JOURNAL OF VIROLOGY, May 1986, p. 694-699 Vol.58, No. 2 0022-538X/86/050694-06$02.00/0
Copyright © 1986,American Societyfor Microbiology
Localization of
Temperature-Sensitive Transformation Mutations
and Back Mutations in the Rous Sarcoma Virus src Gene
VALERIE J. FINCHAMANDJOHN A. WYKE*Imperial CancerResearchFund Laboratories, St. Bartholomew's Hospital, Dominion House, London ECIA 7BE, UnitedKingdom
Received25November 1985/Accepted 3 February 1986
Cloning and sequencing oftwotemperature-sensitive transforming mutations of Roussarcomavirusreveal thattheir lesions are due to distinct but close single amino acid changes near the carboxy terminus of the v-src geneproduct. Back mutations to wild type result from second mutations at either nearby or distant sites.
Temperature-sensitive (ts) transformation mutations of Rous sarcomavirus (RSV) have proved invaluable in
corre-lating the complex phenotype of the transformed cell with the functioning of the viral src oncogene (17). Different v-src mutations have different consequences for the infected cell,
suggesting a pleiotropism in v-src behavior (1, 3, 34), but
ignoranceof the precise mutations has made it impossible to correlate these behavioral differences with defined
alter-ations in the v-src gene product. Site-directed mutagenesis
(5-9, 28, 29, 35) and the use of anti-peptide antibodies (13, 26, 33) are now popular in analyzing the relationship be-tweenv-src structure andfunction,but these procedures are
initially eclectic and can be coarse. A knowledge of the molecular basis of ts mutations has three benefits. (i) It
these mutants were divided, on the basis of their ability to
recombine with one another to yield wild-type virus, into four cooperative transformation groups (37, 38): members within a group recombine with one another at a low but
detectable frequency while they recombine at higher fre-quency with representatives of other groups. We con-structedacoarsemapof thets mutantsbased on their ability to recombine to wild type with v-srcdeletion mutants (11, 12). This mapping, which was consistent with previous
mapping based on recombination frequencies (2), has had twoconsequences. Itshowed that tsmutations are dispersed widely within src, promptingasuccessful searchfor
pheno-typicdifferences between mutantsthat define different func-tional domainsinv-src(30). Moreimportantly for this paper,
LA24 LA31 6862 7129
l
l
I
I ynv(gp37) ||dr| |8706 9058
|
drl
lU31Pstl
I I I
Psil Pstl agli Pstl BgII Pstl
FIG. 1. Salient features of the 3,096-base-pairEcoRIBfragment of thePraguestrain of RSV. Thisfragment, fromnucleotide6144(in env) to9238(inU3), encompasses thewholev-src gene anditsflanking directrepeats (dr) (25). Molecularclones of this fragmentfrommutants, backmutants, and wild-typePrA wereobtained in pAT153 after first cloning Hirtsupernatant DNAfrom infected chick cells (10, 14), or integrated proviral fragments from transformed chickorratcells, in bacteriophageA vectorsbystandardtechniques (18, 19).Majorrestriction enzyme sites used in subcloning as a preliminary to sequencing are indicated. Subcloning in bacteriophage M13 (21) was followed by sequencing bythechaintermination method (4, 24), (see Fig. 2foranexample). Sequences of regions of interestwere confirmed by the Maxamand Gilbert method (20) aftersubcloning in pAT153. The barabove theline showsthepredicted location ofthe mutations inLA24 andLA31(11). Thesolidarrowbelowtheline shows the minimumregion sequenced in itsentirety for allthevirusesstudiedhere. complements these other procedures, defining sites in the
gene where mutations conditionally affect function. (ii) It may identify otherregions ofthe gene as targets for addi-tional analyses;thepropensity oftsmutants toreverttowild typeisofparticularinterestsince thelocationofsecond-site backmutationsmay indicateparts ofthegene productthat interact withoneanother. (iii)Itprovidesusefulinformation
onmutantsthat havebeenusedextensivelyforphysiological studies.
