0022-538X/87/030755-09$02.00/0
Derivation
and Characterization of
POJ
Cells, Transformed Human
Fetal
Glial Cells
That Retain Their
Permissivity for JC Virus
CHRISTIAN
MANDL,lt
DUARD L. WALKER,2 AND RICHARD J. FRISQUE1*Departmentof Molecular and Cell Biology, The Pennsylvania State University, University Park, Pennsylvania
16802,1
and Departmentof Medical Microbiology, University of Wisconsin Medical School, Madison, Wisconsin 537062
Received 25 August1986/Accepted 10November 1986
The study of the medically importantpolyomavirus JC virus is limited to only a few laboratories, primarily because thepermissive cell system most often used, primary human fetal glial cells, isdifficultto obtain and propagate. We have introduced mutations at the origin of DNA replication of JC virus and transformed glial cells with thereplication-defective genomes. Although normal glial cell cultures rapidly lose their permissivity for the virus after subculture, the transformed cells (designated POJ) had a greatly expanded life span and remained permissive for JC virus even after 30 passages in vitro. POJ cells constitutively express a functional Tprotein that complements the replication defect of lethal early-region mutations in JC virus. We expect that these cellswill greatly facilitate the study of this human virus.
JC virus (JCV), a human polyomavirus, infects most people early in life and in some immunocompromised
indi-viduals causes the fatal demyelinating disease progressive
multifocal lqukoencephalopathy. In spite of the medical
importance
If
JCV, many laboratories have beendiscour-aged from studyingit because a convenient cell system has not been available. Primary human fetal glial (PHFG) cells, used in the initial isolation of JCV in 1971 by Padgett and co-workers (30), have been the cells usually used for the growth ofthe virus. These cells pose a number of serious problems for the virologist. (i) Fetal brain tissue is difficult to obtain insufficient quantitieson aregular basis. (ii) Cultures
of PHFGcells are a mixedpopulationof cell typescomposed
primarily ofastrocytes and spongioblasts. The latter cell is
presumedtobethe precursoroftheoligodendrocyte, the cell
in which JCV multipliesmostefficiently(28).(iii) The ratio of astrocytes to spongioblasts varies considerably between cultures prepared atdifferent times, and specialhandlingis required toobtain the preferred spongioblast-rich cultures.
Once "established" inculture, these cellsrapidlylose their
abilitytosupport JCVreplication. (iv)The lytic activity of JCV, evenin PHFGcells, isinefficient;thegrowth cycle is
prolonged, and cytopathic effects are difficult to recognize
(28). Typically, infectedcells are maintained in culture for 4 to5 weeks before virus is harvested.
From theverybeginning, major effortshave beendirected
at finding amore suitable cell system forpropagating JCV. Many kinds of cells, both
primary
and established lines,havebeentestedwithlittle success(39a).Afewhumancells
(e.g., embryonic kidney, amnion, andurine-derived
epithe-lial cells) do support JCV replication tovarious degrees (1, 27,36);however,theyalso suffer fromrestrictedavailability
and difficulthandling.Inaddition, largenumbers of defective
virionsaredetected in viruspools prepared fromthese cells. The needfor a useful cell system for thegrowthof JCV in vitroprompted us to attempt to derive apermissive cell line
by transforming PHFG cells with
replication-defective
(Ori-) mutants ofJCV. Thisapproach
has been usedsuc-cessfully for simian virus 40
(SV40)
(COS
cells[11])
and*Correspondingauthor.
tPresent address: Institute ofVirology, University ofVienna, Vienna, AustriaA-1180.
mousepolyomavirus (COP cells and Polyoma COS cells [2,
39]). In these instances, the objective was to obtain a
complementing cell system to propagate viral mutants and vectors. In our case, however, the primary goal was to derive apermanent cell line for propagating wild-type JCV.
RecentlyMajoretal. (21) transformedPHFG cellswith the sameSV40mutantsused togenerate COS cells (12).
Signif-icantly, these cells (designated SVG cells) support the
rep-lication ofJCV to approximately the same degree as the parent PHFG cells. While offering animportant alternative forthegrowth ofJCV,wefeel these cells have two serious drawbacks.First, fromthestudies ofMajor et al., it appears that the SV40 T antigen constitutively expressed in SVG
cells does not interact effectively with the JCV regulatory
machinery. A more serious obstacle to the practical use of
SVGcellsinvolvesthepossibility of recombination between
theintegrated SV40 sequences and input JCV DNA. Inthis paper we describe the first deletion and insertion
mutants of JCV constructed by site-specific mutagenesis techniques. Several ofthese mutants have beensequenced
and theirbiological functions investigated.Themutantshave allowedus toidentifysequencesrequiredfor the replication oftheJCV genome.
Wealso report theuseofOri-mutantsofJCVtoestablish transformed PHFG cell lines (designated POJ, for PHFG cellstransformedby Ori- JCV)that support thelytic growth of JCV. Becausethey constitutively express afunctional T
protein,POJ cellscomplementthereplication defect of JCV early-region mutants. Details of the derivation and
charac-terization ofthis cellline arepresented.
MATERIALS ANDMETHODS
Cells and virus. PHFG cells were obtained from 10- to
16-week-old abortusesasdescribed earlier(28). BothPHFG
and POJ cells were grown in Dulbecco modified
Eagle
(DME) mediumsupplemented
with 10% fetal calf serum.Duringthe DNA
replication
and virusproduction
assays,the serum concentrations were reduced to 3 to 5% to prevent cells frombecoming
toodense andsloughing
off theplates.
JCV (Madl
strain)
waspropagated
in PHFG cells andpurifiedasdescribed
by
Padgett
etal.(28).
Mutant construction and analysis. The
plasmid pMITC
(ABam)
containing
thecomplete
genome ofJCV inserted 755on November 10, 2019 by guest
http://jvi.asm.org/
M ITOC 9 1 Hindm(4498)
477' t pM TCABam) pBR322
co
l624 np
FIG. 1. PlasmidPMITC(ABam)usedto constructJCVmutants.
pMITC(ABam) contains the entire genome of prototype JCV
(Madi)inserted into the EcoRI site ofpBR322. pBR322 sequences between the unique Sall and Clal restriction sites have been
deleted, thereby eliminating the single Hindlll site in the vector.
