0022-538X/06/$08.00⫹0 doi:10.1128/JVI.02720-05
Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Rinderpest Virus Blocks Type I and Type II Interferon Action:
Role of Structural and Nonstructural Proteins
Sambit K. Nanda and Michael D. Baron*
Institute for Animal Health, Ash Road, Pirbright, Surrey GU24 0NF, United Kingdom
Received 27 December 2005/Accepted 15 May 2006
Rinderpest virus(RPV) is a paramyxovirus closely related to the human pathogenMeasles virus. It causes
severe disease in cattle, buffalo, and some wild animals; although it can infect humans, it does not cause disease. Here, we demonstrate that RPV blocks the action of both type I (␣) and type II (␥) interferons (IFNs) by blocking the phosphorylation and nuclear translocation of STAT1 and STAT2 and that this block is not related to species specificity. In addition, both wild-type virulent and vaccine strains of the virus blocked IFN action. Unlike the case with some other paramyxoviruses, neither STAT1 nor STAT2 is degraded upon virus infection. STAT1 is bound by both the viral structural protein P, and thereby recruited to concentrations of viral protein in the cell, and the nonstructural protein V. Although both P and V proteins bind to STAT1 and can block IFN action when expressed in transfected cells, the IFN antagonist activity of the P protein is weaker than that of the V protein. The viral C protein also seems to weakly block IFN-induced activation of STAT1 in transfection experiments. However, studies with knockout viruses showed that the viral V protein appears to be the dominant inhibitor of IFN signaling in the context of virus infection, since prevention of viral V expression restored the IFN sensitivity of infected cells. Although a change in the distribution pattern of STAT2 was observed in virus-infected cells, STAT2 was not bound by any viral protein.
Rinderpest is a highly contagious viral disease that affects even-toed ungulates of the order Artiodactyla, primarily do-mestic and wild bovids. The causative agent isRinderpest virus
(RPV), which belongs to the genusMorbillivirusof the family
Paramyxoviridae (55). Though there is only one serotype of
RPV, strains vary greatly in their pathogenicity, from those causing 100% mortality, as seen with the Saudi/81 strain, to others in which infection causes no detectable clinical signs (74, 77). The molecular basis of this variation is not known.
The aim of the host innate immune response is to eliminate the pathogen or to avoid the spread of the disease until an orchestrated adaptive immune response is launched to clear the infection. During virus infection, one of the most promi-nent groups of cytokines produced are the interferons (IFNs), named for their ability to interfere with virus replication. In addition, IFNs have profound immunomodulatory functions and promote an adaptive immune response (21, 59, 61). The onset of type I (primarily IFN-␣and -) synthesis and secre-tion occurs in response to the detecsecre-tion of virus by Toll-like receptors and/or detection of viral proteins or RNA by cyto-plasmic pathways (20, 31, 36, 41, 65, 79, 80). The type I IFNs mediate their biological function by binding to the IFN-␣ re-ceptor on the target cells, resulting in the autophosphorylation and activation of the receptor-associated Janus kinases (JAKs), JAK1 and tyrosine kinase 2 (TYK2), and then the phosphory-lation of the signaling molecules STAT1 and STAT2 (13, 29). The phosphorylated STATs heterodimerize and translocate to the nucleus, where they associate with interferon regulatory factor 9 (IRF-9) (p48), forming interferon-stimulated gene
factor 3 (ISGF3) (12, 27), which activates the transcription of a large number of genes involved in establishing a virus-resis-tant state in the cell (15, 19). The importance of IFN-␣/ in protecting the host against virus infection has been demon-strated by the finding that knockout mice lacking type I IFN receptors or components of the JAK-STAT pathway are ex-tremely susceptible to virus infections (17, 50, 76).
While type I IFNs are produced by most cell types, type II IFNs (IFN-␥) are mainly produced by Th1 CD4⫹helper T cells and CD8⫹T cells after stimulation by antigen-presenting cells. IFN-␥activates antiviral genes through a different receptor, leading to phosphorylation of STAT1 but not STAT2; ho-modimers of phosphorylated STAT1 are translocated to the nucleus where they activate transcription of a different set of genes.
It has become clear from the evidence found in last few years that paramyxoviruses have adopted different strategies to an-tagonize IFN function by blocking the signal transduction pathway mediated by IFN, to their replicative advantage. The nonstructural proteins of the members of the family
Paramyxo-viridae, which includes important human and animal viruses
like measles virus (MV), mumps virus (MuV), parainfluenza viruses, respiratory syncytial viruses, Newcastle disease virus (NDV), and also viruses of zoonotic importance like Hendra and Nipah viruses, have been implicated in blocking IFN ac-tion (reviewed in references 21, 23, and 26). The P genes of paramyxoviruses are known to encode one or more nonstruc-tural proteins through utilization of overlapping open reading frames (ORFs) (giving rise to the proteins called C) and a process known as RNA editing (9, 53) which gives rise to the V and W proteins. The V protein has the amino-terminal half of the P protein followed by a highly conserved cysteine-rich domain that binds zinc, while the W protein (usually produced in very small amounts) consists of just the amino-terminal half
* Corresponding author. Mailing address: Institute for Animal Health, Ash Road, Pirbright, Surrey GU24 0NF, United Kingdom. Phone: 44 (0) 1483 231024. Fax: 44 (0) 1483 232448. E-mail: michael .baron@bbsrc.ac.uk.
7555
on November 8, 2019 by guest
http://jvi.asm.org/
of P in most paramyxoviruses (40, 54), although the W protein of the recently identified henipaviruses has a substantial (45 amino acids) W-specific sequence downstream of the editing site (78). Recent studies have demonstrated that a number of paramyxoviruses inhibit IFN signaling pathways by various mechanisms involving one or more of their nonstructural pro-teins (23, 83). The V propro-teins of simian virus 5 (SV5), MuV, and NDV target STAT1 for proteasomal degradation (16, 28, 34, 81), whereas the human parainfluenza virus type 2 V pro-tein targets STAT2 for degradation (44, 49). Affinity purifica-tion and yeast two-hybrid studies have suggested that the STAT1-targeting machinery consists of the V protein-depen-dent degradation complex that includes the V protein, STAT1, STAT2, P127 UV-damage-specific-DNA binding protein 1 (DDB1), and cullin 4A (1, 39, 75). A recent study has shown that the V protein of MuV also directly inhibits STAT1 and STAT2 phosphorylation (35). The V protein of NDV antago-nizes IFN signaling more efficiently in avian cells, suggesting a role in species specificity (51).
The V proteins of Hendra and Nipah viruses do not cause the degradation of STAT proteins, but they have been found to sequester STAT1 and STAT2 to the cytoplasm, preventing activation and nuclear translocation (57, 58). The C protein of Nipah virus along with P, V, or W proteins was also found to have anti-IFN activity (52, 64).
In theMorbillivirusgenus, the V and P proteins of MV were both found to inhibit IFN signaling (45) without degrading STAT1 or STAT2, and V has been suggested to block the transport of type I or type II IFN-activated STAT molecules to the nucleus without interfering with their tyrosine phosphory-lation (47). Other studies, however, found that the V protein of MV blocked only type I IFN-stimulated STAT1 and STAT2 phosphorylation, without an effect on IFN-␥action (71), find-ings supported by the observation that both C and V proteins of MV suppress JAK1 phosphorylation by association with the IFN-␣receptor complex (82). The C protein of MV has also been implicated in blocking the type I IFN response, although the exact molecular mechanism is not known (62).
Initial studies in our laboratory showed that both virulent (Saudi/81) and vaccine (RBOK) strains of RPV grow in cells treated with IFN-␣ and can block induction of Mx protein production in response to IFN treatment (data not shown), suggesting that RPV has mechanisms to circumvent the action of IFN. To analyze these mechanisms, we decided to look at the initial events in the IFN signaling pathway, i.e., activation of STAT1 and STAT2. We have found that the viral C, P, and V proteins can block IFN action when expressed in cells. In the context of viral infection, however, the V protein appears to be the important functional effector. This block of the IFN sig-naling pathways by RPV does not appear to be related to host species specificity or sufficient for virulence.
MATERIALS AND METHODS
Cells and viruses.Vero (African green monkey kidney) and A549 (human lung carcinoma) cells were grown in Dulbecco’s modified Eagle’s medium con-taining 5% fetal calf serum (FCS), 25 mM HEPES buffer, penicillin (100 U/ml), and streptomycin sulfate (100 g/ml). B95a cells (Epstein-Barr virus-trans-formed tamarin B-lymphoblastoid cell line) were maintained in RPMI 1640 medium (GIBCO-Invitrogen) containing 10% FCS. Bovine skin fibroblast (BSF) cells were prepared earlier in the lab (5) following the protocol described by Childerstone et al. (11). The cells were maintained in Iscove’s modified Dulbecco’s
modified Eagle’s medium (GIBCO-Invitrogen) supplemented with 10% FCS, penicillin, and streptomycin. FCS for culturing BSF cells was rechecked in-house for bovine viral diarrhea virus contamination. The following recombinant viruses rescued in the laboratory have been used for this study: recombinant RPV (RBOK strain) rRPV 2C (14), recombinant RBOK V-knockout virus (rRPV V⫺), C-knockout virus (rRPV C⫺), and double (V and C)-knockout virus (rRPV V⫺C⫺) (5). Virus stocks were grown and had their titers determined in Vero cells. The wild-type RPV strain Saudi/81 was grown in B95a cells, as it had not been observed to induce IFN production (unpublished data). All cells and virus stocks were negative for mycoplasma (4⬘,6-diamidino-2-phenylindole [DAPI] staining).