Ourwork has concentratedon asuiteofts mutantsin the
v-src geneof the Prague strain of RSV, subgroup A (PrA). Soon afterisolationfollowing5-azacytidine mutagenesis (36)
*Corresponding author.
theroughlocalization of the causative mutations heldoutthe hope that they could be identified and distinguished from otherinconsequential mutations that the mutagenesis might have caused.
We considered ts LA24 and ts LA31 ideal tests ofthe feasibility of identifying ts mutations. They belong to the
samecooperative transformationgroup(38) andmapwithin about100 basepairstowards the3' endofv-src(11) (Fig. 1).
Both induceamarkedlytsphenotypeand have been used for
many physiological studies. They arenot leaky, so we can
isolate stable spontaneous back mutants from them, as
described previously (39). The v-src genes from LA24, LA31, three spontaneous back mutants, and thePrA
progen-itorweremolecularlycloned(10, 14, 18, 19) (Fig. 1). Chick 694
6269
l l
on November 10, 2019 by guest
http://jvi.asm.org/
[image:1.612.154.467.401.491.2]TABLE 1. Nucleotide andpredicted aminoacidsequencecomparisons ofPrAanditsmutants
Change from PrC in: Presenceofchangein:
Nucleotide
Base Aminoacid' PrA LA31A 31A.1.4 31A.3.4 LA24A 24A.7b2
6517 T C L-* L(gp37,83) + + NDb ND ND ND
6957 C T Noncoding (indirectrepeat) + + ND ND ND ND
7018 A C Noncoding + + ND ND ND ND
7059 A G' Noncoding - + + + + +
7082 G A Noncoding - + + +
7185 A *G' H R(src,16) + + + + + +
7332 C-T T T(src,69) + + + + + +
7742 A-*G Y Cd(src,205) + + + + + +
7749 C T R R(src,217) + - - - -
-7852 G Ae A T(src,242) + + + + + +
7855 A-C N H(src,243) + + + + + +
7991 A Ge D G(src,288) + + + + + +
8257 G A V M(src,377) - - +
8561 G A G D(src,478) - + + +
8567 G-A R H(src,480) - - - - + +
8602 C T H Y(src,492) - - - + - +
8632 G A D N(src,502) +
8706 G A E E(src,526) + + + + + +
a Data in parentheses are the affected gene and amino acid.
bND, Not determined.
'This change isalso seen in the sequences of SRA and c-src (31, 32).
dSRA andc-srcdifferfrom PrC at nucleotide 7741,substituting arginine for tyrosine(31, 32).
e This change isseeninthe sequences of SRAandc-src (31, 32) as well assomevariants of PrC(25.)
A
PrA 31A.1.4 31A.3.4 24A.7b28558 ;-
AA
C C Tf,I
_
-B570
A C G T G
- /
C-/ A" <
- . . T
A
..~~~C
..c*. .._.C-c A / T
A C G T A C G T
T -3
C, Nn
C a
* * T I _
.5.~ ~ .^ 6
T __
T
G
-""A
M _s.- oT
--AA
LA 31A
8260C T C C
G T G T
82 54 T.
A
31A.1.4
G T G C A
G As
T
T w .
T
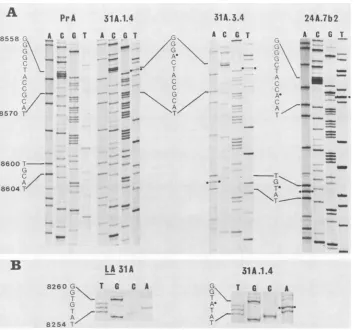
FIG. 2. Locationofmutations andback mutationsin derivatives of PrA. Allsequenceladders in panel A show anti-parallel strands. The sequences inwhich mutations(0)arelocatedareindicated beside the ladders. (A) In this region (approximately nucleotides 8540to8630), tsLA31A(notshown) is identical insequenceto itsback mutant, 31A.1.4.tsLA24A (not shown) shares with its backmutant,24A.7b2, the
G-to-Atransition atnucleotide8567 butdoes notdisplaythe C-to-Ttransition at nucleotide 8602. (B) In this region (approximately nucleotides 8250to8265), allthe viruses not shownareidenticaltotsLA31A.
