0022-538X/89/020819-09$02.00/0
Copyright © 1989, AmericanSocietyforMicrobiology
Herpes
Simplex Virus Glycoprotein
D
Mediates Interference with
Herpes
Simplex Virus Infection
RAYMOND M. JOHNSON1AND PATRICIA G. SPEAR2*
Department ofMolecularGenetics and Cell Biology, The Universityof Chicago, Chicago, Illinois60637,1 and DepartmentofMicrobiology-Immunology, Northwestern University Medical and Dental Schools,
303 EastChicagoAvenue, Chicago, Illinois606112 Received 12 August 1988/Accepted 2 November 1988
We showedthattheexpression ofasingle protein, glycoproteinD (gD-l),specifiedby herpessimplex virus type1(HSV-1)renders cellsresistanttoinfectionbyHSVbutnottoinfectionby otherviruses. Mouse (LMtk-) and human (HEp-2) cell lines containing the gene for gD-l under control of the human metallothionein promoterIIexpressedvarious levels of gD-l constitutively and could be inducedtoexpresshigher levels with heavy metal ions.Radiolabeled virusesbound equally welltogD-l-expressingandcontrol cell lines. Adsorbed viruseswereunableto penetratecellsexpressing sufficient levels of gD-l, basedonlackofanycytopathiceffects of thechallenge virus andonfailuretodetecteither theinductionofviralproteinsynthesisortheshutoffofhost protein synthesisnormally mediatedbyavirion-associatedfactor. The resistance toHSV infectionconferred by gD-l expression was not absolute and depended on several variables, including the amount of gD-l expressed,the dosageofthechallengevirus,theserotypeof thechallengevirus,andtheproperties of the cells themselves.The interference activity of gD-1isdiscussed inrelationtotherole ofgD-lin virioninfectivity and itspossible role in permittingescapeofprogenyHSV from infected cells.
The last stepof successful viral replication is the egress of progeny viruses from infected cells. A progeny virus must escapefromthe cell that produced it withoutsuperinfecting
thatcell. Superinfection would result in eclipse ofprogeny
virions and a decreasein the infectious yield.
Different viruses may usedifferentmechanisms to prevent
eclipse of progeny virions by host cells. Members of the
paramyxovirus and orthomyxovirus families express a
re-ceptor-destroyingenzyme, neuraminidase. Thisenzyme re-moves sialic acid, the virus receptor, from viral and cell
glycoproteins and glycolipids(35, 52).Similarly, some
coro-naviruses express a receptor-destroying esterase with a
unique specificity (29). Cells producing retrovirusgene
prod-ucts are resistant toinfection by closely related strains of retroviruses and presumably to superinfection by progeny
virusreleasedfromthecells (49). Themechanism(s)
respon-siblefor this interference isnotfully understood.Ithas been shown that the CD4 receptor for the human immunodefi-ciency virus isreduced in amount on the surfacesofinfected
cells,atleast in part becauseofsequesteringof the receptor
withinthe cell by the env gene product (18, 47). Expression
oftheenvgene product alonecanreduce the levels ofCD4 onthe surfaces of transformed cells(46).
Superinfection exclusion or interference has not been
documented forherpesviruses, in part becauseof difficulties associated withdetectingsuperinfection ofcellsinfectedby
alytic virus. As shownhere, expression of herpes simplex virustype 1(HSV-1) glycoproteinD(gD-1)in the absenceof lytic infectionissufficienttorenderpermissive cells resistant
toHSVinfection, justasexpressionofaretroviral envgene
product can make cells resistant to infection by closely related retroviruses. Our studyfollows onthe observations of Campadelli-Fiume et al. (5), who found that a baby hamsterkidney cell line transformed by theBamHI J
frag-mentoftheHSV-1 genome and some subclones of the cell linewere resistanttoHSV infection. The cells were shown
* Correspondingauthor.
to express gD-1 but may also have expressed other HSV proteins, inasmuchas the DNAfragment usedfor
transfor-mation containsfourcompleteopenreadingframesencoding
membrane glycoproteins (gG, US5, gD, and gI) and trun-catedforms ofthe openreading frames foraprotein kinase and gE (32). In the subclones of the transformed cell line
analyzed by Campadelli-Fiume etal. (5), it was possibleto detectexpression of
gD-1,
butnotgI-1orgE-1,by immuno-precipitation; attempts to detect the other potentiallyex-pressed HSV proteinswerenotreported.