Plasmids and antibodies.Except where indicated, all DNA manipulation was done following standard methods. Plasmids were cloned and grown in Esche-richia coli(DH5␣strain) and purified on CsCl gradients or using QIAGEN midi prep kits. The C ORF was obtained from pGST-C (70) by digestion with BamHI and inserted into the multiple-cloning site of pcDNA3.1⫹plasmid (BamHI cut) to make pcDNA-C. We constructed expression plasmids expressing either P or V protein (but not C protein) by introducing the same mutations used for prevent-ing expression in RPV C virus (5), with an additional replacement of C by A at position⫺3 relative to the start codon of the P/V ORF, thereby strengthening the use of this start codon (33) to reduce the use of downstream start codons in the C ORF. The insert PCE (RBOK P gene in which the start codon of the C gene has been changed from AUG to ACG) from the plasmid pKS-PCE (5) was introduced into pcDNA at the EcoRI site. To generate pcDNA-PCSTOP or pcDNA-VCSTOP (expressing either P or V protein only), PCR amplification was done by using MDBPVOPTSTART (5⬘-CGATTATGAATTCAAGATGG CAGAGGAGCAAG) with CSTOPM1 (5⬘-CTTAGAGCTTTTATGCATTCCA GGCCTTTGT) to amplify a 60-bp fragment at the start of the P/V ORF. PCR amplifications were done to amplify the rest of the ORFs with CSTOPM2 (5⬘-CATAAAAGCTCTAAGAGCCCGACCCCTCAG) and KATPEND (5⬘-TC TCGCGGCCGCCTAGTTCTTTAGAATTTTGACC) using pcDNA-PCE as the template (P ORF) or CSTOPM2 and KATVEND (5⬘-TCTCGCGGCCGC TCAATGTTACTCTGGGATAT) using pcDNA-V (an existing cDNA clone of RBOK V mRNA) as the template (V ORF). These reactions generated⬃ 1.5-kbp and⬃1-kb fragments, respectively. Overlapping PCRs were done with the 60-bp and⬃1.5-kbp products generated above using the MDBPVOPTSTART and KATPEND primer pair and with the 60-bp and⬃1-kb V-specific fragments using MDBPVOPTSTART and KATVEND primer pairs. The final products were cloned into pcDNA3.1(⫹) using the EcoRI and NotI sites at N and C termini, respectively, giving pcDNA-PCSTOP and pcDNA-VCSTOP. These plasmids were used in all experiments where P or V was expressed in transfected cells. Plasmids pcDNA-C-V5 and pcDNA-W-V5 were made by amplifying, re-spectively, the C ORF from pcDNA-C (using primers MDBV5CFOR (5⬘GCT CGAGAATTCAAGATGGGGTCAACAAAGGCCTGG) and MDBV5CREV (5⬘-CTGGTCTCTAGACTGTTTCAACATCGGAGGCT) or the W ORF from pcDNA-PCSTOP (using primers MDBWV5FOR (CAGATGGAATTCAAGA TGGCAGAGGAGCAAG) and MDBWV5REV (5⬘-CGGTTGTCTAGACGT CTGTGCTGCCTTTTTAATGGG), digesting the resultant PCR products with EcoRI and XbaI and ligating into EcoRI-XbaI–cut pcDNA6-V5HISa. Plasmid pcDNA-N was made by transferring the N ORF (EcoRI cut) from pKS-N (4) to pcDNA3.1(⫹). pcDNA-HAL, expressing a HA-tagged L protein of RPV, has been previously described (66). pcDNA-IRES-eGFP was obtained from K. Mof-fat (Division of Immunology, Institute for Animal Health, Pirbright, United Kingdom). Plasmids pJAT-lacZ, pGAS-luc, and pISRE-luc (16) were the kind gifts of S. Goodbourn, St. George’s Hospital Medical School, London, United Kingdom. All PCRs were performed using proofreading polymerase (KOD; Novagen). All the PCR products introduced into plasmids were sequenced en-tirely following manufacturer’s protocols (Beckman Coulter).
Mouse monoclonal antibodies against STAT1 and phosphotyrosine 701-STAT1 were purchased from BD Biosciences. Polyclonal antibodies against STAT1, STAT2, and phosphotyrosine 689-STAT2 were obtained from Upstate. Mouse monoclonal antibodies, against proliferating cell nuclear antigen (PCNA) and STAT2, and polyclonal antibodies against IRF-1 and c-Myc protein (A-14) were obtained from Santa Cruz Biotechnology. Rat monoclonal HA anti-bodies (with and without horseradish peroxidase [HRP]) were obtained from Roche. Rabbit polyclonal antibody against green fluorescent protein was from Abcam. Rabbit polyclonal antibodies against RPV P (MB18), N (MB2), and C (MB36) proteins were previously prepared in our lab (4, 70). Mouse monoclonal antibodies (U32) (against an epitope common to RPV P and V proteins) and 2-1 (recognizing specifically the RPV P protein) were the kind gift of M. Sugiyama, Department of Veterinary Public Health, Faculty of Agriculture, Gifu Univer-sity, Gifu, Japan (69). The rabbit anti-V antiserum recognizing the carboxy terminus of the RPV V protein was the kind gift of D. Briedis, Department of
on November 8, 2019 by guest
http://jvi.asm.org/
Microbiology and Immunology, McGill University, Montreal, Quebec, Canada. Rabbit polyclonal antiserum against the Mx protein was the kind gift of I. Julkkonen, National Public Health Institute, Helsinki, Finland. Mouse monoclo-nal antibody against the V5 epitope tag was the kind gift of R. Randall, St. Andrews University, Scotland. The Alexa Fluor 488 or 568 antimouse or anti-rabbit secondary antibodies used for immunofluorescence experiments were purchased from Invitrogen.
Virus infection, IFN treatment, cell extraction, and immunoblotting.BSF, A549, and Vero cells were plated at an initial seeding density of 2⫻105
cells/well in 6-well plates. The cells were mock infected or infected with RPV (Saudi/81) at a multiplicity of infection (MOI) of 5. The virus-infected cells were incubated for 1 to 2 h before removing the virus inoculum and replacing it with fresh growth medium. The infected cells were further incubated for 18 h and treated with 1,000 IU/ml human IFN-␣A or 5 ng/ml bovine (BSF) or 1,000 IU/ml human (A549, Vero) IFN-␥. Recombinant human IFN-␣A and recombinant human IFN-␥were obtained from Calbiochem; bovine IFN-␥was obtained from C. Howard, Institute for Animal Health, Compton Laboratories. Cells were treated for 30 min or mock treated. The cells were harvested and lysed with 100l of 1⫻ sodium dodecyl sulfate (SDS) sample buffer (NEB) and heated immediately for 5 min at 95°C. Required volumes of the protein samples were analyzed by SDS-polyacrylamide gel electrophoresis on 8% acrylamide gels using the buffer system described by Laemmli (37), electrotransferred onto polyvinylidene diflu-oride membranes (Immobilon-P, Millipore), blocked for at least 1 h in 50 mM Tris/Cl (pH 7.5), 150 mM NaCl, and 0.1% (vol/vol) Tween 20 (TBST) containing 5% (wt/vol) dried milk (“Marvel”), and immunoblotted with specific primary antibodies. Specific proteins were detected using HRP-conjugated goat anti-mouse or anti-rabbit antibodies (Amersham) or HRP-conjugated antirabbit TrueBlot (eBioscience) and the Super-Signal West-Pico chemiluminescence re-agent (Pierce) or the Immobilon Western detection rere-agent (Millipore).
Immunofluorescence.Immunofluorescent staining and confocal microscopy were carried out essentially as described previously (66). Briefly, BSF and Vero cells grown on 13-mm-diameter coverslips (Fisher) to approximately 60% con-fluence were either mock infected or infected at an MOI of 0.1. Eighteen hours postinfection, the cells were treated with IFN for 30 min or mock treated. In order to look at STAT1P or STAT2P, cells were then fixed/permeabilized for 5 min in methanol:acetone (1:1) at room temperature; for staining with all other antibodies, cells were fixed with 3% paraformaldehyde and then permeabilized with 0.4% Triton X-100. The fixed and permeabilized cells were blocked with 10% goat serum for at least 30 min before processing for antibody staining. Initial confocal microscope images were obtained using the manufacturer’s soft-ware (Leica Confocal Softsoft-ware) and were resized for printing and overlays using Adobe Photoshop. All experiments were performed at least three times, and representative images are shown.