8600 -r-G C
8604T
B
VOL.58, 1986 NOTES 695
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.58.559.81.279.2] [image:2.612.131.485.344.674.2]696 NOTES
cells transfected with cloned ts mutant proviral DNA pro-duced transformants with ts morphology and protein kinase activity. As anticipated, transformants induced by cloned back mutant DNA lacked temperature dependence (data not shown).
To determine the extent of spontaneous and mutagen-induced variation, we sequenced (4, 20, 24) the entire EcoRI B fragment of LA31 and our current stock of wild-type PrA and compared them with the published sequence of the Prague strain of RSV, subgroup C (PrC [25]). Nucleotide numbering of this EcoRI fragment B followed that of Schwartz et al. (25). Table 1 summarizes the differences between PrA and its derivatives and PrC.
PrA and LA31 show common differences from PrC at 10 sites. Two are noncoding and another three affect the third bases of codons without altering coding. The other five changes alter the coding assignment of v-src at amino acids 16, 205, 242, 243, and 288. PrA differs at two further sites from PrC, LA31, LA24, and their derivatives; one of these differences changes the coding of amino acid 502 of v-src. These latter changes presumably reflect variation that has occurred in our stock of PrA after the isolation of the ts mutants in 1971.
LA31 shows an alteration at nucleotide 8561, changing amino acid 478 of v-src from glycine to aspartic acid. Likewise, in LA24, a change at nucleotide 8567 converts amino acid 480 from arginine tohistidine (Table 1 and Fig. 2). We conclude that these differences account for the temperature sensitivity of the two mutants because they are the only changes within v-src unique to each mutant, they are within the region in which the mutations were located by recombination mapping (Fig. 1), and they are very close to one another, explaining the allocation of LA31 and LA24 to the same cooperative transformation group. Moreover, the mutation in each case is a G-to-A transition, the type of change expected with the use of 5-azacytidine.
These conclusions are strengthened by examination of back mutations to a wild phenotype. The sequences of two of these, LOILA31A.3.4 andLOILA24A.7b2, show that they are identical to their parent ts viruses with one exception; both show the same C-to-T transition at nucleotide 8602, changing amino acid 492 of v-src from histidine to tyrosine (Table 1 and Fig. 2). Thus, an alteration to this amino acid appears to compensate for mutations affecting eitheramino acid 478 or 480. However, this is not the only way bywhich backmutations can occur, forLO/LA31A.1.4 does not differ from parental ts LA31A in this region but shows instead a mutation atnucleotide 8257, changing amino acid 377 from valine to methionine (Table 1 and Fig. 2).
Several interesting points emerge from this work. (i) The sequences of LA24 and LA31 are an impressivevalidation of earlier genetic studies that predicted that LA24 and LA31 bear distinct but closely linked mutations (37, 38). The mutations are 6 nucleotides apart, and this remarkable degree oflinkage can therefore be discerned by examining recombination frequencies. Moreover, the mapping of Fincham et al. (11) of these mutations has been proven accurate, increasing our confidence that the other 12 ts src mutants of PrA that we have studied by genetic means indeed have distinct mutations whose lesions span a large partof the src gene. We suspect that the allocation of these mutants to cooperative transformation groups does not reflect primarily the existence of hot spots for genetic
recombination between groups but gives anindicationof the physical spacing of these mutations in the src sequence.
(ii) Although LA24 and LA31 were obtained after
muta-genesis, itis noteworthy thatextensive silent mutations are not presentintheir genomes, and themutations could have been identified without prior knowledge of their location.