In the experiments reported here, HSV DNA fragments encodinggD-1 plus gI-1 (the gI-1 transcription unit is
con-tained totally within the gD-1 transcription unit) or gD-1
alone were cloned into expression vectors and used to
transform cells. In all plasmid constructs, the endogenous
promoter ofgD-1 was replaced by the human
metallothio-nein promoter II. Because this promoter is inducible by heavy metal ions (22), we could assess the effects on
interference of increased levelsofgD-1expressionwithin a cellline.
gD isoneofsevenvirionenvelope
glycoproteins
knownto be encodedby herpessimplex viruses;theothersaregB,gC,
gE,gG,gH, andgI (1, 9,14,28, 38, 40, 44).Geneticanalyses
have demonstratedthatgD, gB, and gHare all
required
for virion infectivity (4, 6, 26, 41). The other four HSV glyco-proteinsappeartobedispensible
for viralreplication
in cell culture (15, 28, 33, 51).Monoclonal antibodies specific for gD can be strong neutralizers of HSVinfectivity. Theseantibodies block the
penetration stepratherthan
adsorption
of virus tocells(11,12, 16, 36).Inaddition,somemonoclonalantibodies
specific
forgDcan, athigh concentrations,inhibitstable attachmentofthe virustocells(11).
Anti-gD
monoclonal antibodiescanalsoblock virus-induced cell-cell fusion(34).
Finally,
mutant virions devoid of gDcanadsorbtocells but failtopenetrate thecells(26). These data suggest thatgDplays
animportant
role inviral entry atthefusion-penetration
step.819
on November 10, 2019 by guest
http://jvi.asm.org/
820 JOHNSON AND SPEAR
MATERIALS ANDMETHODS
Viruses and cells. HEp-2 cells and mouse LMtk- fibro-blasts were grown in Dulbecco modified Eagle medium
(DMEM;
GIBCO Laboratories) supplemented with 10%fetal calfserum
(FCS;
Hyclone and GIBCO). African greenmonkey kidney
cells (Vero cells) were grown in Hanks 199supplementedwith5% FCS.Thevirus strains used for these
experiments
wereHSV-1(KOS), HSV-1(F), HSV-2(G),
HSV-2(333),
vaccinia virus(WR), and vesicular stomatitisvirus (VSV)(San Juan). Virus stocks were prepared by low-multiplicitypassageinHEp-2cells,and their titerswere determinedon Vero andHEp-2 cells.
Plasmid construction. The
coding
sequences forgD-1 andgI-1 weretakenfroma
plasmid,
pDW15, whichcontains the3,641-base-pair (bp)
SmaIsubfragmentofBamHIfragment
Jof
HSV-1(KOS)
DNA inserted into the SmaI site ofpUC8
(produced
by
Darrell WuDunn). Theendogenous
gD-1 pro-moter wasremovedbydigestion
of theplasmid
withHindlll and SinaI. TheHindlIl site lies very close tothetranscrip-tion initiation site forgD-1 mRNA(7, 19, 50). The2,900-bp Hindlll-Smal fragment
containing
the gD-1and gI-1 codingsequences was filled in with the Klenowfragment of DNA
polymerase
andinsertedintotheSmaI site ofplasmid
pHSIcontaining
the humanmetallothionein promoter 11(22;plas-mid
kindly
provided by E.Kieff).
The resulting plasmid,with the insert oriented such that gD-1 was immediately
downstream of the metallothionein promoter, was
desig-nated pRJ20. An NruI-NruI 813-bp deletion within pRJ20removed the translation initiation codon and the N-terminal
coding
sequences ofgIdowntoits transmembranedomain.gD-1
was theonly
remaining
intact HSVcoding
sequencewithin thisplasmid, which was designated pRJ41.