JCV DNAcontainsthree Hindlllsites: onesiteatnucleotide 4498
occurs within theuniquecoding regionof small-tprotein,asecond site at nucleotide4914 lies withinthe shared coding sequences of bothearly proteins,andthe third site at nucleotide 5112 is found in the control region ofJCV adjacentto the center of thepresumed origin of DNA replication. Closed bars represent pBR322
se-quences,andopen bars indicate viral sequences thatare translated
intothelargeandsmall Tproteins.Thenarrowlinerepresentsviral
sequences. Numbers refer to nucleotidepositions on thegenomic
mapofMadl(9).Numberingstarts within the25-np dyad symmetry thoughtto include theoriginofreplicationandproceedstowards the latecoding region.The entireplasmidis8,868np inlength,including 5,130 np of JCV DNA.
into theEcoRI siteofpBR322 (Fig. 1)wasusedto construct
deletionand insertion mutants ofJCV. Briefly, the plasmid
waslinearizedby partial digestionwith therestriction
endo-nucleaseHindlIl (20 ,ug ofDNAwas digestedwith 20 Uof
the enzyme for 20 min at 37°C). The HindIII recognition sequence occurs three times within the viral DNA: at nucleotide4498intheregion encodingthecarboxyterminus
of the small tprotein,at nucleotide 4914in theoverlapping
sequences specifying the amino termini of the large and
small T proteins, and at nucleotide 5112 adjacent to a
twofold symmetrical sequence assumed to be the center of
theJCVoriginof DNAreplication (9).Afterextraction with phenol and chloroform, the DNA was precipitated with
ethanol and suspendedin S1nucleasebuffer(280mMNaCl,
50 mM sodiumacetate, pH 5.2,1 mMZnCl2).Portions of the
sample weredigestedwith different amountsof Si nuclease (0.5 to 10U/Iug ofDNA)for20minat 20°C. This treatment
causedanincreasingnumberofnucleotides to beremovedat
both ends ofthe linear DNA. To create insertion mutants,
thelinearizedpMITC(ABam)DNAwassuspendedinbuffer
(50mMNaCl,10mMMgCl2,1mMdithiothreitol) containing all four deoxynucleotides (1 mM each) and ATP (0.1 mM). Five units of the large fragment of DNA polymerase I (Klenow reagent)wasadded,andthemixturewasincubated
at roomtemperaturefor 30min. Following S1 nucleaseand
Klenowtreatments,full-lengthlinearplasmidwasseparated
fromcircularforms andsmaller linearfragments by
fraction-ation on a 1% low-melting-point agarose gel at 4°C. After
purification, DNA was recircularized by ligation with T4
DNA ligase at aDNA concentration of less than 10 ngl,ul. Competent Escherichia coli DH-1 cells were transfected with this DNA andplatedontoLB agarplatescontaining 30
jig
ofampicillin per ml (23). Small-scale plasmid prepara-tions of individual ampicillin-resistant colonies (14) were digested with HindlIl, and mutants were identified by the absence ofoneof the three HindIll sites.Following S1 nuclease treatment approximately 10% of the plasmids screened contained deleted HindIll sites.
Al-though allthree types ofmutants were obtained, mutations affecting the HindIII site atnucleotide 4498 occurred about 10 times more frequently than at the other two sites. A number of mutants were chosen for further study. To estimatethe sizes of the deletions in these mutants, restric-tion enzyme analysis was performed, and the sizes of the cleavage products from mutated and wild-type DNAs were comparedon agarose and polyacrylamide gels. DNAs con-taining relatively small deletions were sequenced by the Maxam and Gilbert technique (24). Each mutant in this serieswasdesignated by the letter S (forS1nuclease) anda number. These mutants are characterized in Table 1.
Mutations arising from treatment with Klenow reagent weredetected at nucleotides 4498 and 4914, but not 5112. As expected, the DNAs from these mutants contained an inser-tion(duplication) of the sequence 5'-AGCT-3'. This series of mutantswasdesignated by the letter K (for Klenow reagent) and anumber (Table 1).
This reportfocusesonthecharacterization anduseof the mutantsalteredattheHindlIl siteatposition 5112, particu-larly the mutants S-15 (35-nucleotide-pair [np] deletion), S-19 (11-np deletion), and S-27 (10-np deletion). In the complementation experiments outlined below, the mutant S-1, which has a 67-np deletion around nucleotide 4914 (affects large and small T proteins), will be discussed.
DNA transfection. Transfection of mammalian cells with viral DNA was done by either the modified calcium phos-phate (40) or the DEAE-dextran (34) procedure. The first transfectiontechniquewasused in thetransformation
exper-TABLE 1. Mutantsof JCV affected in theoriandearly codingsequences
Mutant Mutated Mutation Mutation T-Ag Transformation
regiona sizeb boundaryc expressiond of PHFG cells
K-1 t +4 4499-4502 + NTe
S-6 t -72 4467-4538 +
-S-21 t -26 4486-4511 +
-S-44 t -8 4498-4505 + NT
K-2 T/t +4 4915-4918 -
-S-i T/t -67 4871-4937 - NT
S-18 T/t -15 4908-4922 +/-
-S-36 T/t -13 4912-4924 NT NT
S-3 ori -59 5072-5130 +
S-8 ori -64 5072-5135 +
S-15 ori -35 5093-5127 +
S-19 ori -11 5108-5118 + +
S-27 ori -10 5107-5116 + +
aMutations wereintroducedat theHindIll sites atnucleotides4498, 4914,
and5112,which affectedsmall t,sharedlarge and small T,andorisequences,
respectively.
bSize in nucleotide pairs; +,insertion; -,deletion.
cBased on thenumberingscheme ofFrisqueet al. (9).
dTantigen (T-Ag) expression monitoredin PHFG cells 3 days
posttransfec-tion. A few cells transfected with S-18 DNA gave a weak, questionable nuclearfluorescence.
eNT,Nottested.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.62.300.69.241.2] [image:2.612.318.557.484.649.2]T-binding I T - binding II
| S 17np | P17n
1
5n ~ A5060 5070 5080 5090 5100 5110 5120 5130 10 20 29
5 -GAAAAACAAG GGAATTTCCC T;GCCTCCTA AAAAGCCTCC AC CCCTTACTACTT GAG TAAGCTTGA GGCGGAG GCGGCCTCGGCCT CCTGTATATA TAAAAAAAA
3-CTTTTTGTTC CCTTAAAGGG ACCGGAGGAT TTTTCGGAGG TGCGGGAATGATG CTC ATTGAAC CT CCGCCTC CGC CGAGCCGGAGGACATATAT ATTTTTTTT
HindM
S-27 10Onp _4 -27
S-I19 Ilnp S-l9
S- 15 '35np S -15
S-3 -59np -S-3
[image:3.612.65.541.65.206.2]S- 8 64np S- 8
FIG. 2. Deletion mutations in the regulatoryregion of JCV. The regulatory region of JCV includes 393 np located between the coding regionsforearly and late proteins. This stretch of DNA includesa number of control elements including promoter and enhancer sequences andthe origin of DNAreplication. Only a portion of this region is depicted in the figure, and earlyand late coding information is to the left and right,respectively, of the sequences shown. The 25-np dyad symmetry (S) is assumed to be the center of the replication origin andspans nucleotides 5118 to 12. This symmetry differs from those ofSV40 and BKV at a single position. This nucleotide substitution disrupts the symmetry of the two human viralori signals (indicated by asterisks). Adjacent to this symmetry is theHindlllrecognition site (nucleotides 5112 to 5117) used in constructing the JCV origin-defective mutants. The arrows point to the cleavage positions. Also shown are two additionalsymmetries (17 np and 19 np) and a 17-np palindromic sequence. Two T-antigen-binding sitesare indicated by open bars and include severalcopies of the contact consensus sequence 5'-(G>T)(A>G)GGC-3' (6, 37).It is likely that the actual binding sites involve sequences extending to both sides ofthese pentanucleotide sequences. The sizes and borders of the deletions in five differentori mutants areindicated. In someinstances short repeats at the deletion boundaries make positioning of the borders ambiguous. To remain consistent, borders were always drawn as far to the left (i.e., towards the early coding region)as possible. The AT-rich region containing the TATA box of the early viralpromoter was not affected in any of the deletion mutants.
iments; the second method was used in the DNA replication assays.