However, mapping studies arevaluable in examining other
mutantsthatdifferfrom thewildtypeatseveralsites (A. W. Stoker, unpublished data). Furthermore, attempts to map certain ts mutants by crosses with deletion mutants have proved equivocal(J. A.Wyke, V. J. Fincham,R. Friis, and
M.Weber,unpublished data).Notably,NY68(15)andsome mutants of the GI and CU series (1, 34) recombine to the wild type with deletionmutants that lack src sequences 5' to nucleotide 8200 (approximately) but cannot do so with mutants lacking sequences 5' to nucleotide 8500
(approxi-mately).This is primafacieevidence that crucial mutations
are located between nucleotides 8200 and 8500 but the wild-typerecombinants either retain somephenotypic pecu-liarities or are ofirregular occurrence. The present study
makes us confident that mapping by recombination is a
powerful andprecise procedure, and wenowconsiderthat these equivocal results may bedue to additional mutations
elsewhere in src thatalsoinfluencethephenotype of infected
cells. Recentfindings (22) confirm this prediction forNY68. (iii) The deduced amino acid sequence ofour wild-type
PrAdiffers from that of PrC (25) at several sites. Some of
these differences are shared withtheSchmidt-Ruppin strain
A(SRA)of RSV andchickenc-src(31, 32)(Table1and
Fig.
3),but overall PrAresembles PrCmore closelythan itdoes
SRA. LA24and LA31 resemble PrA inmostrespects,so our wild-type clonedoes not represent aminorgenotype in the
population.
(iv) The mutations in LA24 and LA31 affect amino acids outside theregions of pp60v-src thatare most
strongly
con-servedamongtyrosinekinases,
thepresumed active siteandATP-binding region (Fig. 3). This is not
surprising,
for although mapping has located several ts mutations in the strongly conserved region, othermutations,
as inLA29,
affect pp60v-src kinase activity but map even closer to the
carboxy terminus of the
protein
than those of LA24 and LA31 (11, 30). This accords with thefindings
ofWilkerson andcollaborators (35), who havestudiedportions
ofv-srcby
in vitro mutagenesis and propose that the
carboxy
terminus oftheproteincomprisesaregulatory
domainthat influences the adjacent, conserved catalytic domain (23). The muta-tions in LA24 and LA31 are near thejunction
of thesepostulated domains.
The altered amino acids at
positions
478 and 480 arethemselves
reasonably
conserved in differenttyrosine
kinases (Fig. 3), but the
significance
of their alterations is unclear. Wild-type PrAalso shows achange
ina conservedamino acid, atposition 502
(Fig.
3), butthis does not affect its transforming activity. Furthermore, while it is easy to appreciate thatan alteration fromglycine
toaspartic
acid at position478could result in thechanges
inprotein
stability
or function expected inaconditional mutant, otherchanges
in forwardorback mutations are moreconservativeand donot conform to a discernible pattern.(v) The significance of these mutations should be more
apparent ifinformation on the
tertiary
structure ofpp60v-src
is obtained. Inthe
meantime,
perhaps
the most informative part of this study is the location of spontaneous back mutationstowild type. It mayonly
befeasibletoobtain such back mutations when the forward mutation is asingle
basechange, andthis seems a
particular
advantage
ofts mutants obtained in vivo. All three back mutants retain theoriginal
forward mutation
(Fig. 2),
and thecompensating