A
general
purpose vector, pRJ40,containing
both thehumanmetallothioneinII promoterand theneomycin select-ablemarker from TnSwasproducedby
inserting
the BamHIfragment (containing
theresistancecartridge)frompBKneo(kindly provided
by R.Manservigi)
into the Narl site ofpHS1
afterfilling
bothfragments
in withT4DNApolymer-ase.The metallothionein-HSV
transcription
unitsfromplas-mids pRJ20 and pRJ41 were transferred into the pRJ40
vector by exchanging a HindIIl-EcoRI fragment with a
HindIII-EcoRI
fragment
in pRJ40. The new plasmidscon-taining
thegD-1andgI-1
genesoronlythegD-1gene,alongwith the metallothionein promoter and the neomycin select-able marker, were
designated
pRJ35 and pRJ42,respec-tively.
Restriction enzymes were from New EnglandBio-Labs.
Transfections. Transfections were done by the calcium
phosphate precipitation
procedure describedby
Spandidos
and Wilkie
(43).
All vectors were linearized outside of the sequencesof interestbefore transfection. Mouse LMtk- orHEp-2
cellswereplated24hbefore transfectionatdensitiesthat
yielded
monolayers about75% confluent atthe time oftransfection. Calcium phosphate precipitates of plasmid
DNA (0.5 to 1.0
p.g)
along with salmon sperm DNA as the carrier were added to the monolayers in25-cm2
tissue cultureflasksorin 60-mm-diameter dishescontaining5 mlofDMEM,
1.0%
FCS, and gentamicin (GIBCO) at 25pg/ml.
The medium containing DNA was replaced with fresh me-dium 17 to 20 h after transfection was initiated. After
allowing
24 h morefor expressionof the selectable marker,the medium was changedto DMEM-10% FCS with
geneti-cin
(GIBCO)
at 400p.g/ml.
Selection medium was changedevery 3 to4days,andindividual colonies werepicked 10 to 15
days
after transfection. LMtk- cells transformed veryefficientlyand werelimitingdilutedto assureclonal
popula-tions. The cell line designated CA35gD10 was obtained by cotransfectingpRJ20withpSV2neoat a5p.g:1p.g ratio. The parent L cell line forCA35gD1Owas CA14.11.35(30).
Induction ofgD-1 expression. Cell lines were induced to express higher levels ofgD-1 by incubation with DMEM-10% FCS containing either 2 FM cadmium chloride or 100
F.M zinc chloride for 6 to 18 h. The metal chloride stock solutions wereacidified with HCItodissolve the metal ions and thenadjusted to pH 5.0.
Southernblotting and Western immunoblotting. For South-ern blots, cells from two 25-cm2 flasks were detached with EDTA and pelleted. DNA was isolated as described by
Maniatis et al. (31). Proteinase K and RNase A were from Boehringer Mannheim Biochemicals. A
10-Rg
sample of DNA from each cell line was digested with PvuII and separated on a 0.8% agarose gel. Before denaturation, the DNA in thegel was depurinated by two 15-min incubations in 0.25 M HCl. The DNA was denatured with NaOH and transferred to nitrocellulose (BA85; Schleicher & Schuell,Inc.) with a Vacublot apparatus (American Bionetics). Pre-hybridization and Pre-hybridization conditions were those out-lined by Gatti et al. (13). The probe used was a purified BamHI-NruI fragment nick translated (Bethesda Research Laboratories, Inc.) with 32P dCTP (Amersham Corp.). The probe encompasses the entire gD coding sequence. Washes were done at 60°C.