Indirectimmunofluorescence assay. Atvarious times after DNAtransfectionorvirusinfection, cells growing on 12-mm coverslipswerefixed for90 s in a 1:1mixture of acetone and
methanol. To detect T antigen, serum from a hamster
bearingaJCV-orSV40-inducedtumor wasadded to cells for 30 minat37°C. Afterthecells were rinsedwith
phosphate-buffered saline (PBS), fluorescein-conjugated goat
anti-hamsterimmunoglobulin G (IgG) serum was added. Thirty
minutes later the cover slips were rinsed with PBS and distilled water and mounted in buffered Gelvutol (10 g of polyvinyl alcohol in40 ml of PBSand 20 ml ofglycerol)on
microscope slides. The same procedure was followed for
capsid antigenexceptthatrabbitanti-capsidserum wasused as the first antibody and fluorescein-conjugated goat anti-rabbit IgGserum wasthe second antibody.
Viral DNA replication assay. Plasmid DNA containing a mutant orwild-type viralgenomewas digestedwithEcoRI andHhaItoyieldonelargefragment, representingtheentire
viral genome, and several small plasmid DNA fragments.
The linear viral DNA was separated fromthe smaller
frag-ments on a 1% agarose gel and isolated by electroelution. Viral DNA was recircularized during an overnight
incuba-tionwith T4 DNA ligaseat 14°C withlow DNA
concentra-tions (1to10 ng/,l). Aportion oftheself-ligated DNAwas
analyzed on agarose gels to determine the efficiency of
ligation.DNApreparations usedtotransfectcellscontained
atleast 50% circular forms. Cells were plated onto 35-mm
tissue culturedishestheday before transfection atdensities
that would yield nearly confluent monolayers after one
doubling. Transfections were
performed
byincubating
thecells for 75 min at
37°C
with 0.5 ml of DMEcontaining
DEAE-dextran(250,ug/ml)
and 100 ng ofcircularized DNA.Viral DNA was extracted by the method of Hirt
(13)
at severaltimepoints,ranging
from0to21days
posttransfec-tion. Aportion ofeachextract wasdigested
withDpnI
andEcoRIby the scheme of Peden et al. (31).DpnIcleaves DNA
of bacterial origin (input DNA) but does not cut DNA
replicated ineucaryotic cells due toits sensitivity to
differ-ences in DNA methylation patterns in the two cell types.
EcoRI cuts JCV DNA once independent of methylation. Thus, inthese experiments,input DNA was cut into several smallerfragments while newly synthesized viral DNA was
linearizedby the singleEcoRI cleavage. Digestion products were fractionated on 1% agarose gels and transferred to
nitrocellulose filters bythe methodofSouthern(35). Follow-ing hybridization at68°C to a
32P-nick-translated
(32) JCVDNA probe, viral DNA on the filter was detected by
autoradiography.
HAtiters.Thehemagglutination(HA) testwasconducted
asdescribed by Padgettand Walker (29). Briefly, doubling dilutions ofthe virus preparations were made in PBS. An
equal volume of 0.5% human type 0 erythrocytes from
donors lacking antibodies to JCVwas added, and the mix-ture wasincubatedfor3 hat4°C. The HAtiterwastakenas thereciprocal ofthehigher dilution ofthe virus suspension showing complete
agglutination
ofthe erythrocytes.ViralDNAintegration patterns. Confluent monolayers of
cells from several 100-mm tissue culture plates were col-lected and incubated with pronase in the presence of0.5% sodium dodecyl sulfate for 4 h at 45°C.
High-molecular-weightDNAwaspurified byseveralextractions withphenoland chloroform, followedby
precipitation
withethanol. Allmanipulations were done as gently as
possible
to avoidshearing the chromosomal DNA. RNA was removed
by
RNase treatment and ethanol
precipitation.
Cellular DNA (10jig)
wasdigestedwith restriction enzymesthat cleavedviral DNA sequences onlyalimited numberof times
(0
to3cuts).This DNAwasloadedonto1or0.8% agarose
gels and,
after
electrophoresis,
transferred tonitrocellulose filtersby
the Southern
technique
(35). Viral DNA was visualizedby
autoradiography afterhybridization
to viralprobes
labeled with 32P by nicktranslation(32).on November 10, 2019 by guest
http://jvi.asm.org/
S-19 97
M~ A 0C. 7 2 x 7 2
_m
FIG. 3. Replicative activityofJCV mutant DNAs in PHFG cells. PHFG cells were transfected with 0.1 ,ug of wild-type (w.t.) or
mutant (S-15, S-19, and S-27) JCV DNA. Each DNA had been separated from vector sequences and recircularized by ligation.
Viral DNA was extracted from the cells at 0, 7, and 21 days posttransfectionanddigestedwiththe restrictionenzymesDpnIand EcoRI.DNArepresenting 1/4of eachdigested sample (1/60of the
21-day wild-type DNA sample) was electrophoresed on a 1%
agarosegelandsubjectedto Southern blotanalysis (35).MlandM2
were wild-type inputDNAcleaved withDpnI plusEcoRI(1 ng of DNA) and EcoRI alone (0.3 ng of DNA), respectively. Newly synthesizedviral DNAmigratedasasingle5.1-kbfragment (sizeof
M2)andwasvisualizedby autoradiographyafterhybridizationtoa
nick-translated(32) pMITC(ABam) probe. Filmwasexposedfor5
dayswithoutan intensifyingscreen.
RESULTS
JCV mutants with deletions around nucleotide 5112 are
replication defective. A dyad symmetry is located near the replication origin of SV40; nearly identical symmetries are
found inthecorresponding regionsof the JCV and BK virus (BKV)genomes(9, 38). The three viralsequences differata
single position (nucleotide 5124 in JCV). This base change
disturbs the otherwise perfect symmetry in the JCV and
BKV sequences and accounts for the absence of a BglI
restriction site inthese two DNAs. Gluzmanand co-workers
(12) tookadvantageof theBglI sitepresentwithin the SV40 origin region to construct the Ori- mutants of SV40 that
were used to derive the COS cell line (11). The most
convenient restriction site nearest to the center of the
presumedJCVoriginsequences(the symmetryextendsfrom
nucleotide 5118 to 12) is the Hindlll recognition sequence
encompassing nucleotides 5112 to 5117. A diagram of this region, (Fig. 2) indicates the boundaries of the deletions of
thefive mutations obtained atthis HindlIl site. Itshould be
notedthatS-27retainedthecomplete 25-nptwofold
symme-try, while mutants S-19 and S-15 were missing the
5'-terminal nucleotideand the 5' halfof the symmetry,
respec-tively. To test thereplicativeabilityof thesemutants,0.1 ,ug ofthe S-15, S-19, and S-27 DNAs(plasmid sequences were
removed and the viral DNAs were recircularized) were
transfectedintoPHFG cellsbyamodification of the
DEAE-dextran method (34). Low-molecular-weight DNA was
ex-tracted fromthecells atseveraltime points following
trans-fection andsubjectedto theDpnI replicationassay (31) (see
Materialsand Methods). Newly synthesizedDNAcould be
detected as early as 3 days after wild-typeJCV DNA was
added to the cultures (Fig. 3). The amount of replicated
DNA increased steadilyuntilthe final time pointat 21days
posttransfection. Viral DNA synthesis was not detected at
anytimein cellstransfected with the mutantDNAs. It was
concluded thatmutantsS-15,S-19,and S-27werereplication
negative (Ori-).