second-sitechanges substitute amino acids that are
peculiar
to these J. VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
231
V-0 V-0 0 0 00o0 0 0o * 000* 0o
,SKHADGLCHR,LANVCPTSKP,QTQGLAKDAW,EIPRESLRLE,AKLGQGCFGE,VWMGTWNDTT,
291
V
00 0 0000000 0 0 0 0 0 0 00000 0 000
,RVAIKTLKPG,TMSPEAFLQE,AQVMKKLRHE,KLVQLYAVVS,EEPIYIVIEY,MSKGSLLDFL,
351
v
00 0 0 00 0 0000 00 0 0000
,KEMGKYLRL
*PQLVDMAAQI,ASGMAYVOERM,
NYVOH;RDLAA,NILVGENLVC,KVADFGLARLISR-A PrC PrA
31A
31A.1.4
31A.3.4
24A 24A. 7b2 SR-A PrC
411
G-G-0000 00 0 0
,IEDNEYTARQ,GAKFPIKWTA,PEAALYGRFT,IKSDVWSFGI,ILLTELTTKGR,VPYPGMVNRE
471
R R E
00C00 000 00 00 00 00 0
,VLDOVERGYR MPCPPECPES,LHDLOMCQCWR,KDPEERPTFK, YLQAQLLPAC,VLEVAE
PrA 31A
31A.1.4
31A. 3.4 24A 24A.7b2
FIG. 3. Predictedsequencesof thecarboxy-terminal300 amino acids ofpp60v-src comparingPrC(25)withSRA(31)and PrAanditsmutant derivatives. Amino acids thatareconserved between v-src,v-yes, andv-fps (0)andmore stronglyconserved residuesthatarealsofound
incyclicAMP-dependent proteinkinase(-)areindicated(dataarethose of Kitamura et al. [16]andShibuyaetal.[27]). Atposition295,the lysineattheATP-bindingsite(A) isindicated;theputativephosphoacceptor tyrosineatposition416(A)is alsoidentified.
SR-A
PrC PrA 31A 31A. 1.4
31A. 3.4
24A
24A.7b2
SR-A
PrC PrA
31A
31A. 1.4
31A. 3.4 24A 24A.7b2
SR-A PrC
PrA 31A
31A. 1.4 31A.3.4
24A 24A. 7b2
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.146.471.79.658.2]698 NOTES
viruses and are thus not a consequence of recombination with c-src. Back mutations affecting amino acid 492 may affect the same domain or portion ofa domain as the ts forward mutations. However, the back mutation in LOILA31A.1.4. alters an amino acid on the other side of the kinase-active site. This implies a direct or indirect
interac-tion betweentheregionsaroundamino acids377 and 478. A
novel interaction of this kindhas been revealed after
inves-tigating only three spontaneous back mutants. The facility
with which spontaneous back mutants can be obtained from avariety of different ts src mutants suggests that they may
provide useful and unique ways to analyze
structure-function relationships in this gene.
We aregrateful to David Gillespie for advice and discussion, Paul Scotting for commentsonthe manuscript, and Andrea Sterlini for secretarial assistance.
LITERATURECITED
1. Anderson, D. D., R. P. Beckmann, E. H. Harms, K. Nakamura, and M.J. Weber. 1981. Biological properties of "partial" transformation mutants ofRous sarcomavirus and character-izationof their pp6Osrc kinase.J. Virol. 37:445-458.
2. Balduzzi, P., J. A. Beamand, J. R. Christensen, Y. M.Pearson, andJ.A. Wyke. 1978. Provisional mappingof transformation defectivetemperature-sensitivemutantsofRous sarcomavirus, p. 112-121.InS. Barlati and C. deGiuli-Morghen (ed.), Avian RNA tumourviruses. Piccin MedicalBooks, Padua, Italy. 3. Becker, D., R. Kurth, D. Critchley, R. R. Friis,and H. Bauer.
1977. Distinguishable transformation-defective phenotypes amongtemperature-sensitivemutantsofRous sarcomavirus.J. Virol. 21:1042-1055.
4. Biggin, M. D., T. J. Gibson, and G. F. Hong. 1983. Buffer gradient gels and 35S label as an aid to rapid DNA sequence determination. Proc. Natl. Acad. Sci. USA 80:3963-3965. 5. Bryant, D.,andJ.T. Parsons. 1982. Site-directedmutagenesis
of the src gene of Rous sarcoma virus: construction and characterization ofadeletionmutanttemperaturesensitive for transformation. J. Virol.44:683-691.