Plasmid transfection.BSF cells grown on 13-mm-diameter coverslips to ap-proximately 60% confluence were transfected with 1g of specific plasmid using FuGENE6 (Roche) as described by the manufacturer, using a ratio of 3l of transfection reagent perg of plasmid DNA. For transfecting A549 and Vero cells, the cells were grown to approximately 90% confluence and then transfected with 1g of plasmid using Lipofectamine 2000 transfection reagent (Invitrogen) following the manufacturer’s protocol. Two hours after transfection, the trans-fection mix was removed and the normal growth medium was added to the cells. Twenty hours posttransfection, the cells were incubated with or without IFN-␣A or IFN-␥for 30 min and then fixed and processed for immunofluorescent stain-ing. All experiments were performed at least three times, and representative images are shown.
Coimmunoprecipitations.Vero cells were seeded at a density of 4⫻105
cells/well in 6-well plates; 20 h later, they were transfected with 4g of the desired plasmid using Lipofectamine 2000 (Invitrogen) as specified by the man-ufacturer. After 48 h of transfection, the cells were lysed using low-salt lysis buffer (50 mM Tris/Cl [pH 7.5], 150 mM NaCl, 2 mM EDTA, 1% (vol/vol) Nonidet P-40) containing protease inhibitor cocktail set III (Calbiochem) at a final dilution of 1/200; for the studies of C protein binding to STATs, the lysis buffer contained 500 mM NaCl, as we have found these conditions release more C protein from transfected cells. The lysates were centrifuged at 10,000⫻gfor 15 min, and the supernatants were subjected to immunoextraction with 1 to 2l of the appropriate antibody and protein G-agarose beads (Upstate) as previously published (66). The immunoextracts were analyzed by SDS-polyacrylamide gel electrophoresis and immunoblotting with specific antibodies.
IFN-induced-luciferase assays.Vero cells (3⫻105per well) were plated in
6-well plates. One day later they were transfected with (per well) 0.7g pJAT-lacZ plus 0.7g of either pISRE-luc or pGAS-luc plus 1.0g of either pcDNA, pcDNA-PCSTOP, pcDNA-VCSTOP, or pcDNA-C. Transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
After 24 h, IFN-␣A or IFN-␥was added to the medium at 1,000 U/ml and the cells were incubated for a further 18 h. The cells were then lysed in 400l lysis buffer (120 mM NaCl, 50 mM Tris/Cl [pH 7.5], 0.05% Nonidet P-40) and the luciferase activity was measured using luciferase assay reagent (Promega) and a Bio-Orbit 1237 luminometer.-Galactosidase activity in the same sample was measured as previously described (32). Each transfection was carried out in triplicate in each experiment, and each experiment was performed at least twice. Variations in the transfection efficiency were accounted for by calculating the ratio of luciferase activity to-galactosidase activity in any one sample (termed the relative luciferase units [RLU]); the ratio of the mean RLU in IFN-treated samples to the RLU in mock-treated samples was taken as the induction (n-fold) of the IFN-responsive promoter.
RESULTS
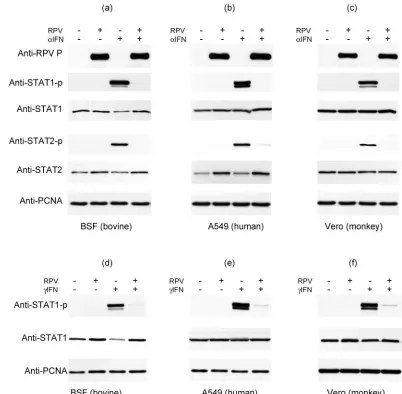
RPV blocks IFN-mediated phosphorylation of STAT1 and STAT2 without causing degradation of either.To determine the mechanism by which RPV infection suppresses IFN action, we first investigated the early events of the IFN signaling path-way, the phosphorylation of STAT1 and STAT2. Since obser-vations in related viruses have shown that STAT1 or STAT2 can be degraded by proteasomes in response to viral infection (16, 34, 49), we also examined the levels of STAT1 and STAT2 in RPV-infected cells. Bovine primary cells (BSFs) were mock infected or infected with wild-type RPV Saudi/81 at an MOI of 5. Under these conditions, almost all cells were infected, as checked by immunofluorescence (data not shown). Eighteen hours postinfection, the infected cells were treated with IFN-␣ for 30 min and the levels of phosphotyrosine 701-STAT1 (STAT1P) and phosphotyrosine 689-STAT2 (STAT2P), as well as the total STAT1 and STAT2, were measured in the same samples by immunoblotting (Fig. 1a). There was no de-tectable STAT1P or STAT2P in either the mock-infected or infected cells in the absence of IFN-␣treatment. In the mock-infected cells, STAT1P and STAT2P were detected following treatment with IFN-␣ (Fig. 1a). However, no STAT1P or STAT2P was detected in the cells infected with RPV. No reduction in the levels of STAT1 was seen in infected cells; in contrast, while uninfected cells showed a decrease in total STAT1 after treatment with IFN-␣, infected cells did not show this, so that IFN-treated, RPV-infected cells actually appeared to have more STAT1 than their uninfected counterparts. In-terestingly, there was a slight but reproducible increase in the amount of STAT2 in cells infected with the virus (Fig. 1a) visible in both IFN-treated and untreated cells. These data suggest that RPV completely blocks the type I IFN-induced phosphorylation of STAT1 and STAT2 but does not lead to the degradation of either STAT1 or STAT2.
The block of IFN-␣signaling by RPV is not species specific.
In respiratory syncytial virus, SV5, and NDV, the block of type I IFN signaling has been suggested to determine the species specificity of the virus (8, 48, 51). In order to address the question of whether the block of STAT1/2 phosphorylation by RPV is species specific and thereby possibly a factor involved in host specificity, similar experiments were done to look at levels of baseline STAT1 and STAT2 and IFN-induced STAT1P and STAT2P in RPV-infected A549 and Vero cells (Fig. 1b and c). There was a complete block of IFN-induced phosphorylation of both STAT1 and STAT2 in the cells in-fected with RPV, similar to the result found in BSF cells. In contrast to the findings with BSFs, the levels of STAT1 re-mained unchanged after IFN treatment in both A549 and Vero
on November 8, 2019 by guest
http://jvi.asm.org/
cells (Fig. 1b and c), and infection again had no effect on the level of STAT1 detected. Interestingly, the level of STAT2 was again slightly elevated in virus-infected A549 cells (Fig. 1b), as found in BSF cells; the level of STAT2 in RPV-infected Vero cells, on the other hand, was unchanged (Fig. 1c).
RPV also blocks IFN-␥action.We performed similar studies to determine if RPV infection could also block the IFN-␥ -stimulated phosphorylation of STAT1, a process involving a different membrane receptor and different receptor-associated kinases. Treatment of BSF cells with bovine IFN-␥, or A549 or Vero cells with human IFN-␥, for 30 min led to strong stimu-lation of STAT1 phosphorystimu-lation, a process that was almost completely blocked by RPV infection (Fig. 1d through f). We again saw a reduction in STAT1 in IFN-treated BSF cells, a process that was also blocked by RPV infection. Thus, RPV
infection blocks the initial steps in the signaling pathways of both type I and type II IFNs.
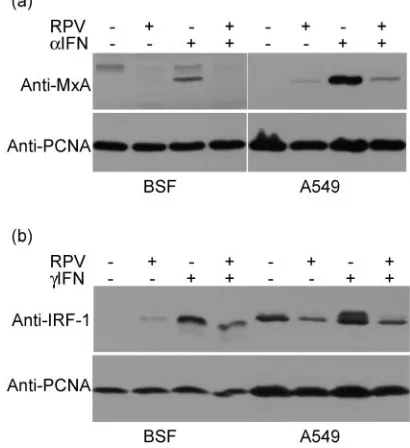
[image:4.585.86.488.68.462.2]Expression of IFN-induced proteins is also blocked in RPV-infected cells.We also determined whether the block of STAT phosphorylation was sufficient to block IFN action, as assessed by the induction of specific proteins by type I or type II IFNs. We chose the highly specific type I IFN-inducible protein Mx to measure response to IFN-␣ (68) and interferon response factor 1 (IRF-1) to measure response to IFN-␥ (21). Unin-fected BSF cells showed clear synthesis of Mx in response to overnight incubation with IFN-␣, while the infected cells showed none (Fig. 2a), indicating that the prevention of STAT1 and STAT2 phosphorylation had indeed blocked the type I IFN signaling pathway. A549 cells express much more Mx in response to type I IFN stimulation than observed in BSF
FIG. 1. RPV infection inhibits the tyrosine phosphorylation of STAT1 and STAT2 in response to IFN-␣and IFN-␥. BSF cells (a, d), A549 cells (b, e), or Vero cells (c, f) were infected with RPV Saudi/81 at an MOI of 5 or left uninfected. Eighteen hours postinfection, the cells were treated with or without IFN-␣(a to c), bovine IFN-␥(d) or human IFN-␥(e to f) for 30 min, and the cells were harvested and the levels of STATs and tyrosine-phosphorylated STAT proteins determined by Western blot analysis with the corresponding specific antibodies, as indicated to the left of each panel. PCNA levels served as a recovery/loading control; the levels of PCNA were checked for every experiment, although only representative data sets are presented here. Samples were also probed for the presence of viral protein (P) to confirm infection (a to c). The figure shows results from one of three experiments which all yielded consistent results.
on November 8, 2019 by guest
http://jvi.asm.org/
cells, and we observed a slight induction of Mx expression in infected cells not exposed to exogenous IFN-␣ (Fig. 2a); the stimulation of Mx expression due to added IFN-␣ was again blocked by RPV infection (Fig. 2a). Similarly, BSF and A549 cells infected with RPV did not express IRF-1 in response to IFN-␥, unlike the mock-infected cells (Fig. 2b); on the con-trary, RPV-infected A549 cells showed decreased expression of IRF-1.