Samples for Western blots were prepared by lysing cell monolayers with 1% Nonidet P-40-10 mM Tris
hydrochlo-ride (pH 8)-0.15 M NaCI-2 mM phenylmethylsulfonyl fluo-ride-8 mM iodoacetamide at 4°C for 30 min. The lysates were collected and centrifuged for 5 min in a water-cooled microcentrifuge. The supernatant was collected, and 4x sodium dodecyl sulfate (SDS) sample buffer was added to yield 1% SDS-0.01 M phosphate buffer (pH 7.0)-0.1 M dithiothreitol-0.02% bromophenol blue-10% glycerol. Sam-ples were loaded onto an 8.5% polyacrylamide gel cross-linked with N,N'-diallyltartardiamide (Bio-Rad Laborato-ries). At thecompletion of the separation, the proteinswere transferred to nitrocellulose (BA85) inTris-glycine-methanol buffer. Nitrocellulose was blocked with phosphate-buffered saline(PBS)-5%milk and incubatedwith diluted polyclonal antiserum (hyperimmune rabbit serum, Rb58/6, kindly pro-vided by I. Halliburton) inPBS-1% bovine serum albumin-0.2% sodium azide for 2 to 4h at room temperature or4°C overnight. Detection wasaccomplished with[125I]protein A (Amersham). Washes were performed with PBS-5%
milk-0.2% Tween 20. Blots were wrapped and placed against Cronex film(Du Pont Co.) withanintensifyingscreenfor 12 to 72 h.
Infectionof cells andlabeling with [35S]methionine. Mono-layers of HEp-2 and LMtk- cells grown in 24-well tissue culture dishes (Corning Glass Works) were infected with HSV in 0.1 mlofPBS-1% heat-inactivated FCS-0.1% (wt/ vol) glucose for 2hat37°Cwith frequent shaking. After 2 h, the inoculum was removed and replaced with 1.5 ml of Hanks 199 with 1% heat-inactivated FCS. At 5 h after infection, the monolayers were washed three times with methionine-free medium 199-1% heat-inactivated FCS and labeled for 30 min in DMEM lacking cold methionine and containing 2% heat-inactivated FCS and [35S]methionine
(Amersham) at 15 p.Ci/ml. After removal of the radioactive medium, the monolayerswere harvestedin lx SDS sample
buffer and separated on an 8.5% polyacrylamide gel. Gels werefixed and treated withAmplify (Amersham), dried, and placed against Cronex film for12to 24 h.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
HSV gD-INDUCED INTERFERENCE 821
Adsorption ofradioactive virustocells.Radiolabeled HSV-1(KOS) was prepared from HEp-2 cells infected at 4 PFU per cell and incubated with [3H]thymidine at 0.02 mCi/ml from 4 to 36 h after infection. Virions were purified on
dextran T10 (Pharmacia) gradients as previously described
(45). Binding assays were done in 96- and 24-well tissue
culture plates byexposing the cellstovariousconcentrations of purified labeled virus in PBS containing 0.1%glucose,1% heat-inactivatedFCS, and bovineserumalbuminat 1mg/ml in atotal volume of30,ul for 96-well plates and 100 ,u1 for 24-well plates. Forassaysdoneat37°C, bindingwasallowed
to proceed for 30 min. At 4°C, binding was allowed to
proceed for 2 h. After the adsorption period, the cell monolayers were washed twice with PBS containing 0.1% glucose and1%heat-inactivated FCS andoncewith PBS and then harvested in 50 ,u1 of PBS containing 1% SDS-1% Triton X-100. The lysateswereaddedto4ml of Econolume scintillation fluid(ICN Pharmaceuticals Inc.) and counted in a scintillation counter (LS3133T; Beckman Instruments, Inc.).
Flow cytometry. Cells were dislodged with EDTA and suspendedinPBS containing 5%heat-inactivated FCS-0.1% sodium azide.The cellswereincubated with 1:10 dilutions of ascites fluidcontaining III-114 (monoclonal antibody specific forgD) orpurified monoclonal antibody II-125 (specific for gB) for 30 min on ice. These monoclonal antibodies were previously described (36). The second-step antibody was fluorescein isothiocyanate-coupled goat anti-mouse immu-noglobulin G from Southern Biotechnology. Stained cells werefixed inPBS-1% paraformaldehyde and analyzedon an EPIX flowcytometer.