6. Bryant,D. L.,andJ.T. Parsons. 1984.Amino acid alterations within a highly conservedregion ofthe Rous sarcomavirussrc geneproductpp6Osrc inactivatetyrosine protein kinase activity. Mol. Cell. Biol.4:862-866.
7. Cross,F.R.,E.A. Garber,and H. Hanafusa. 1985. N-terminal deletions in Rous sarcoma virus p6Osr: effects on tyrosine kinase and biological activity, and on recombination in tissue culture withthecellularsrcgene. Mol. Cell. Biol. 5:2789-2795. 8. Cross,F.R.,E. A.Garber,D.Pellman,and H.Hanafusa. 1984. Ashort sequence in thep6Os'cNterminus isrequired for p6Osrc myristylation and membrane association and for cell transfor-mation. Mol. Cell. Biol.4:1834-1842.
9. Cross,F.R.,and H. Hanafusa.1983. LocalmutagenesisofRous sarcomavirus: themajorsites oftyrosineand serine phosphor-ylationaredispensible for transformation. Cell34:597-608. 10. DeLorbe, W. J., P. A. Luciw, H. M.Goodman, H. E.Varmus,
andJ. M.Bishop. 1980. Molecularcloningandcharacterization of avian sarcoma virus circular DNA molecules. J. Virol. 36:50-61.
11. Fincham,V.J.,D.J.Chiswell,andJ.A.Wyke. 1982. Mapping of nonconditional and conditional mutants in the src gene of Prague strainRous sarcomavirus.Virology 116:72-83. 12. Fincham, V.J., P. E. Neiman, andJ. A. Wyke. 1980. Novel
nonconditionalmutants in thesrcgene ofRous sarcomavirus: isolationandpreliminary characterization. Virology103:99-111. 13. Gentry, L. E., L. R. Rohrschneider, J. E.Casnellie, and E. G. Krebs. 1983.Antibodiesto adefinedregions ofpp6Osrc neutral-ize the tyrosine-specific kinase activity. J. Biol. Chem. 258: 11219-11228.
14. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmousecellcultures. J.Mol. Biol.26:365-369. 15. Kawai, S., and H. Hanafusa. 1971. The effects of reciprocal
changes in temperature on the transformed state of cells in-fected withaRoussarcomavirusmutant.Virology 46:470-479. 16. Kitamura, N., A.Kitamura, K. Toyoshima, Y. Hirayama, and M. Yoshida.1982. AviansarcomavirusY73 genome sequence and structuralsimilarityof itstransforminggeneproducttothat ofRoussarcomavirus.Nature(London) 297:205-208. 17. Linial, M., and D. Blair. 1984. Genetics of retroviruses, p.
649-783. In R.Weiss,N.Teich,H.Varmus,and J.Coffin(ed.), RNAtumorviruses, 2nd ed.Cold SpringHarborLaboratory, ColdSpring Harbor, N.Y.
18. Loenen, W. A., and W. J. Brammar. 1980. A bacteriophage lambda vector for cloning large DNA fragments made with several restriction enzymes. Gene 20:249-259.
19. Maniatis, T.,E. F. Fritsch, andJ.Sambrook. 1982. Molecular cloning:alaboratorymanual. ColdSpringHarborLaboratory, ColdSpring Harbor, N.Y.
20. Maxam,A. M., and W. Gilbert. 1980. Sequencingend-labeled DNAwithbase-specificchemicalcleavages.MethodsEnzymol. 65:499-560.
21. Messing, J.,andJ.Viera. 1982.AnewpairofM13vectorsfor selecting either DNA strand ofdouble-digest restriction
frag-ments.Gene 19:269-276.
22. Nishizawa, M., B. J. Mayer, T. Takeya, T. Yamamoto, K. Toyoshima,H.Hanafusa,andS. Kawai. 1985. Twoindependent mutations arerequiredfortemperature-sensitive cell transfor-mationbyaRoussarcomavirustemperature-sensitivemutant.