STAT1 is sequestered to the cytoplasm by binding to viral phosphoprotein.Previous studies have shown that the P/V/W proteins of Nipah virus (57, 64) and the V protein of MV (47) interact with STAT molecules without causing their degrada-tion, and this interaction prevents the nuclear translocation of these molecules and thereby antagonizes IFN action. In Sendai virus (SeV), interaction of the C protein with STAT1 provides the underlying mechanism of the block of IFN signaling (72). Based on these observations, we wished to investigate the effects of the RPV nonstructural proteins V and C on IFN action. We have previously created mutant RPVs which ex-press no C (RPV C⫺), no V (RPV V⫺), or neither V nor C (RPV V⫺C⫺) (5). However, these viruses do not grow to a sufficient titer to achieve 100% infection of tissue culture cells (MOI ⱖ 5), so immunofluorescence was used to study the phosphorylation of STAT1 and STAT2 in infected cells. BSF cells were mock infected or infected with RPV at an MOI of ⱕ0.1 for 18 h and then processed for antibody staining to look first at total STAT1 and STAT2. In the uninfected cells,
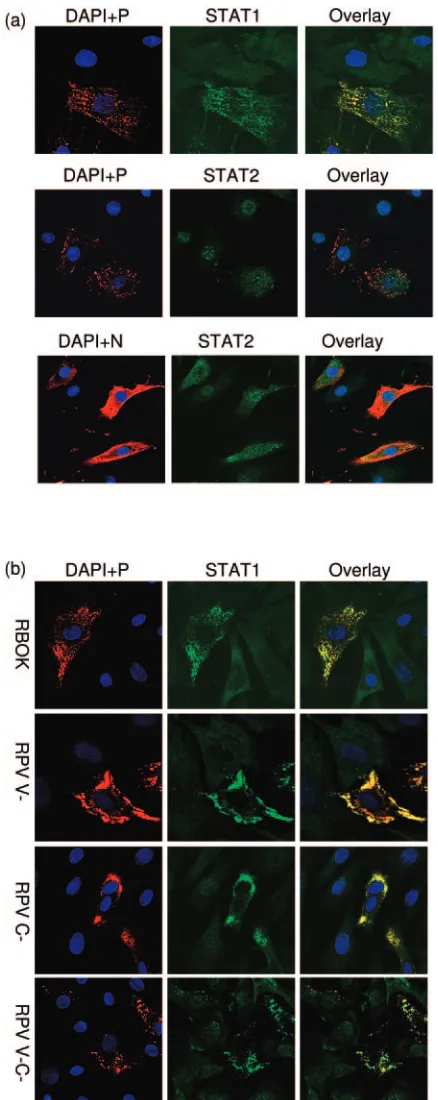
STAT1 and STAT2 are found predominantly in cytoplasm, although both the transcription factors shuttle between the nucleus and cytoplasm in unstimulated cells (2, 42, 60). In the cells infected with RPV, there was change in the distribution pattern of both the STAT proteins (Fig. 3a). STAT1 was found to colocalize with the viral P protein (and therefore also with the viral N, L, C, and possibly V proteins [70]) (Fig. 3a). These data suggest that STAT1 is sequestered in the cytoplasm by binding to some viral protein. In contrast, STAT2 was found in small inclusions in the nuclei of infected cells, in addition to a general distribution in the cytoplasm (Fig. 3a). Although there have been reports of morbillivirus N protein accumulation in the nuclei of infected cells or cells expressing N protein (46, 67), we did not observe N in the nuclei of most RPV-infected cells or any specific colocalization of N with STAT2 in either the cytoplasm or the nuclei of infected cells (Fig. 3a).
We observed similar distribution patterns of STAT1 and STAT2 in Vero and A549 cells infected with RPV (data not shown).
[image:5.585.59.264.74.298.2]In order to investigate the possible roles of the V or C protein in the subcellular distribution of STAT1, similar immunofluorescence experiments were done in BSF cells using the RPV V⫺, RPV C⫺, and RPV V⫺C⫺viruses. Since these mutants are based on the RBOK vaccine strain of RPV, we also demonstrated that STAT1 was bound to cy-toplasmic viral protein in cells infected with this strain. No difference was seen between the effects of the Saudi/81 and RBOK strains (Fig. 3a and b); in cells infected with the V-and/or C-knockout viruses, the STAT1 still colocalized with the viral protein complexes, suggesting that neither C nor V protein is individually or in combination responsible for the recruitment of STAT1 into these complexes (Fig. 3b). Again, no difference was seen between bovine (BSF) and primate (A549, Vero) cells. Since this effect on STAT1 must therefore be a direct or indirect effect of the viral P, N, or L protein, or a combination of these proteins, we looked to see if any of these proteins bound STAT1 directly. Vero cells were either mock transfected, transfected with blank vector, or transfected with mammalian expression plasmids encod-ing the viral N, P, or L proteins. The P gene ORF was modified to eliminate expression of the C protein (see Ma-terials and Methods). Cell lysates were immunoextracted with the corresponding specific antibodies for the viral pro-teins (in the case of the L protein, this was an antibody against the HA epitope tag cloned in frame at the amino terminus of the L protein). Then the immunoprecipitated proteins were analyzed by Western blotting with anti-STAT1 antibody. As a control, anti-STAT1 was immunoprecipi-tated with a specific anti-STAT1 antibody and analyzed on the same Western blots. STAT1 protein was detected nor-mally in cells transfected with empty plasmid vector, show-ing that transfection alone had no effect on STAT1 levels. No STAT1 was detected in immunoprecipitated N or L; however, STAT1 was detected in the immunoprecipitated P protein (Fig. 4). Interestingly, in a similar pull-down assay, viral V protein was also found to be interacting with STAT1 (Fig. 4). These data indicate that STAT1 is bound in the viral protein complexes in the infected cells by interaction with the viral P protein (since it also takes place in cells infected with RPV V⫺or RPV V⫺C⫺) and that the binding
FIG. 2. RPV infection blocks the expression of type I or type II IFN-inducible proteins. (a) BSF and A549 cells were mock infected or infected with RPV Saudi/81 at an MOI of 5 as indicated on the figure; 24 h postinfection the cells were treated with IFN-␣overnight or left untreated, and the level of MxA protein was determined by Western blotting. PCNA acted as the loading control. (b) BSF and A549 cells were infected as in the legend for panel a; 24 h postinfection, the cells were treated with bovine or human IFN-␥as appropriate or left un-treated. After a further 18 h incubation, the cells were harvested and the levels of IRF-1 were determined by Western blotting. The figure
shows results from one of two consistent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
site for STAT1 resides in the amino-terminal half of P, since this is shared with the V protein. We confirmed this by expressing the viral W protein, which in RPV is the amino-terminal half of P followed by just four amino acids (RAQT); only a few percent of P gene transcripts give rise to W (6). RPV W also bound STAT1 (Fig. 4), confirming that the STAT1 binding site lies in the first half of the P protein. The immunoprecipitates from these experiments were also screened for the presence of STAT2; in agreement with our immunofluorescence experiments, no STAT2 was found with any of the immunoprecipitated viral proteins (Fig. 4). The viral C protein did not appear to bind either STAT1 or STAT2 (Fig. 4). Treatment of transfected cells with IFN before lysis and immunoextraction had no appar-ent effect on the association of STAT1 or STAT2 with P or V (data not shown).