RESULTS
Isolation of transformed cell lines expressing gD-1. The plasmids used to transform cells eithercontained the neo-mycin phosphotransferase selectable marker (Fig. 1B) or were cotransfected with pSV2neo (Fig. 1A). The plasmids carried thegD-1 transcriptionunitfromHSV-1(KOS) DNA, modified such that the endogenous viral promoter was replaced by the human metallothionein promoter II. Two
openreadingframesarecontained in thegD-1 transcription unit; the downstream one encodes gI-1. To produce a plasmid capable of expressing gD-1 but unable to express gI-1, most of the gI-1 open reading frame, including the translation initiationcodon,wasdeleted inpRJ42 (Fig. 1C). The controlplasmid used,pRJ40, was essentially as shown in Fig. 1B except that no HSV DNA sequences were inserted.
MouseLMtk-cells and HEp-2 cellsweretransfectedwith theplasmids described above,and stabletransformantswere selected for the ability to replicate in medium containing G418. Table 1 lists the cell clones characterized here, the plasmids usedfor thetransfections, andshorthand designa-tions for the clones. Themousecell linesareprefixedwithan Land human cell lines areprefixed withan H,followed by the HSV glycoprotein-coding sequences potentially ex-pressed by the clone and an arbitrary number to identify different clones. Control cell lines were transfected with pRJ40, whichcontains noHSV DNA, andwere selected in mediumcontainingG418 inparallel with the other clones.
Asexpected,Southern blotanalysisrevealed thepresence
ofappropriate plasmid DNA sequences in the transformed clones. Figure 2A shows the results of hybridization ofa labeledprobespecificfor HSV DNAsequences(the BamHI-NruI fragment indicated in Fig. 1A) with PvuII digests of
A B
gD
C
HMTP gD gI
...rlr .. ._ ... . ...= a 4 pRJ20
oIyA pRJ35
HMTP
-,I
I pRJ42 polyA
Nrula
FIG. 1. Plasmids used for transfection to isolate mouse and humancelllinesexpressing HSV-1 gD. (A) Plasmid cotransfected with pSV2neo. (13)Plasmid incorporating the neomycin selectable marker. (C) Transcription unitsofthedifferentvectors,showing the human metallothionein promoter II (HMTP), the glycoprotein-codingregions for gD and gI (open boxes with black shading to
denote the membrane-spanning regions of each), and transcripts. The promoterfor gDwasreplaced byHMTPin thesevectors.Most ofthecoding region forgI wasdeletedfrompRJ42.
DNAisolatedfrom the transformed clones. Fordigests from all except thecontrolclones, theprobedetected HSV DNA sequences in fragments of sizes similarto those present in digestsoftheplasmidusedfortransfection. Acoupleof the transformed clones (L-gDgI-1 andL-gDgI-2) also contained
extensively rearranged forms ofthese HSV DNA sequences. The clonestransformed bythe plasmidfrom whichmost of thegI-1 sequencesweredeleted(pRJ42)canbe identifiedby
loss ofthe2,883-bp band and appearance ofa newband at 2,070bp.