T-antigen expression and transforming activity of JCV
mutants. A major reason for constructing Ori- mutants of JCV was to see whether they could be used to transform PHFG cells intopermanent cell lines that retained theability
to support the growth of JCV. To determine whether this approach wasfeasible, itwasfirstnecessaryto testwhether the mutants induced Tantigen, since thisprotein isrequired
for stable transformation. In addition to the five mutants altered in the origin region, several mutants affectedat the other twoHindlIl sitesweretested. PHFG cellswereseeded onto coverslips in 16-mm wells and transfected with 0.5plg
of intact plasmid by the modified calcium phosphate tech-nique (40). Three days later cells were stained for Tantigen byanindirect immunofluorescence assay (Table 1). All Ori-and small-t mutants induced T antigen in theglial cells, while the T/t mutantsfailed to do so.
Once we had determined that some of the mutantswere capable of inducing T antigen in PHFG cells, our efforts were directed to establishing permissive transformed cell lines. PHFG cellswere seeded onto 60-mm dishes (5 x 105 cells perdish) and transfected the following day with 2 ,ug of intact mutant plasmid DNA (Table 1). The medium (DME
plus10% fetal calf serum) was changed every 4days, andas cultures reached confluence, cells were trypsinized and transferred to new dishes at a ratio of 1 to 2. At 8 weeks posttransfection (passage 4), patches of T-antigen-expressing cells were observed in cultures infected with mutants S-19 and S-27. By 10 weeks (passage 6), cells in thesetwoculturesbegantodemonstrateanaccelerated rate ofgrowth andanaltered shape (cells became elongated and weremuchlargerthan uninfectedglial cells). More than 50% of the cells in each culture were producing T antigen. Cells transfected with the other mutants remained indistinguish-able from those in mock-infected cultures; cells did not contain viralproteins and their growth rate declined steadily until passage 8, when cell division ceased. On the basis of these observations, we concluded that the cells transfected with S-19 and S-27 DNA were transformed (Table 1). We named these transformants POJ-19 andPOJ-27, for PHFG cells transformed by origin-defective JCVmutants S-19 and S-27. Furthercharacterizationindicated that these cells had apartiallytransformed phenotype.By passage 8, essentially
100% of the POJ-19 andPOJ-27 cells expressed T antigen. Thecells hadatendencytopileupin small clusters and were able to grow in medium supplemented with low (1%) con-centrations ofserum. Although POJ cells grew faster than their normal counterparts,they still grew slowly relative to
fullytransformed cells. Furthermore, the cells grew poorly if subcultured at low cell densities. We found that POJ cells canbetrypsinizedandpassedat aratio of 1 to 4about once aweek. As aresultof their sensitivity to growth at low cell
densities, wehavehaddifficultycloning POJ-19 and POJ-27
cells;
cloning efficienciesonplasticwith medium containing 10% fetal calfserum range from 0.1 to 1.0%. However, we succeeded in obtaining a number of clones of POJ-19 and POJ-27 cells that have been called POJ-1 and POJ-2,respec-tively. Thegrowth properties and appearance of the cloned cell lineswere indistinguishablefrom those of the uncloned POJ cells. After approximately 70 cell generations, both cloned anduncloned POJ cells entered crisis; the growth rate decreased dramatically and the cells enlarged, became
rounded,anddetachedfromthe culture dishes. Attempts to nurse these cells through this period continue; however, becauselarge numbersof cells from earlier passages can be
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.94.281.69.210.2]0 3 7 14 21
r---I I---~-'-~-~I
a N OJ N' N7 7
° I O 0 0 0
MiCL L - a -,:,CL -m aLL
M 20a t 0 1 0 m 0 = 0 I: 2 C a- a- a EL aI a a a~ a
0 3 7 14 21
L r-
r-b
77 7-
r-2-IN N (M N N U C} C\lN
0 0 0 0 0 0 0 0 0 0
MI M2 a a a a a a a a a a
4,
*
4WO ~ ...
[image:5.612.133.489.69.239.2]..>
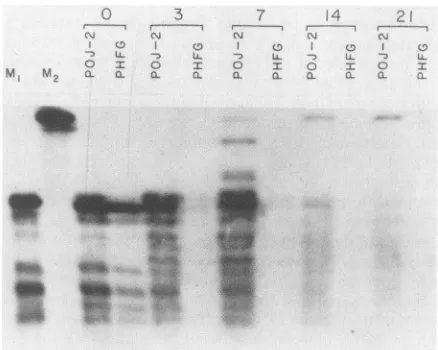
FIG. 4. Replication of wild-type JCV DNA in POJ and PHFG cells. Viral DNA was extracted fromPOJandPHFG cells, 0, 3, 7, 14, and 21days after the cells were transfected with 0.1 ,ug of wild-type JCV DNA. DNA (1/4 of the POJ-2 and POJ-27 samples, 1/12 of the PHFG sample) wasdigested withDpnIplusEcoRIand analyzed by the technique ofSouthern(35). Amounts of the PHFG samples were reduced threefold to account for the difference in cell number at the time of transfection. An additional threefold reduction was made in all 21-day samplestoavoidpossible overexposure of these bands. Shown arecomparisons of the accumulation of replicated viral DNA in POJ-2 and PHFG cells (a) andin POJ-2 and POJ-27 cells (b). Input JCV DNA cleaved with DpnI andEcoRI (M1)orEcoRIalone(M2) served as markers. Mlrepresents 3 ngand 1 ng of DNA in panels a and b, respectively; M2 represents 1 ng and 0.3 ng of DNA in panels a and b, respectively. Films were exposed for 6 days (a) or 7 days (b).
frozenfor future use, their entry into crisis is not considered aseriousproblem.
