tion of immunoglobulinmRNA with premature translation-termination codons requires the 5⬘-half of the VDJ exon. Nucleic Acids Res. 32:
3304–3315.
4.Buzina, A., and M. J. Shulman.1999. Infrequent translation of a nonsense codon is sufficient to decrease mRNA level. Mol. Biol. Cell10:515–524. 5.Carter, M. S., J. Doskow, P. Morris, S. Li, R. P. Nhim, S. Sandstedt, and
M. F. Wilkinson.1995. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J. Biol. Chem.270:28995–29003.
6.Chandran, B., C. Bloomer, S. R. Chan, L. Zhu, E. Goldstein, and R. Horvat.
1998. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glyco-proteins generated by spliced transcripts. Virology249:140–149.
7.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore.1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science266:1865–1869.
8.Donis-Keller, H.1979. Site specific enzymatic cleavage of RNA. Nucleic Acids Res.7:179–192.
9.Fakhari, F. D., and D. P. Dittmer.2002. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quan-titative PCR. J. Virol.76:6213–6223.
10.Frilander, M. J., and J. A. Steitz.1999. Initial recognition of U12-dependent introns requires both U11/5⬘splice-site and U12/branchpoint interactions. Genes Dev.13:851–863.
11.Graveley, B. R.2000. Sorting out the complexity of SR protein functions. RNA6:1197–1211.
12.Graveley, B. R., K. J. Hertel, and T. Maniatis.2001. The role of U2AF35 and U2AF65 in enhancer-dependent splicing. RNA7:806–818.
13.Gruffat, H., S. Portes-Sentis, A. Sergeant, and E. Manet.1999. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) encodes a homo-logue of the Epstein-Barr virus bZip protein EB1. J. Gen. Virol.80:557–561. 14.Gunzl, A., Z. Palfi, and A. Bindereif.2002. Analysis of RNA-protein com-plexes by oligonucleotide-targeted RNase H digestion. Methods26:162–169. 15.Hamm, J., and I. W. Mattaj.1990. Monomethylated cap structures facilitate
RNA export from the nucleus. Cell63:109–118.
16.Hayward, G. S. 2003. Initiation of angiogenic Kaposi’s sarcoma lesions. Cancer Cell3:1–3.
17.Hwang, D. Y., and J. B. Cohen.1996. Base pairing at the 5⬘splice site with U1 small nuclear RNA promotes splicing of the upstream intron but may be dispensable for slicing of the downstream intron. Mol. Cell. Biol. 16:
3012–3022.
18.Inacio, A., A. L. Silva, J. Pinto, X. Ji, A. Morgado, F. Almeida, P. Faustino, J. Lavinha, S. A. Liebhaber, and L. Romao.2004. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J. Biol. Chem.279:32170–32180.
19.Izumiya, Y., S. F. Lin, T. Ellison, L. Y. Chen, C. Izumiya, P. Luciw, and H. J. Kung.2003. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregula-tor of K-Rta: physical association and promoter-dependent transcriptional repression. J. Virol.77:1441–1451.
20.Izumiya, Y., S. F. Lin, T. J. Ellison, A. M. Levy, G. L. Mayeur, C. Izumiya, and H. J. Kung.2003. Cell cycle regulation by Kaposi’s sarcoma-associated herpesvirus K-bZIP: direct interaction with cyclin-CDK2 and induction of G1growth arrest. J. Virol.77:9652–9661.
21.Liao, W., Y. Tang, S. F. Lin, H. J. Kung, and C. Z. Giam.2003. K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/ HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactiva-tion. J. Virol.77:3809–3815.
22.Lin, C. L., H. Li, Y. Wang, F. X. Zhu, S. Kudchodkar, and Y. Yuan.2003. Kaposi’s sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: identification of theori-Lytand association of K8 bZip protein with the origin. J. Virol.77:5578–5588.
23.Lin, S. F., D. R. Robinson, G. Miller, and H. J. Kung. 1999. Kaposi’s sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol.73:1909–1917.
24.Lukac, D. M., J. R. Kirshner, and D. Ganem.1999. Transcriptional activa-tion by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol.73:
9348–9361.
25.Maquat, L. E.2004. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol.5:89–99.
26.Miller, G., M. O. Rigsby, L. Heston, E. Grogan, R. Sun, C. Metroka, J. A. Levy, S. J. Gao, Y. Chang, and P. Moore.1996. Antibodies to butyrate-inducible antigens of Kaposi’s sarcoma-associated herpesvirus in patients with HIV-1 infection. N. Engl. J. Med.334:1292–1297.
27.Moore, P. S., S. J. Gao, G. Dominguez, E. Cesarman, O. Lungu, D. M. Knowles, R. Garber, P. E. Pellett, D. J. McGeoch, and Y. Chang.1996. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J. Virol.70:549–558.