Transformed clones carrying the gD-1 gene expressed
differentconstitutive levelsofgD-1,asdeterminedby West-ernblot analysiswith apolyclonalrabbit antiserum. Figure
[image:3.612.309.549.74.272.2]2B shows theconstitutive levels ofgD-1
expression
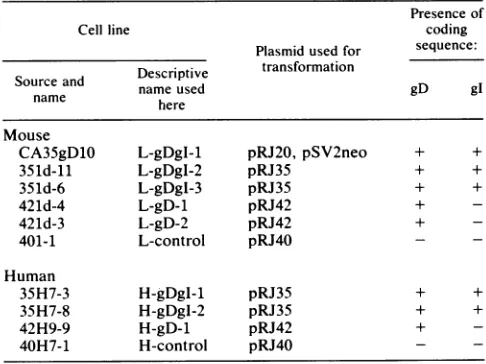
forsomeTABLE 1. Transformed mouseand human celllines
Presenceof
Cellline coding
Plasmidused for sequence: Descriptive transformation
Source and Decitvnameused gD gI
name here
Mouse
CA35gD1O L-gDgI-1 pRJ20, pSV2neo + +
351d-11 L-gDgI-2 pRJ35 + +
351d-6 L-gDgI-3 pRJ35 + +
421d-4 L-gD-1 pRJ42 +
-421d-3 L-gD-2 pRJ42 +
-401-1 L-control pRJ40
Human
35H7-3 H-gDgI-1 pRJ35 + +
35H7-8 H-gDgl-2 pRJ35 + +
42H9-9 H-gD-1 pRJ42 +
-40H7-1 H-control pRJ40
VOL. 63, 1989
p
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.306.549.542.724.2]822 JOHNSON AND SPEAR
_
C? CN 2 7 c4&- -I.--4_ _I 0
O3
aCa,
In> -iuJ
ICDw I L ,j! -j _:.a = =I I=23100- 9416-
6557-
4361-
2322-
2027-Copy Number 0.1 1 10 100
-0 0 CN
o o - 0) 0
_ . _ I I I 1
Cd+*--+ - + - -
-_.,*.
4'4'
[image:4.612.117.529.70.313.2]
564-A B
FIG. 2. (A) Southern blot showing HSV DNAsequences incorporated into the transformedcell lines and theapproximatecopynumber
per cell genome. DNAs extracted from the cell lines indicated were digested with PvuII, and equivalent samples of the digests were
fractionated by electrophoresis on anagarosegel and then blotted ontoanitrocellulose filter. The labeledprobeusedwas aBamHI-NruI fragment of pRJ20 containing the gD-1-coding sequences(Fig. 1). The plasmid usedforthecopynumber controlwaspRJ20digestedwith PvuIl. Lambda HindIll digest markersareshownonthe leftmargin.(B)Western blot of severalmouseandhumancelllines, showingbasal levels of gD-1 expression andincreased levels following induction with cadmium.Anequivalentamountofproteinwasloaded in each lane. Molecular weight (inthousands) markersareindicatedontheright margin.
of theclones, along withthe induced levels detected 8 h after additionof 2,uM cadmiumchloride. Zinc chloride could also be usedtoenhance gD-i expressionin these celllines(data notshown). Theheavy metal ion inductionwas more effec-tive for the human clones than for the mouse clones. A possible explanation is that the promoter is a human pro-moterorthe mouse cells arethymidine kinase negative (or both). Thymidine kinaseisbelievedtobepart of the metal-lothionein induction pathway (25).
For the human clones, the maximal inducible levels of gD-i expression seem to correlatewith the estimated num-bers ofgD-i gene copies present. The same does not hold true for the mouse cell lines. It is uncertain whether the transformed clones carryingthe gI-l gene actually express thisglycoprotein. All of the bands detectedonthe Western blots by the rabbit polyclonal antiserum were removed by immunoprecipitation of the Western blot samples with a monoclonal antibody specificfor gD-i (datanotshown).
Plaqueformationontransformed celllines. Becauseplaque formationbyHSV ispoor onmouseLcells, only thehuman HEp-2 cell clones were testedfor ability to supportplaque formation after challenge with HSV-1(KOS), HSV-2(G), VSV, and vaccinia virus(WR) (Table 2). All of the gD-1-expressing HEp-2 cell transformants tested (including H-gDgI-2, in addition to those shown in Table 2) failed to supportHSV-i plaque formation. Infact, little ifany
cyto-pathic effectwas observed when 106 PFU of HSV-1(KOS)
were plated on 25-cm2 monolayers (about4 x 106 cells) of clone H-gDgI-1 or H-gD-i. By this test, cells expressing gD-i wereresistant tocytopathic effects of HSV-1whether or nottherewas potential for gI-l expression.