[image:6.585.54.273.70.620.2]P, V, and C proteins individually inhibit phosphorylation of STATs and/or their nuclear translocation. This binding of STAT1 to both viral P and V proteins might be related to the block of the IFN signaling pathway by the virus. In order to investigate this possibility, indirect immunofluorescence exper-iments were carried out to look at the effect of individually expressed P and V on the phosphorylation of STAT1 and subcellular distribution of STAT1P after IFN stimulation. BSF cells were transfected with plasmid expressing P, V, or other proteins. The P and V expression plasmids were again those modified to prevent expression of the C protein. Expression of green fluorescent protein (GFP) was taken as the control to study the effect of transfection on STAT1 activation. The transfected cells were either fixed/permeabilized directly or stimulated with IFN-␣for 30 min prior to fixation. Cells with-out IFN-␣stimulation did not show any STAT1P in the cyto-plasm or the nucleus, so transfection or expression of the proteins did not induce any STAT1 phosphorylation by itself (Fig. 5a through c) (negative control data shown only for GFP), while the cells transfected with plasmid expressing GFP and then stimulated with IFN-␣showed efficient phosphoryla-tion of STAT1 and translocaphosphoryla-tion of STAT1P to the nucleus (Fig. 5d through f). Similar activation of STAT1 was seen in cells expressing the viral N protein (Fig. 5g through i). After treatment with IFN-␣, cells expressing smaller amounts of P protein showed clear activation of STAT1; in cells with more P protein, STAT1P was not observed (Fig. 5j through l). Observ-ing 100 randomly selected transfected cells showed that 71% of P-expressing cells showed no detectable nuclear STAT1P. The RPV V protein was much more effective at blocking the acti-vation of STAT1, and we did not detect phosphorylated STAT1 in cells expressing even low levels of V, while the neighboring untransfected cells showed clear nuclear STAT1P (Fig. 5m through o); 100% of observed V-expressing cells were
FIG. 3. RPV virus infection alters STAT protein distribution in the cell. (a) BSF cells were infected with RPV Saudi/81 at an MOI of 0.1 for 18 h. The cells were fixed and stained with monoclonal antibody against either viral phosphoprotein (P) or nucleocapsid protein (N) and rabbit polyclonal antibody against either STAT1 or STAT2. The viral N or P was visualized using Alexa Fluor 568 (red) and the STAT1 or STAT2 using Alexa Fluor 488 (green) secondary antibodies. Nuclei were counterstained with DAPI (blue). (b) BSF cells were infected with RBOK, RPV V⫺, RPV C⫺, or RPV V⫺C⫺at an MOI of 0.1 for 18 h, as indicated on the left hand side of each panel. Cells were fixed and stained with monoclonal antibody against viral
phos-phoprotein (P; red) and rabbit polyclonal antibody against STAT1 (green); DAPI was used to stain nuclei (blue). Images were acquired by sequential laser scanning on the confocal microscope. For all the images, the left-hand panel represents merged images of the nucleus (blue) and viral protein (red) showing the infected cells, the middle panel shows the specific STAT1 or STAT2 protein stain, and the right-hand panel is an overlay of all three colors.
on November 8, 2019 by guest
http://jvi.asm.org/
negative for detectable nuclear STAT1P. Interestingly, similar experiments to look at the effect of the RPV C protein on STAT1 phosphorylation showed that this protein also appears to block STAT1 activation, albeit much less effectively than the V or P proteins (Fig. 5p through r). Only 22% of C-expressing cells were negative for nuclear STAT1P. We observed that, expressed alone, the C protein is mostly found in the nucleus (Fig. 5p), while the C protein in RPV-infected cells is predom-inantly found in the cytoplasm (70). This altered localization of the protein may have altered its interaction with host cell components. Given that virus infection seems able to block phosphorylation of STAT2, we also checked to see if the viral P, C, or V proteins also blocked this process. Cells transfected with P, C, or V expression plasmids were treated with IFN-␣ (or left untreated) and stained for the presence of the viral protein and STAT2P. As seen for STAT1, both P and V blocked the IFN-␣-stimulated phosphorylation of STAT2, but only V was completely effective, since no activated STAT2 was seen in cells expressing even the smallest detectable level of V but STAT2P was seen in the majority of cells expressing P (Fig. 5s through dd) (only 34% of P-expressing cells showed no detectable STAT2P). The C protein had an even more limited effect on STAT2 phosphorylation in response to IFN treat-ment (Fig. 5ee through jj), with 85% of C-expressing cells
showing at least some nuclear STAT2P. Similar results were obtained using transfected A549 cells (data not shown).
V and P proteins also block type II IFN action.BSF cells transfected with expression plasmids encoding RPV P, V, N, or C, or GFP, were treated with bovine IFN-␥to study the effect of the individual viral proteins on type II IFN action. As seen for type I IFN action, V protein was the most potent inhibitor of activation of STAT1 (Fig. 6m through o), and 100% of V-expressing cells observed had no detectable nuclear STAT1P. The P protein also blocked STAT1 phosphorylation to a degree similar to that blocked in response to IFN-␣(Fig. 6j through l) (69% of P-expressing cells blocked), while cells expressing C again showed a limited reduction in STAT1P levels (Fig. 6p through r) (18% of C-expressing cells blocked). Expression of RPV N or GFP had no observable effect on STAT1 phosphorylation in response to IFN-␥(Fig. 6d through i).
[image:7.585.89.496.69.297.2]In order to obtain quantitative measures of the inhibition of IFN action by each protein, we used reporter constructs in which expression of a reporter protein (luciferase) is governed by either an interferon response element-containing promoter or a gamma-activated-sequence-containing promoter. Vero cells were transfected with one of these plasmids along with a plasmid encoding RPV C, V, or P (or empty vector) plus
FIG. 4. STAT1 is bound to viral P, W, and V proteins. Vero cells were transfected with either empty plasmid vector or plasmid encoding the RPV P PCSTOP), N N), L (with an amino-terminal HA tag) HAL), V (with an amino-terminal c-Myc tag) (pcDNA-c-Myc-V), W (with carboxy-terminal V5 tag (pcDNA-W-V5), or C (pcDNA-C-V5), as indicated at the top of the figure. Forty-eight hours posttransfection, the transfected cells were lysed and the lysates were immunoextracted with mouse anti-P, rabbit anti-N, rat anti-HA, rabbit anti-c-Myc, or mouse anti-V5 antibodies. Cells transfected with empty vector were lysed and STAT1 or STAT2 was precipitated with the respective rabbit polyclonal antibody. The immunoprecipitated materials were probed by Western blotting for the presence of STAT1 (top panel) or STAT2 (middle panel). Rabbit polyclonal antibody against STAT1 was used to detect STAT1 in all the immunoprecipitated material. The primary antibody was detected with TrueBlot anti-rabbit immunoglobulin G (precipitates with anti-STAT1, anti-P, and anti-N) or HRP-antirabbit (precipitates with anti-HA, anti-c-Myc, or anti-V5). Mouse monoclonal anti-STAT2 antibody was used to detect STAT2 in the immune precipitates with polyclonal anti-N or anti-c-Myc antibodies, while polyclonal rabbit-anti-STAT2 was used to detect STAT2 in immunoprecipitates with mouse anti-P, mouse anti-V5, or rat anti-HA antibodies. The bottom panels show that the immunoprecipitates contained the expected viral protein, as shown by probing with rabbit anti-P, mouse anti-N, rat anti-HA, mouse anti-P/V, or mouse anti-V5, respectively. Note that samples precipitated with anti-V5 and probed with the same antibody showed clear immunoglobulin G-derived bands (the light chain visible in the blot showing C protein expression). i.p., immunoprecipitation; w.b., Western blotting.
on November 8, 2019 by guest
http://jvi.asm.org/
pJAT-lacZ to normalize transfection efficiency (see Materials and Methods for details); 24 h posttransfection, the cells were treated with IFN-␣or IFN-␥overnight to induce the expres-sion of luciferase, followed by lysis and determination of lucif-erase activity. Expression of individual viral proteins led to alterations in the base (uninduced) expression of luciferase
[image:8.585.75.511.65.540.2]and-galactosidase, so we compared the increase (n-fold) in RLU induced by IFN in each case. Figure 7 shows that expres-sion of the RPV V protein completely or almost completely blocked the induction of luciferase activity in response to ei-ther type of IFN, and expression of the P protein led to a similar, though not quite as strong, inhibition. Expression of
FIG. 5. RPV V, P, and C all inhibit IFN-induced phosphorylation of STATs. BSF cells were transfected with plasmids expressing GFP or N-, V-, P-, C-, or C-V5-tagged proteins as indicated. Twenty hours posttransfection, STAT1/STAT2 phosphorylation was induced where indicated by treatment with 1,000 U/ml of IFN-␣A for 30 min. The cells were fixed/permeabilized with methanol:acetone and stained with either the relevant rabbit polyclonal antibody to the expressed protein and mouse monoclonal antibody against pY701-STAT1 (a to r), mouse monoclonal antibody recognizing both P and V proteins and rabbit polyclonal (s to dd), or mouse monoclonal anti-V5 tag and rabbit anti-pY689-STAT2 (ee to jj). Expressed proteins were detected using Alexa Fluor 568 (red) except for GFP, which was detected with Alexa Fluor 488 (green). STAT1 and STAT2 were detected using Alexa Fluor 488 (green) for all except for cells transfected with plasmid expressing GFP, when Alexa Fluor 568 (red) was used. Nuclei were stained with DAPI (blue). All images were obtained by sequential laser scanning with the confocal microscope. For all the images, the top panel shows the overlay of nuclear staining (DAPI) and the expressed protein, the middle panel shows the phosphorylated STAT, and the bottom panel shows the overlay of phosphorylated STATs and expressed (exp.) protein.
on November 8, 2019 by guest
http://jvi.asm.org/
the RPV C protein failed to block the action of either type of IFN by this assay. It was apparent that the base levels of luciferase expression were particularly depressed in C-protein-expressing cells, and it is possible that this protein has a more general effect on cells which leads to a reduction in STAT1 phosphorylation.