POJ cells support the lytic growth ofJCV.The next step in our studies was to ask whether POJ cells had retained the
ability oftheparental glial cells to support the replication of
JCVDNAandthepropagationof JCV. To address the first part of this question, POJ-2 cells and PHFG cells were
transfected with0.1 ,ugof circularJCVDNA, and at several
time points low-molecular-weight DNA was extracted from
the cellsandsubjectedtotheDpnI replicationassay (Fig. 4). Thecultures ofprimarycells weregenerally more dense than
those ofthelarger POJ cellsatthe startofeach experiment,
and therefore the total numberof cells per dishwas
deter-minedpriorto eachtransfection. These values were used to
adjust the amount of sample applied to the agarose gels
(indicated in each figure legend) so that band intensities
could be considered torepresent the DNA froman equiva-lent number of cells. Extractions at day 0 represented the amountof viralDNAstill associatedwith the cells at the end
of the transfection procedure. Results indicated that a greater amountofDNA wasrecoveredfromthePOJ-2cells
thanfrom the PHFG cells. Thisfindingwasreproducible and
may reflect the larger surface area of the POJ cells or
possible differences in the efficiency ofDNAuptakein the two celltypes. The amount of residual input DNAquickly decreasedin the PHFGcells,but wasstilldetected inPOJ-2 cells aslate as 14days posttransfection. Fragments ofinput
DNA generatedby DpnI cleavage became more diffuse on the Southern blots with time,
indicating
slow but steadydegradation
ofinput
DNAby
the cell. Thepersistence
ofDNA inPOJ-2 cells may be partlyexplained by the
possi-bility
that more DNA enteredeach POJ cellinitially. Newly
synthesizedDNA(uncutbyDpnI,
linearizedby
EcoRI)wasfirst
detected
in each cell type as early as 3 days posttransfection. By 7days
the bandrepresenting
DNAreplicated
in POJ-2 cells was stronger than its counterpartfrom PHFG cells. At 14
days
the band intensities wereroughly equal for the POJ-2 and PHFG
samples.
At about this timeadecreasewasobserved in the cell number of bothDNA- andmock-transfected POJ cultures, and this decline was very apparent by 3 weeks. The data show that the amount ofDNA synthesized by 21 days had continued to
increaseinbothcell types; however, by this time the amount of DNAextracted from thePHFG cellshad begun to surpass thatobtained fromPOJ-2 cells.
ThePOJ-2 cells used in this experiment had a relatively
highpassagenumber(ca. passage 35)and were within a few
generations of crisis. We suspected that POJ cells from earlier passagesmighthave a betterchance of surviving the entire course of the replication experiment and that such cells would presumably accumulate greater quantities of
replicatedviral DNA at the latertime points. To pursuethis possibility,POJ-2 cells(ca.passage35)andunclonedPOJ-27 cells(passage12)weretransfectedwithwild-type JCVDNA and testedfortheirabilityto supportviral DNA replication (Fig. 4b). Results similar tothose obtainedfor the
POJ-2-PHFGcellcomparisons (Fig. 4a)wereobtained; JCVDNA
replicatedmost efficientlyinPOJ-2 cells onlyatearly times posttransfection. In this experiment the amount of viral DNAinPOJ-2 cellsdecreasedslightly between
days
14and21, again reflecting a significant loss of cells from these
cultures. POJ-27 cells, however, maintained a healthy ap-pearance throughout the experiment, and cell numbers
re-mainedconstant.The amountofreplicatedDNAdetected in POJ-27 cells inweeks2and 3oftheexperimentfarexceeded
thatfoundin POJ-2 cells. ViralDNAaccumulatedto an even greater extent in POJ-19 cells (ca. two- to threefold more than in POJ-27 cells; data not shown). These studies indi-cated that the uncloned POJ cells
supported
JCV DNAreplication better than
permissive
PHFGcells.After
establishing
that POJ cellssupported
JCV DNAreplication, experiments were
designed
todemonstrate thatinfectious virions were
produced by
these cells. Confluent cultures ofPOJ-19,
POJ-27,
and PHFG cells were estab-lished in 24-well(16-mm-diameter)
clusterplates
containing 12-mm cover slips. Each well was inoculated with 100 HAunits ofprototype MadlJCV. Virus
production
was moni-tored by immunofluorescentstaining
of infected cells toon November 10, 2019 by guest
http://jvi.asm.org/
TABLE 2. JCV multiplication inPOJ-19, POJ-27,and PHFG cells
CellsDays after Meanno.ofnuclei
Cells
Dayioatio
containingcapsid HAunitsbinoculation atgn-Santigena ± SD
PHFG 7 87 11 93
14 607 ± 125 587
21 715 ± 73 1,696
28 1,045 + 99 2,240
POJ-19 7 649 ± 118 267
14 434 ±42 747
21 333 ± 61 427
28 341 ± 20 427
POJ-27 7 92± 27 120
14 132 ± 16 373
21 158 ± 51 320
28 254± 41 533
aMeannumber(±standarddeviation) ofantigen-containingnucleionhalf
ofa12-mm coverslip. Basedonelevencountsfrom threeexperiments. bTotalyield;meanofthreeexperiments.
detect viral capsid antigens and measuring HA
activity
in lysatesoftheinfected cells. Atweekly intervals coverslipswere removed for infected-cell counts and cells were
har-vested for virus assays (Table 2). Virus from the 28-day
harvest of one experiment was passed through four
addi-tional serial passages ofPOJ-19 and POJ-27 cells in 25-cm2 flasks. The final virus yield was a 10,000-fold increase in
POJ-19and a5,000-foldincrease in POJ-27overthe
original
virusinoculum,demonstratingthatthe cellsproduced com-pleteinfectious virus.POJcellssupport DNAreplication ofJCVcarryingalethal early-region mutation. The primary reasonfor
deriving
POJ cellswasto obtaina permanentcellline that could be used topropagatewild-type JCV. However, if these cellsconsti-tutively
expressed
afunctionalTprotein, theywouldalsobe usefulfor complementingthe growthofJCV mutantscarry-ingalethalearly-region mutation. Toinvestigate this possi-bility, we made useofthe JCV mutantS-1, which failed to
induce detectableTantigen productioninPHFG cells(Table
1). The DpnI assay was used to determine the replicative ability ofS-1 DNAinPHFG and POJ-2cells. As expected, S-i DNA didnot replicate in PHFG cells; however, newly synthesized viralDNAwasdetected inPOJ-2 cells, indicat-ingthatafunctionalTprotein was presentin thetransformed
cells(Fig. 5). Althoughreplicated viral DNAwasdetectedin the POJ-2cells, the amount of DNA was significantly lower than that following transfection with wild-type JCV DNA.
Substituting POJ-19 or POJ-27
celis
for the POJ-2 cells inthese experiments did not raise the levels of replicated S-1 DNA observed atthelatertime points (data not shown).
Integration patterns of JCV DNA in cloned POJ cells. The presence ofS-19 and S-27 DNA in POJ-1 and POJ-2 cells,
respectively, was examined by the approach of Botchan et al. (3) and Ketner and Kelly (17). Results from these
experiments revealed complex integration patterns of JCV DNAin the cellular genomes of these two cell lines. When restriction enzymes were used that did not cleave the parental
DNAs
(e.g.,Bcll andEcoRV), each band resolved onthe blotrepresented at least oneindependent integration event.BothPOJ-1andPOJ-2 cells had viral DNA integratedintothe cellular DNA at four or more sites (Fig. 6a). DNA
fragments comigrating with the supercoiled and nicked cir-cular forms of the parental plasmids were not detected,
suggesting thatfree
episomal
viral DNA was absent in the cells. At least three oftheintegrated
sequences in POJ-1 cellsandoneinPOJ-2 cellswereshorter than thetransform-ing
DNA.Toinvestigate
thenatureoftheinsertionsfurther,
cellular DNA was
digested
with restriction enzymes thatcleavedthe
parental
DNAsonce(Fig.