J.Virol. 56:743-749.
23. Parsons, J.T.,D.Bryant,V.Wilkerson,G.Gilmartin,and S.J. Parsons.1984.Site directedmutagenesisofRoussarcomavirus pp6Osr': identification of functional domainsrequiredfor trans-formation, p. 36-42. In G. F. Vande Woude, A. J. Levine, W. C. Topp, and J.D. Watson (ed.), Cancer cells: oncogenes and viralgenes. Cold Spring HarborLaboratory, Cold Spring Harbor,N.Y.
24. Sanger, F.,S.Nicklen,and A. R.Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
25. Schwartz, D. E., R. Tizard, and W. Gilbert. 1983. Nucleotide sequenceofRoussarcomavirus. Cell32:853-869.
26. Sefton,B.M.,andG. Walter. 1982. Antiserumspecificforthe carboxyterminusofthetransforming proteinofRoussarcoma
virus.J.Virol.44:467-474.
27. Shibuya, M., and H. Hanafusa. 1982. Nucleotide sequenceof Fujinamisarcomavirus: evolutionary relationshipof its trans-forminggenewithtransforminggenesofothersarcomaviruses. Cell30:787-795.
28. Snyder,M.A., J.M.Biship,W. W.Colby,and A. D. Levinson. 1983. Phosphorylation oftyrosine-416 is not required for the transforming properties and kinase activity of pp6o0-sr. Cell 32:891-901.
29. Snyder, M. A., J. M. Bishop, J. P. McGrath, and A. D. Levinson. 1985.A mutationattheATP-bindingsiteofpp6oV-src abolishes kinase activity, transformation, and tumorigenicity.
Mol. Cell. Biol. 5:1772-1779.
30. Stoker,A.W.,P.J. Enrietto,andJ.A.Wyke.1984.Functional domains of the pp60v-src protein as revealed by analysis of temperature-sensitive Rous sarcomavirusmutants. Mol. Cell. Biol.4:1508-1514.
31. Takeya, T.,and H.Hanafusa.1982. DNAsequence of theviral and cellular src gene of chickens. II. Comparison of the src genesoftwo strainsofavian sarcomavirusand of thecellular homolog.J. Virol.44:12-18.
32. Takeya, T., andH. Hanafusa. 1983. Structure and sequence of the cellular gene homologous to the RSV src gene and the mechanismforgenerating thetransforming virus. Cell 32:881-890.
33. Tamura, T.,H.Bauer,C.Birr,and R.Pipkorn.1983. Antibod-iesagainstsynthetic peptidesas atoolfor functionalanalysisof thetransformingproteinpp6Osrc.Cell 34:587-596.
34. Weber, M.J., andR. R. Friis. 1979. Dissociation of transfor-mation parameters using temperature-conditional mutants of Roussarcomavirus. Cell16:25-32.
35. Wilkerson,V.W.,D. L.Bryant,andJ.T.Parsons. 1985. Rous J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
sarcomavirus variants that encode src proteins with an altered carboxy terminus are defective for cellular transformation. J. Virol. 55:314-321.
36. Wyke, J. A. 1973. Theselective isolation of temperature sensi-tive mutants of Rous sarcoma virus. Virology 52:587-590. 37. Wyke, J. A. 1973. Complementation of transforming functions
by temperature-sensitive mutantsof avian sarcoma virus. Virol-ogy54:28-36.
38. Wyke, J. A., J. G. Bell, and J. A. Beamand. 1975. Genetic recombination among temperature-sensitive mutants of Rous sarcoma virus. Cold Spring Harbor Symp. Quant. Biol. 39: 897-905.
39. Wyke, J. A., and R. Kurth. 1978. Reversion of temperature-sensitive mutants of Rous sarcoma virus andits effecton the expression of tumour specific surface antigen. J. Gen. Virol. 40:701-704.