V protein is the main inhibitor of STAT1 activation in in-fected cells.Having demonstrated that individually expressed P, V, and C proteins can each block, or partially block, the type I and type II IFN signaling pathways, we went on to validate our results for the involvement of these proteins in the antag-onism of IFN action in the context of virus infection, using the previously described V- and C-knockout viruses. In immuno-fluorescence experiments, phosphorylated STAT1 was not de-tected in unstimulated cells while, in contrast, uninfected cells treated with type I or type II IFN showed intense nuclear staining indicative of phosphorylation and nuclear transloca-tion of STAT1 (Fig. 8). As expected from the Western blotting experiments, there was no detectable STAT1P in the Saudi/ 81-infected cells after treatment with either IFN-␣or IFN-␥, while the neighboring uninfected cells have clear nuclear STAT1P (Fig. 8a through i). Cells infected with the RBOK vaccine strain also showed a complete absence of STAT1P after stimulation with either IFN-␣or IFN-␥(Fig. 8j through r). In comparison to the RBOK-infected cells, RPV V-infected cells (P⫹C⫹V⫺) showed clear STAT1P in their nuclei after treatment with IFN, although sometimes less than in the un-infected cells (Fig. 8s through aa). In contrast, cells un-infected with RPV C⫺virus (P⫹C⫺V⫹) showed little or no nuclear
STAT1P (Fig. 8bb through jj). These data suggest that, in the virus-infected cells, the V protein is required for a maximally efficient blockade of type I or type II IFN action. As expected, in RPV V⫺ C⫺-infected cells (P⫹ C⫺ V⫺) we found clear nuclear STAT1P at a level similar to that of neighboring un-infected cells; some STAT1P was also found colocalizing with viral protein (presumably P protein) in the cytoplasm. The binding of STAT1 (and STAT1P) by the P protein therefore appears not to be sufficient to prevent STAT1 activation and accumulation in the nucleus. Identical results were obtained in Vero cells, again showing that the ability of RPV to block the action of type I and type II IFNs is not species specific.
DISCUSSION
The establishment of viral infection is a battle between the virus and the host. Since the virus and host have coevolved, the host organism is armed with mechanisms to defeat the foreign pathogens and, likewise, the virus has adopted evasion strate-gies to circumvent the host response. The innate immune re-sponse represents the nonspecific first line of host cell defense against invading microorganisms, characterized by the produc-tion of various cytokines. IFNs are one of the most important groups of cytokines, having antiviral properties as well as play-ing a crucial role in shapplay-ing the pathogen-specific adaptive immune response (7).
[image:9.585.72.514.69.304.2]Several viruses of the subfamilyParamyxovirinaehave been shown to express proteins that inhibit the cellular response to type I IFN (and sometimes type II IFN) (21, 23). In the present
FIG. 6. RPV V, P, and C all inhibit IFN-␥-induced phosphorylation of STAT1. BSF cells were transfected with plasmid expressing GFP or N, V, P, or C proteins (as indicated at the bottom of the figure) for 20 hours. STAT1 phosphorylation was induced by treatment with 5 ng/ml of bovine IFN-␥for 30 min. The cells were fixed/permeabilized with methanol:acetone and stained with the relevant rabbit polyclonal antibody to the expressed protein and mouse monoclonal antibody against pY701-STAT1. Expressed proteins were detected using Alexa Fluor 568 (red) except for GFP, which was detected with Alexa Fluor 488 (green). Phosphorylated STAT1 was detected using Alexa Fluor 488 (green) for all except cells transfected with plasmid expressing GFP, when Alexa Fluor 568 (red) was used. Nuclei were stained with DAPI (blue). All images were obtained by sequential laser scanning with the confocal microscope. For all the figures, the bottom panels show the overlay of phosphorylated STAT1 and expressed protein. The top panel shows the overlay of nuclear staining (DAPI) and the expressed protein.
on November 8, 2019 by guest
http://jvi.asm.org/
study we have shown that RPV blocks the phosphorylation of STAT1 and STAT2 in response to stimulation with IFN-␣or -␥, although without causing the degradation of either STAT protein. This shows that the mechanism of inhibition of IFN signaling by RPV is different from those used by the members of the genera Rubulavirus and Avulavirus, where the viruses cause degradation of STAT proteins by proteasomes (16, 28, 49). Interestingly, the level of STAT2 is slightly up-regulated by RPV infection of some types of cells (A549 and BSF) in comparison to the mock-infected cells, as shown by Western blotting experiments. Up-regulation of STAT2 during virus infection has previously been reported in dendritic cells of mice infected with MV (25), a process thought to involve virus-induced type I IFN. Both A549 and BSF cells can make IFN, while Vero cells, which showed no increase in STAT2 levels, do not make type I IFN (43). Further evidence for low levels of IFN induction by RPV came from the studies on Mx induction. A549 cells produced much more Mx in response to IFN-␣than did BSF cells (Fig. 2), and this enabled us to see that low levels of this protein were being induced in the in-fected A549 cultures, presumably in the 4% to 5% of cells uninfected. If endogenous production of IFN is the cause of
the observed increase in STAT2 and the induction of Mx, the level of IFN involved must be very low, since we were unable to detect IFN in the supernatants of cells infected with RPV Saudi/81. We sometimes also observed an increase in STAT1 levels in infected cells (e.g., Fig. 1d), but this was not consis-tent. In uninfected cells, STAT2 is found mainly in the cyto-plasm, while we observed the accumulation of STAT2 in small nuclear bodies in RPV-infected cells. This STAT2 did not colocalize with viral P or N proteins, nor with promyelocytic leukemia nuclear bodies (data not shown), and further work is required to identify the nature of these structures and their biological significance. It is possible that this accumulation of STAT2 in the nucleus leads to a decrease in the turnover of the protein and hence an increase in steady-state levels.
RPV does not cause clinical disease in human beings but causes severe disease in even-toed ungulates, primarily cattle. To address the question of this species barrier to pathogenesis, we looked at the block of IFN signaling by RPV in human (A549) and monkey (Vero) cell lines, as the block of IFN signaling pathways by several members of paramyxoviruses has been suggested to be related to the species specificity of patho-genesis (48, 51). To our surprise, we found that both virulent and vaccine strains of the virus block the activation of STAT1 and STAT2 in both A549 and Vero cells. These data suggest that the block of IFN signaling by RPV is not a major factor governing species specificity of the virus. Given that cell entry is not a factor limiting the spread of these viruses, since they all seem to use the highly conserved CD150 protein (3, 73), the underlying mechanism for species specificity of disease contin-ues to be an interesting qcontin-uestion.
Interestingly, all the IFN-antagonist proteins in the
subfam-ily Paramyxovirinaeappear to be encoded by the viral
phos-phoprotein (P) gene. The members of the generaRubulavirus
andAvulavirusexpress P and V proteins but not a C protein
(38), and all the members of these two genera have been shown to antagonize IFN action through the V protein, causing the degradation of STAT1 or STAT2 (16, 28, 49). The significance of the unique cysteine-rich carboxy terminus of the V proteins in degradation of STAT proteins has been shown in V protein expression and also in knockout-virus infection studies (28, 34, 44). In a recent report, it has also been demonstrated that cysteine residues in the carboxy terminus of MuV V protein are not only required for the degradation of STAT1 but also for blocking the phosphorylation of both STAT1 and STAT2, indicating block of IFN signaling by multiple mechanisms (35). Although the carboxy terminus of the V protein is highly con-served among different paramyxoviruses, the ways these vi-ruses antagonize the IFN signaling pathway appear to vary. The V proteins of RPV (work reported here), of MV (45, 47, 71), and of Nipah and Hendra viruses (57, 58) do not cause degradation of STAT proteins; instead, they inhibit the acti-vation and/or nuclear translocation of STAT1 and STAT2. The essential region appears to be the amino-terminal sequence shared by the V, P, and W proteins (45, 64), although there is some evidence that carboxy-terminal residues of V may play an additional role (45). We found that both P and V of RPV inhibit interferon signaling, also suggesting that the amino-terminal part of these molecules is important. As seen in MV-infected cells (47), STAT1 was found to colocalize with viral proteins. We used a combination of knockout viruses and
co-FIG. 7. RPV V and P inhibit the induction of expression from IFN-responsive promoters. Vero cells were transfected with plasmids encoding the RPV P, V, or C proteins or blank vector, along with pJATLacZ and (a) pISRE-luc or (b) pGAS-luc. After 24 h of trans-fection, the cells were treated for 18 h with (a) IFN-␣or (b) IFN-␥, lysed, and the luciferase and-galactosidase activities measured. The ratio of these two activities was taken as the relative luciferase activity (in RLU). Shown are the data from a representative experiment; error bars represent one standard deviation. The induction (n-fold) (RLU⫹ IFN/RLU⫺IFN) is shown for each⫺IFN/⫹IFN pair.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:10.585.82.245.67.369.2]FIG. 8. Effect of RPV knockout virus on IFN-induced STAT1 activation. BSF cells were infected with the indicated virus (shown on the left of the panels) at an MOI of 0.1 for 18 h. The cells were treated with either 1,000 IU/ml of IFN-␣A or 5 ng/ml of bovine IFN-␥, or left untreated, prior to fixation/permeabilization using methanol:acetone (1:1). The cells were stained with rabbit anti-P antibody to detect infected cells and mouse anti-pY701-STAT1 antibody followed by Alexa Fluor 568 (red) antirabbit plus Alexa Fluor 488 (green) antimouse. The nuclei were counterstained with DAPI (blue). All images were obtained by sequential laser scanning with the confocal microscope. For all combinations, the left panels show the overlay of DAPI and P protein staining, the middle panels show phosphorylated STAT1, and the right panels show the overlay of STAT1P and P protein staining.