6b).
BstXIandBamHI
cutwithin the
early
coding region
ofJCV,
whileBstEIIcutswithin the late coding sequences. Scal cleaves the
pBR322
vectoronly. The large number of bands observed on
blots
representing
the BstXI- andBamHI-digested
samples
sug-gested
that most of the viral inserts retained these tworestriction sites. It is
possible
that several of these inserts contained anintact copy ofthe JCVearly
region
andmight
thereforespecify
functionalTproteins.
The BstEII and ScaIdigests were less
complex,
suggesting
that several of theintegrated viral copies were
missing
these restriction sites. Onthe basis of these results withsingle-cutting
enzymes, it appears that theintegration
of viral DNA in POJ cellsfrequentlyinvolved sequencesfromthelatecoding
region
of the virus orfrom theplasmid
vector.Digestion
of S-19 and S-27plasmid
DNAs with PvuII yielded four fragments, one ofwhich contained the entireJCVearly codingregion (2.5 kilobase [kb] fragment).
Cleav-age of POJ-1 and POJ-2 cellular DNA with this restriction
enzyme gave a strong 2.5-kb band (data not shown),
sup-porting
theassumption
thatseveralintactcopies
of the entireearly
region
are present in the POJ-1 and POJ-2 cellular genomes.A second cloned cell line derived independently from POJ-27 cells
(designated
POJ-2c)gaveJCV DNAintegration
profiles
that were identical to those seen in POJ-2cells,
indicating that both cell lines arosefrom the sametransfor-mationevent(datanotshown). Thisresult may indicatethat
the original transformation was a rare occurrence and that the whole population of uncloned transformed cellswas the progeny of only a few individual transformants. Our diffi-culty in deriving the POJ cell lines would support this suggestion. Alternatively, the precursor cells ofthe cloned
0
0
o
MMMM
a f3
0
o T
a- a0
7
N 0
I
o I:
a-
a-i4
0 -) IL
o fr
a- a
2
r~~
C0
-~
U-o lZ
a a
46I
FIG. 5. Replication activity of a JCV early-region mutant in POJ-2 andPHFGcells Thereplicative activityofS-iDNA inPOJ-2 andPHFG cellswasinvestigatedasdescribed in thelegendtoFig. 4exceptthatno further reduction was made in the amountof the 21-day samples. Additional bands in thedigest of the7-day POJ-2 extract resulted fromincomplete Dpnldigestion of the DNA. The amountsandidentities of the
Ml
andM2markersare asgivenin thelegend toFig. 3.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.327.546.467.642.2]-{cl EcoRV
Ml M22M3 i1 2 2'M4 Ml NM2
*BstX BOmH BstEI1 Sco
Ml M2M3 2 2 2 21 Ml
a
formSn
form
8.9kb
--7.1kb Om
b
89kb - o 7.1kb
-5.1 kb
_..1mI
_a
_aa
p
40 _ of _
_1
*
_-U
_U.
on
U
5.1kb-X 18kb- t
Af
4
FIG. 6. Integration ofviral DNA in the cellular genomes of POJ-1 and POJ-2 cells. Cellular DNA (10pg) was digested with various restriction enzymes, electrophoresed on 1% agarose gels, and transferred to nitrocellulose filters (35). JCV DNA minus pBR322 vector sequences wasradioactivelylabeled by nicktranslation (32) and used as a virus-specific hybridization probe. The following DNAs were used assize markers: linearizedpMITC(ABam), theparental plasmid(M1;8.9 kb),pMITC(ABam)cleaved withHindIII(M2; 7.1, 1.8, 0.4, and 0.2 kb), linearized JCVDNA (M3; 5.1 kb), and uncut form I and formIIpMITC(ABam)(M4; 8.9 kb). Theamounts of marker DNAs loaded on the gelrangedfrom5 x 10-6 to 2 x 10-5 pug(1 x 10 ,ug of DNA is equivalent to the amount of viral DNA in 10 ,ug of cellular DNA if 1 copyofthe genome is present in each cell). Thefilms were exposed for 2 days with an intensifying screen. (a) Digestion of POJ-1 (lanes 1) and POJ-2(lanes 2) cellular DNAs withBcIIandEcoRV,which do not cut S-19 and S-27 DNAs. (b)Digestion of POJ-1 (lanes 1) and POJ-2 (lanes 2) cellular DNAs with BstXI,BamHI,BstEII,andScaI, each of which cleaves S-19 and S-27DNAs once.
POJ cell lines may have had a selective growth advantage over other transformants in the culture. Because of this advantage, such cells may have become predominant in the uncloned population prior to cloning or may have had a
higherplating efficiency during the cloning operation.
DISCUSSION
The studies presented here describe our attempts to
es-tablish a permissive cell line for the propagation of the human polyomavirus JCV. Although our approach paral-leled that taken by Gluzman (11) in deriving COS cells, its success wasdifficult to predict because of several problems
inherent to the JCV system. To derive a JCV COS cell
equivalent, three requirements had to be met:
origin-defectivemutantshadtobe constructed,PHFG cells had to be transformed by the mutant DNAs, and the cells had to
retain their permissivity for JCV. Although the JCV ori sequenceshad notbeen defined precisely,weexpected that
thefirststepwouldbe straightforward. Problems associated
with steps two and three were considered more serious
obstacles to the establishment ofa useful cell line. These
problems includedthehighly inefficient transforming activity ofthevirus (10, 15)and theinability of normal PHFG cells,
once passaged a few times in culture, to support JCV
replication (B. L. Padgett and D. L. Walker, unpublished observations).
Basedonits closehomology with the SV40DNA
replica-tionorigin, we assumed that the 25-np dyad symmetry found atnucleotides 5118 to 12 in the JCV genome formed partof
thereplication origin ofJCV(8, 26). A
HindIlI
cleavagesiteisfoundnearthis symmetry, and becauseofthe
infrequent
occurrenceof thisrecognition sequence in the JCV genome
(threetimes),
HindIlI
wasused in thesite-specific
mutagen-esisprocedure. Mutationswereobtainedatall three HindIII sites. Thoseoccurringatnucleotides 4498(within
thesmall-t coding sequences)and 4914(withinsharedcodingsequencesforlarge and smallt) will prove useful in future studies aimed atinvestigatingtheJCVearly proteins. Recent experiments
indicatethat the JCV Tprotein contributes to the restricted lytic and transforming activities of the virus in vitro (4). The
small-tmutantsconstructed in this study will prove useful in
determining the role ofthe small-t protein in the biological
activity of this virus.Themutationsatnucleotide 4498 affect the termination codon of small-t translation or the donor splice site of small-t transcript processing. Most of these mutant DNAs induce T antigen expression in PHFG and BHK-21 cells; the lattercells also become morphologically transformed.
Inthe present studyourprimary interest was in mutants that had deleted the HindIII site at nucleotide 5112. This
cleavage site occurs within a 19-np dyad symmetry lying adjacenttothe25-npsymmetry presumed to be the center of the JCV origin of DNA replication. Three DNAS (S-15,
S-19, and S-27) mutated at this site were tested in aDpnI
assay and were found to be replication defective. The
deletion inmutantS-27wasconfined to the19-np symmetry,
indicatingthatmutations affectingsequences next to the ori center and between
T-antigen-binding
sites I and II were sufficienttointerfere with JCV DNAreplication.Recentlyit has been shown that insertions and deletions in thecorre-sponding region of the SV40 genome abolish viral DNA
replication by disrupting the spacer function of these se-quences(5, 20).
The two origin-defective JCV mutants with the smallest
deletions (S-19 and S-27) transformed PHFG cells to
yield
thecell lines called POJ-19 and POJ-27. Thesecellssupport JCV DNA
replication
and virusproduction
atlevels compa-rable to those observed with the traditional cell system, PHFG cells. It was also noted that the JCVlytic cyclewas faster in the POJ-19 cells than in POJ-27 or PHFG cells.Although the
primary
cellsrapidly
lose theirability
to support thelytic cycle
of JCV(prior
to theirsenescence at passage8), POJ cells remainpermissive
nearly
until the timeof crisis(passages35to
40).
Similar resultswerereported by
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.100.522.70.241.2]Major and co-workers with their SVG cell line (21). While SVG cells do support JCV replication, the SV40 T antigen producedin SVG cells apparently fails to interact effectively with the JCV ori sequences. This is surprising, since this
protein does bind to the JCV origin region (7) and recent results demonstrate that plasmids containing JCV ori se-quences replicate in COS cells, presumably through their
interaction with SV40 T antigen (19; K. Lynch, W. F.
Chuke, and R. J. Frisque, unpublished results). Moreover,
studies with hybrid polyomavirus genomes containing SV40 and JCV DNA sequences indicate that SV40 T protein interacts with the JCV regulatory sequences to yield viable
viruses (4). An additionalproblem withthe SVG cell line is
the possibilityofrecombination occurringbetween the
inte-grated SV40genome and the input JCV DNA. It has been shown thatrecombinantviruses doarisewhenSV40 mutants are propagated in COS cells (11, 16, 18, 33); the extensive homology between JCV and SV40 may also favor these recombination events. Our results with JCV-SV40 hybrid viruses constructed in vitro indicate that, should recombi-nants arise inSVG cells, some will be viable and may have aselectivegrowthadvantage over the input JCV. Because of the immunological cross-reactivity of many of the JCV and
SV40 proteins, it might not be possible to identify such
recombinantswithout closeexamination of the viral DNA by
molecularbiological methods.
POJ cells represent the first permissive cell system for
JCV that constitutively produces a wild-type JCV large-T protein. In combination with COS and SVG cells, POJ cells
form ausefulcollection of cell lines forinvestigating
DNA-protein interactions of the closely related viruses JCV and SV40. This is especially relevant now, since recent studies indicatethat the JCV Tproteininteracts lessefficientlythan
its SV40 and BKV counterparts with homologous and
het-erologous polyomavirus regulatory sequences (4).
As with COS cells, the POJ cell lines will be used to propagate viruses carrying lethal early-region mutations.
Althoughthepresentstudies demonstratedtheability of POJ
cells to support DNA replication of the JCV T-antigen
mutant S-1, the replication efficiency of the DNA was
significantly lower than that ofwild-type JCV DNA. Itwill be necessary to test additional early JCV mutants in these
cellstodeterminewhetherthis resultis aconsequence of the
cell system or the particular mutant. The first possibility seemsunlikely; immunoprecipitation andSi nuclease
anal-ysesindicatethatauthentic-sized JCV T antigen is produced and the proper mRNA start sites are utilized in POJ cells
(22). This information, togetherwith the viralDNA integra-tion data presented in this paper, suggests that POJ cells express significant levels of an unaltered T protein, and
thereforethe cell system is probably notresponsible for the
reduced replication of S-1 DNA. It is possible that this mutant produces a nonfunctional T polypeptide that is not detected in transfected cells by immunofluorescentstaining.
Suchapolypeptide might be capable of interfering with viral DNA replication by forming inactive oligomers with wild-type Tprotein monomers.
Southern blot analysis of POJ-1 and POJ-2 cells revealed that integration of viral sequences into the host genomes occurred at a minimum of four to five unique sites. Similar
complex
patterns
have been reported to occur inJCV-transformed hamster cell lines and tumors (22, 25, 41). In
parallel studies,theSV40 transformantsfrequently exhibited
simpler integrationpatterns. It is possible that JCV
transfor-mation requiresmultipleintegration events; if this is found to
be a prerequisite, it might explain the poor transforming
activity of JCV, since theprobabilityofmultiple integation
eventswould likely be low.
The JCV mutants and POJ cell lines described in this report should allow us to pursue new avenues of study for this poorly understood yet medically important polyoma-virus. We believe that POJ cells will greatly facilitate the study of this virus and that theavailability of these cells will encourageothers to undertake JCV research.
ACKNOWLEDGMENTS
We think Fran Reilly forthe preparation ofthismanuscript. This workwas supported by grants from the American Cancer Society (MV-168), by Public Health Servicegrants from the Na-tional Cancer Institute (CA-38789) and the National Institute of Allergy and Infectious Diseases (AI-11217), and by the Tracy RuhrupMemorial Fund.
LITERATURE CITED
1. Beckman, A. M., K. V. Shah, and B. L.Padgett. 1982. Propa-gation and primary isolationof papovavirus JCinepithelial cells derived fromhumanurine.Infect. Immun.38:774-777. 2. Binetruy, B., G.Rautmann, G. Meneguzzi, R. Breathnach, and
F.Cuzin.1982.Bovinepapilloma virus 1-pBR322 and polyoma-pBR322 recombinants as eukaryotic vectors, p. 87-92. In Y. Gluzman (ed.), Eukaryotic viral vectors. Cold Spring Harbor Laboratory, ColdSpring Harbor,N.Y.
3. Botchan, M., W. Topp, and J. Sambrook. 1976. The arrange-mentof simian virus40 sequences in the DNAof transformed cells. Cell 9:269-287.
4. Chuke,W.-F., D. L. Walker, L. B. Peitzman, and R. J.Frisque. 1986.Construction and characterization ofhybridpolyomavirus genomes.J. Virol. 60:960-971.
5. Deb, S., A. L. DeLucia, C.-P. Baur, A. Koff, and P.Tegtmeyer. 1986. Domain structure of the simian virus 40 core origin of replication. Mol.Cell. Biol.6:1663-1670.
6. DeLucia, A. J., B. A. Lewton, R.Tjian, and P. Tegtmeyer.1983. Topography of simian virus 40 A protein-DNA complexes: arrangementofpentanucleotide interaction sitesattheorigin of replication. J.Virol. 46:143-150.
7. Frisque, R. J. 1983.Regulatorysequences andvirus-cell inter-actions of JC virus. Prog.Clin. Biol.Res. 105:41-59.
8. Frisque, R. J. 1983. Nucleotide sequenceof theregion encom-passing the JC virus origin of DNA replication. J. Virol. 46:170-176.