on November 8, 2019 by guest
http://jvi.asm.org/
immunoprecipitation to show that this effect is due to binding of STAT1 to the viral P protein. Colocalization of STAT1 with viral nucleoprotein has been observed in MV-infected cells (47), and sequences common to P and V proteins of Nipah virus have been shown to bind STAT1 (64). Although the amino-terminal half of P is not well conserved among paramyxoviruses, it seems likely that the STAT1 binding func-tion is common to at least theMorbillivirus and Henipavirus
genera.
Although both P and V bind STAT1, they appear to have slightly different effects, since V blocks phosphorylation of both STAT1 and STAT2 extremely effectively, whereas the action of P appears to be more steric, requiring higher levels of P in the cell to effectively bind all the STAT1 and hence block IFN signaling and being relatively inefficient at blocking STAT2 phosphorylation. In this respect RPV seems to be similar to Nipah virus, where V is also more effective than P at blocking IFN signaling (62). The complete block of STAT1 phosphorylation in the cells expressing V protein might suggest that the V protein has some additional mechanism to inhibit the activation of STAT1, and perhaps the cysteine-rich carboxy terminus of V protein plays an important role in this blockade. Alternatively, the polymeric nature of P may reduce its affinity or capacity for STAT1, making it less efficient generally than V in blocking STAT1 phosphorylation. The interesting observa-tion was that, although RPV P is quite effective in blocking STAT1 phosphorylation in transfected cells, the P-STAT1 in-teraction in RPV-infected cells is not sufficient to prevent the activation of STAT1 by type I or type II IFNs if V is not present (RPV V⫺or V⫺C⫺). This may be due to the available P being concentrated in aggregates in infected cells, where it is less effective at binding the cell pool of STAT1. The P protein in infected cells also interacts with other viral components (N and L), and its role in viral RNA synthesis (10, 56, 63) may make it less available to bind to STAT1/STAT1P.
Different labs have reported that the MV V protein does (71) or does not (47) block the interferon-stimulated phosphor-ylation of STAT1, although both groups identified complexes of V with STAT1 and other host cell proteins. Our observa-tions with RPV proteins are that phosphorylation of STAT1 is very efficiently blocked by the V protein, and, in addition, the V protein binds STAT1. While the binding of STAT1 by V could explain the blockade of both type I and type II IFN action in infected cells, it remains to be determined whether the ability of P to even partially block IFN action has a bio-logical role in the infected animal. It is important to note that many cells infected with RPV V⫺appeared less sensitive to IFN (produced less STAT1P) than neighboring uninfected cells. In addition, cells infected with any of the viruses used here showed, at later stages of infection when the cells had a very high content of viral P protein, reduced or no sensitivity to IFN (not shown). However, this reduced sensitivity may have also been due to other cytopathic effects of infection.
Wild-type virus also blocked activation of STAT2 by type I IFN, and both P and V proteins were able to block the forma-tion of STAT2P when expressed alone. The mechanism of this block remains to be determined. While V protein blocked phosphorylation of both STATs equally well, the P protein was noticeably worse at blocking STAT2 phosphorylation than blocking STAT1 phosphorylation (again suggesting that the V
protein is acting through more than one mechanism). No di-rect binding of STAT2 by any viral protein was observed, and STAT2 phosphorylation does not appear to depend on STAT1 (30). It may be that STAT2P is unstable in the absence of STAT1 or STAT1P, although it is stable enough to be visual-ized in interferon-treated cells lacking STAT1 (30). Alterna-tively, there may be another virally encoded inhibitory mech-anism directly affecting STAT2 activation that we have not yet identified, as indicated by the aggregates of STAT2 observed in the nuclei of infected cells. The fact that, as with STAT1 phosphor-ylation, both P and V proteins were able to block the formation of STAT2P, and V was more effective than P, suggests that the same or similar mechanisms are inhibiting both processes.
In SeV (18, 24), MV (82), and Nipah virus (52), the viral C protein is also an IFN antagonist, although the C proteins of these different genera have no sequence similarity. In SeV, the C protein physically associates with both native and phosphor-ylated STAT1, preventing its nuclear translocation (72). This interaction of SeV C with STAT1 also appears to inhibit acti-vation of STAT2, and this has been shown to be the key event inhibiting IFN signaling (22). The MV C protein has been reported to cause freezing of the type I IFN receptor complex containing RACK1 and STAT1 and thus suppressing of IFN-␣ signaling (82). We have found that RPV C protein partially blocks STAT1 activation on IFN treatment, similar to the partial block observed with Nipah virus C protein (52). How-ever, when assessed with reporter gene assays, the C protein had no measurable effect on IFN induction of protein expres-sion. It must be noted that the RPV C protein was found predominantly in the nuclei of transfected cells, in contrast to the infected cells, where the C protein is found in the cyto-plasms of the infected cells in a complex with other viral pro-teins (70). This might mean that independently expressed RPV C does not block STAT1 phosphorylation completely, as it cannot interact with the components of the early IFN signaling pathway that are mainly found in the cytoplasm, and the longer exposure of the cells to IFN required for the reporter gene assays allows sufficient STAT1P to enter the nuclei of the cells to activate transcription. In the context of viral infection, how-ever, the C protein does not appear to play a role in blocking interferon signaling; our observations on cells infected with RPV mutants that do not express the C protein showed that removal of the C has no detectable effect on type I or type II IFN signaling. These observations are in agreement with find-ings that MV C protein had no effect on IFN signaling (71). It seems more likely, therefore, that the observed effect of C protein expressed in transfected cells is due to a nonbiological effect of C accumulating in the nucleus, underlining the im-portance of correlating transfection studies with studies on virus infection.
ACKNOWLEDGMENTS
S.N. is the recipient of an IAH studentship. The laboratory of M.D.B. is supported by the BBSRC and the Wellcome Trust.
We thank Paul Monaghan and Pippa Hawes for training and assis-tance with confocal microscopy. We thank I. Julkonnen for the anti-Mx antibody, R. Randall for the anti-V5 antibody, and S. Goodbourn for the IFN reporter gene plasmids. We gratefully acknowledge several helpful discussions with S. Goodbourn and R. Randall. S.N. is grateful to R. Randall for the opportunity to spend 2 weeks working in his lab.
on November 8, 2019 by guest
http://jvi.asm.org/
REFERENCES
1.Andrejeva, J., E. Poole, D. F. Young, S. Goodbourn, and R. E. Randall.2002. The p127 subunit (DDB1) of the UV-DNA damage repair binding protein is essential for the targeted degradation of STAT1 by the V protein of the paramyxovirus simian virus 5. J. Virol.76:11379–11386.
2.Banninger, G., and N. C. Reich.2004. STAT2 nuclear trafficking. J. Biol. Chem.279:39199–39206.
3.Baron, M. D.2005. Wild-type Rinderpest virus uses SLAM (CD150) as its receptor. J. Gen. Virol.86:1753–1757.
4.Baron, M. D., and T. Barrett.1997. Rescue of rinderpest virus from cloned cDNA. J. Virol.71:1265–1271.
5.Baron, M. D., and T. Barrett.2000. Rinderpest viruses lacking the C and V proteins show specific defects in growth and transcription of viral RNAs. J. Virol.74:2603–2611.
6.Baron, M. D., M. S. Shaila, and T. Barrett.1993. Cloning and sequence analysis of the phosphoprotein gene of rinderpest virus. J. Gen. Virol.74:
299–304.
7.Biron, C. A.1998. Role of early cytokines, including alpha and beta inter-ferons (IFN-alpha/beta), in innate and adaptive immune responses to viral infections. Semin. Immunol.10:383–390.
8.Bossert, B., and K. K. Conzelmann.2002. Respiratory syncytial virus (RSV) nonstructural (NS) proteins as host range determinants: a chimeric bovine RSV with NS genes from human RSV is attenuated in interferon-competent bovine cells. J. Virol.76:4287–4293.