9. Frisque, R.J., G. L. Bream, and M. T. Cannella. 1984. Human polyomavirus JC virusgenome. J.Virol. 51:458-469.
10. Frisque, R.J., D. B. Rifkin, and D. L. Walker. 1980. Transfor-mation of primary hamster brain cells with JC virus and its DNA.J. Virol. 35:265-269.
11. Gluzman, Y. 1981. SV40transformed cells support the replica-tionof early SV40mutants. Cell23:175-182.
12. Gluzman, Y., R. J. Frisque, and J. Sambrook. 1980. Origin-defectivemutants ofSV40. Cold SpringHarbor Symp.Quant. Biol. 44:293-299.
13. Hirt, B. 1967. Selective extraction of polyoma DNA from infected mousecell cultures.J. Mol. Biol. 26:365-369. 14. Holmes, D. S., and M.Quigley. 1981. A rapid boiling method for
the preparation of bacterial plasmids. Anal. Biochem. 114:193-197.
15. Howley, P. M., F.Rentier-Delrue, C. A. Heilman, M.-F. Law, K. Chowdhury, M. A. Israel, and K. K. Takemoto. 1980. Cloned human polyomavirus JC DNA can transform human amnion cells.J. Virol.36:878-882.
16. Jasin, M., J. deVilliers,F. Weber,and W.Schaffner.1985. High frequency of homologous recombination in mammalian cells between endogenous and introduced SV40 genomes. Cell 43:695-703.
17. Ketner, G., and T. Kelly, Jr. 1976. Integrated simian virus 40 sequences intransformedcell DNA: analysis using restriction endonucleases. Proc. Natl. Acad. Sci. USA 73:1101-1106. 18. Lanford, R. E., and J. S. Butel.1984. Construction and
on November 10, 2019 by guest
http://jvi.asm.org/
terizationofanSV40mutant defectiveinnucleartransport of T antigen. Cell 37:801-813.
19. Li, J. J.,and T. J. Kelly. 1985. Simianvirus 40 DNA replication in vitro: specificity of initiationand evidence forbidirectional replication. Mol. Cell. Biol. 5:1238-1246.
20. Li, J. J., K. W. C.Peden, R. A. F. Dixon, and T. Kelly. 1986. Functional organization of the simian virus 40 origin of DNA replication. Mol. Cell. Biol. 6:1117-1128.
21. Major, E. O., A. E. Miller, P. Mourrain, R. G. Traub, E. deWidt, and J. Sever. 1985. Establishment ofaline ofhuman fetal glial cellsthat supportsJC virus multiplication.Proc. Natl. Acad.Sci. USA82:1257-1261.
22. Mandl, C., and R. J. Frisque. 1986. Characterization ofcells transformed by thehuman polyomavirus JCV. J. Gen. Virol. 67:1733-1739.
23. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: alaboratory manual, p. 86-96, 249-253. Cold Spring HarborLaboratory, ColdSpringHarbor, N.Y.
24. Maxam, A. M., and W. Gilbert. 1980. Sequencingend-labeled DNAwith base-specific chemical cleavages.MethodsEnzymol. 65:499-560.
25. Miller, N. R., W. London, B. L. Padgett, D. L. Walker, and W. C. Wallen. 1983. Thedetection ofJC viral genome in owl monkeytumors. Prog.Clin. Biol. Res.105:271-288.
26. Miyamura, T., H. Jikuya, E. Soeda, and K. Yoshiike. 1983. Genomic structure of human polyoma virus JC: nucleotide sequence of theregioncontaining replication originand small-T-antigengene. J. Virol. 45:73-79.
27. Miyamura, T., K. Yoshiike, and K. K. Takemoto. 1980. Char-acterization of JC papovavirus adapted to growth in human embryonic kidneycells. J. Virol.35:498-504.
28. Padgett, B. L., C. M.Rogers, and D. L. Walker. 1977.JC virus, ahuman polyomavirus associated with progressive multifocal leukoencephalopathy: additional biological characteristics and antigenic relationships. Infect. Immun. 15:656-662.
29. Padgett, B.L., and D. L. Walker.1973.Prevalence of antibodies in human sera against JC virus, an isolate from a case of progressive multifocal leukoencephalopathy. J. Infect. Dis. 127:467-470.
30. Padgett, B. L., D. L. Walker, G. M. ZuRhein, R. J.Eckroade, and B. H. Dessel. 1971. Cultivation ofpapova-like virusfrom human brain withprogressive multifocalleucoencephalopathy. Lanceti:1257-1260.
31. Peden,K. W. C.,J.M.Pipas,S. Pearson-White, and D. Nathans. 1980. Isolation of mutantsofananimal virus inbacteria.Science 209:1392-1396.
32. Rigby, P. W. J., M. Dieckmann, C. Rhodes, and P. Berg. 1977. Labelingdeoxyribonucleic acidtohigh specific activityin vitro by nick translation with DNA polymerase I. J. Mol. Biol. 113:237-251.
33. Shaul, Y., 0. Laub, M. D. Walker, and W. J. Rutter. 1985. Homologous recombination between a defective virus and a chromosomalsequence in mammalian cells. Proc. Natl.Acad. Sci.USA 82:3781-3784.
34. Sompayrac,L.M., and K. J. Danna.1981.Efficient infection of monkey cells with DNA of simian virus 40. Proc. Natl. Acad. Sci. USA 78:7575-7578.
35. Southern,E. M. 1975. Detection ofspecific sequencesamong DNAfragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517.
36. Takemoto, K. K., P. M. Howley, and T. Miyamura. 1979. JC humanpapovavirus replicationin human amnion cells. J. Virol. 30:384-389.
37. Tjian, R.1978.Protein-DNAinteractionsatthe originof simian virus40DNA replication. Cold SpringHarbor Symp. Quant. Biol. 43:655-662.
38. Tooze, J. (ed.).1981. Molecularbiology oftumorviruses,part2, 2nd ed.: DNA tumorviruses. ColdSpring Harbor Laboratory, ColdSpringHarbor, N.Y.
39. Tyndall, C., G. La Mantia, C. M. Thacker, J. Favaloro, and R. Kamen. 1981. Aregion of the polyoma virus genomebetween the replication origin and late protein coding sequences is requiredin cisfor both earlygene expression and viralDNA replication. Nucleic Acids Res. 9:6231-6250.
39a.Walker, D. L., and R. J. Frisque. 1986. Biology and molecular biology of JC virus, p. 327-377. In N.P. Salzman (ed.), The Papovaviridae: the polyomaviruses. Plenum PublishingCorp., NewYork.
40. Wigler, M., A. Pellicer, S. Silverstein, and R. Axel. 1978. Biochemical transfer ofsingle-copy eucaryoticgenesusing total cellularDNAasdonor. Cell 14:725-731.
41. Wold, W. S.M., M. Green,J.K. Mackey,J. D.Martin,B.L. Padgett,and D. L. Walker. 1980. Integration pattern of human JC virussequencesintwoclones ofacell line established from a JC virus-induced hamster brain tumor. J. Virol. 33:1225-1228.