9.Cattaneo, R., K. Kaelin, K. Baczko, and M. A. Billeter.1989. Measles virus editing provides an additional cysteine-rich protein. Cell56:759–764. 10.Chattopadhyay, A., and M. S. Shaila.2004. Rinderpest virus RNA
polymer-ase subunits: mapping of mutual interacting domains on the large protein L and phosphoprotein P. Virus Genes28:169–178.
11.Childerstone, A. J., L. Cedillo-Baron, M. Foster-Cuevas, and R. M. Park-house.1999. Demonstration of bovine CD8⫹T-cell responses to foot-and-mouth disease virus. J. Gen. Virol.80:663–669.
12.Darnell, J. E., Jr.1997. STATs and gene regulation. Science277:1630–1635. 13.Darnell, J. E., Jr., I. M. Kerr, and G. R. Stark.1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signal-ing proteins. Science264:1415–1421.
14.Das, S. C., M. D. Baron, and T. Barrett.2000. Recovery and characterization of a chimeric rinderpest virus with the glycoproteins of peste-des-petits-ruminants virus: homologous F and H proteins are required for virus viabil-ity. J. Virol.74:9039–9047.
15.Der, S. D., A. Zhou, B. R. Williams, and R. H. Silverman.1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA95:15623–15628. 16.Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall.1999. The V
protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol.73:9928–9933.
17.Durbin, J. E., R. Hackenmiller, M. C. Simon, and D. E. Levy.1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell84:443–450.
18.Garcin, D., P. Latorre, and D. Kolakofsky.1999. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J. Virol.
73:6559–6565.
19.George, R. S., M. K. Ian, R. G. W. Bryan, H. S. Robert, and D. S. Robert.
1998. How cells respond to interferons. Annu. Rev. Biochem.67:227–264. 20.Ghosh, S., and M. Karin.2002. Missing pieces in the NF-kappaB puzzle. Cell
109(Suppl.):S81–S96.
21.Goodbourn, S., and L. Didcock.2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol.
81:2341–2364.
22.Gotoh, B., T. Komatsu, K. Takeuchi, and J. Yokoo.2003. The C-terminal half-fragment of the Sendai virus C protein prevents the gamma-activated factor from binding to a gamma-activated sequence site. Virology316:29–40. 23.Gotoh, B., T. Komatsu, K. Takeuchi, and J. Yokoo.2002. Paramyxovirus strategies for evading the interferon response. Rev. Med. Virol.12:337–357. 24.Gotoh, B., K. Takeuchi, T. Komatsu, J. Yokoo, Y. Kimura, A. Kurotani, A. Kato, and Y. Nagai.1999. Knockout of the Sendai virus C gene eliminates the viral ability to prevent the interferon-alpha/beta-mediated responses. FEBS Lett.459:205–210.
25.Hahm, B., M. J. Trifilo, E. I. Zuniga, and M. B. Oldstone.2005. Viruses evade the immune system through type I interferon-mediated STAT2-de-pendent, but STAT1-indeSTAT2-de-pendent, signaling. Immunity22:247–257. 26.Horvath, C. M.2004. Silencing STATs: lessons from paramyxovirus
inter-feron evasion. Cytokine Growth Factor Rev.15:117–127.
27.Horvath, C. M., G. R. Stark, I. M. Kerr, and J. E. Darnell, Jr.1996. Interactions between STAT and non-STAT proteins in the interferon-stim-ulated gene factor 3 transcription complex. Mol. Cell. Biol.16:6957–6964. 28.Huang, Z., S. Krishnamurthy, A. Panda, and S. K. Samal.2003. Newcastle
disease virus V protein is associated with viral pathogenesis and functions as an alpha interferon antagonist. J. Virol.77:8676–8685.
29.Ihle, J. N.1995. The Janus protein tyrosine kinase family and its role in cytokine signaling. Adv. Immunol.60:1–35.
30.Improta, T., C. Schindler, C. M. Horvath, I. M. Kerr, G. R. Stark, and J. E. J. Darnell.1994. Transcription factor ISGF-3 formation requires phos-phorylated Stat91 protein, but Stat113 protein is phosphos-phorylated indepen-dently of Stat91 protein. Proc. Natl. Acad. Sci. USA91:4776–4780. 31.Kato, H., S. Sato, M. Yoneyama, M. Yamamoto, S. Uematsu, K. Matsui, T.
Tsujimura, K. Takeda, T. Fujita, O. Takeuchi, and S. Akira.2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity23:19–28. 32.King, P., and S. Goodbourn.1994. The-interferon promoter responds to priming through multiple independent regulatory elements. J. Biol. Chem.
269:30609–30615.
33.Kozak, M.1989. The scanning model for translation: an update. J. Cell Biol.
108:229–241.
34.Kubota, T., N. Yokosawa, S. Yokota, and N. Fujii.2001. C terminal Cys-rich region of mumps virus structural V protein correlates with block of inter-feron alpha and gamma signal transduction pathway through decrease of STAT1-alpha. Biochem. Biophys. Res. Commun.283:255–259.
35.Kubota, T., N. Yokosawa, S. Yokota, N. Fujii, M. Tashiro, and A. Kato.2005. Mumps virus V protein antagonizes interferon without the complete degra-dation of STAT1. J. Virol.79:4451–4459.
36.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg.2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol.1:398–401. 37.Laemmli, U. K.1970. Cleavage of strutural proteins during the assembly of
the head of the bateriophage T4. Nature227:680–685.
38.Lamb, R. A., and D. Kolakofsky.2001. Paramyxoviridae: the viruses and their replication, p. 1305–1340.InD. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams and Wilkins, Philadelphia, Pa.
39.Lin, G. Y., R. G. Paterson, C. D. Richardson, and R. A. Lamb.1998. The V protein of the paramyxovirus SV5 interacts with damage-specific DNA bind-ing protein. Virology249:189–200.
40.Liston, P., and D. J. Briedis. 1994. Measles virus V protein binds zinc. Virology198:399–404.
41.Malmgaard, L.2004. Induction and regulation of IFNs during viral infec-tions. J. Interferon Cytokine Res.24:439–454.
42.Marg, A., Y. Shan, T. Meyer, T. Meissner, M. Brandenburg, and U. Vinke-meier. 2004. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distri-bution of latent Stat1. J. Cell Biol.165:823–833.
43.Mosca, J. D., and P. M. Pitha.1986. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol.6:2279–2283.
44.Nishio, M., M. Tsurudome, M. Ito, M. Kawano, H. Komada, and Y. Ito.2001. High resistance of human parainfluenza type 2 virus protein-expressing cells to the antiviral and anti-cell proliferative activities of alpha/beta interferons: cysteine-rich V-specific domain is required for high resistance to the inter-ferons. J. Virol.75:9165–9176.
45.Ohno, S., N. Ono, M. Takeda, K. Takeuchi, and Y. Yanagi.2004. Dissection of measles virus V protein in relation to its ability to block alpha/beta interferon signal transduction. J. Gen. Virol.85:2991–2999.
46.Oyanagi, S., V. Ter Meulen, M. Katz, and H. Koprowski.1971. Comparison of subacute sclerosing panencephalitis and measles virus: an electron micro-scope study. J. Virol.7:176–187.
47.Palosaari, H., J. P. Parisien, J. J. Rodriguez, C. M. Ulane, and C. M. Horvath.2003. STAT protein interference and suppression of cytokine sig-nal transduction by measles virus V protein. J. Virol.77:7635–7644. 48.Parisien, J. P., J. F. Lau, and C. M. Horvath.2002. STAT2 acts as a host
range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J. Virol.76:6435–6441.
49.Parisien, J. P., J. F. Lau, J. J. Rodriguez, B. M. Sullivan, A. Moscona, G. D. Parks, R. A. Lamb, and C. M. Horvath.2001. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology283:230–239. 50.Park, C., S. Li, E. Cha, and C. Schindler.2000. Immune response in Stat2
knockout mice. Immunity13:795–804.
51.Park, M. S., A. Garcia-Sastre, J. F. Cros, C. F. Basler, and P. Palese.2003. Newcastle disease virus V protein is a determinant of host range restriction. J. Virol.77:9522–9532.
52.Park, M. S., M. L. Shaw, J. Munoz-Jordan, J. F. Cros, T. Nakaya, N. Bouvier, P. Palese, A. Garcia-Sastre, and C. F. Basler.2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol.
77:1501–1511.
53.Paterson, R. G., and R. A. Lamb.1990. RNA editing by G-nucleotide inser-tion in mumps virus P-gene mRNA transcripts. J. Virol.64:4137–4145. 54.Paterson, R. G., G. P. Leser, M. A. Shaughnessy, and R. A. Lamb.1995. The
paramyxovirus SV5 V protein binds two atoms of zinc and is a structural component of virions. Virology208:121–131.
55.Pringle, C. R.1997. The order Mononegavirales—current status. Arch. Vi-rol.142:2321–2326.
56.Raha, T., R. Kaushik, and M. S. Shaila.2004. Phosphoprotein P of