The results were not so straightforward with HSV-2. HSV-2(G) formed plaques as well orbetter on the H-gD-i
cell line thanontheH-control cell line butproduced consid-erably fewer plaques on the H-gDgI-1 cell line. Curiously, the HSV-2(G) plaques on the H-gD-1 cell line were much larger and had a different morphology than HSV-2(G) plaques on untransformed HEp-2 cells, the H-control cell line, ortheH-gDgI-1 cell line. We have noexplanationfor this observation nor is it known whether presence of the gI-l-coding sequence, as well as expression of gD-i, con-tributes to the reduced plating efficiency ofHSV-2(G) on H-gDgI-1 cells. These results and others described below show that the gD-l-expressing clones are less resistant to HSV-2 thanto HSV-1. The H-control cell line and untrans-formedHEp-2 cell lines didnotdiffer in theabilitytosupport HSVplaqueformation.
The gD-l-expressing clones that failedto support HSV-1 plaqueformationwere able to support plaque formation by
TABLE 2. Plaque formationontransformed humancells
No.ofplaques/plate(%ofcontrol)a
Cell line Vaccinia
HSV-1 HSV-2 virus VSV
H-control 54(100) 69(100) 192(100) 350(100) H-gDgI-1 0(<0.1) 0(0.6) 300(160) 337(96) H-gD-1 0(<0.1) 82(120) 370(190) 70(20)
aSerial dilutions of viruseswereplated(1-mlsamples)onmonolayersin 25-cm2flasks. After incubation for 2 h topermitvirusadsorption,theinocula werereplacedwith mediumcontaining pooledhuman gammaglobulin.After plaqueshaddeveloped (1to3days),thecells werestainedwithGiemsa.The numbers shown are the average ofduplicatecountsfor asingledilutionof each virus.Plaquesof HSV-1 couldnotbedetectedonH-gDgI-1andH-gD-1 cells, even at the next most concentrated serial dilution. Some of the percentagesgivenarebased on countsobtainedatotherdilutions.
-200
-116
- 97
- 66
- 43
J. VIROL.
6ifl:f.:.:,,,f;w
46
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.328.568.590.661.2]TABLE 3. Binding of HSV-1 to transformed human cells
Presence Adsorption at 4 Cb Adsorption at37°C' Cell line of
heparina PFU/cell
cpmb
d PFU/cell bound'H-control + 100 120 ± 40 150 32 ± 8
- 100 2,700 ± 100 150 380 ± 80
- 50 1,500 30 100 280 ±50
H-gDgI-1 + 100 110 ± 10 150 20 ± 10
- 100 3,100 ± 200 150 350 ± 50
- 50 1,700 ± 100 100 270 ± 30
H-gDgI-2 + 100 90 ± 10 NDe ND
- 100 2,970 30 ND ND
- 50 1,800 ±70 ND ND
H-gD-1 + 100 100 ± 10 150 30 ± 10
- 100 2,800 ±100 150 390 ±30
- 50 1,640 ±40 100 230 ±20
aHeparinwas presentduringadsorptionat50
±Lg/ml.
Heparin inhibitsHSVadsorptiontocells (48,53).
bThe virus was added to cellmonolayers in 24-wellplates.
The viruswasadded to cell monolayers in96-wellplates.
d Counts per minute per welladjustedtocompensateforslightdifferences incelldensity (micrograms ofproteinperwell)forthe different cell lines. The valuesrepresent averagesof three(4°C)or five(37°C)wells and the standard deviations.
eND,Notdone.
vaccinia virus and VSV, except that VSV plated with somewhat reduced efficiency on H-gD-1 cells (Table 2). Vaccinia virus actually formed more and larger plaques, with alteredmorphology,onH-gD-1 andH-gDgI-1clones thanon the H-control cellline. Whetherthisrelates togD-1 expres-sionremains to bedetermined.
Adsorption ofHSV totransformedcells.Experimentswere done to determine whether adsorption of HSV-1 to
trans-formed human cells was less efficient than adsorption to control cells. Cells in 24-or96-well plates wereexposed to
purified, labeled virionsat4°C for2h or at37°Cfor 30min, respectively. After the unbound viruswaswashed away, the cells were solubilized for quantitation of the bound virus. The results presented in Table 3 demonstrate that HSV-1 adsorbedasefficientlytocellsexpressing gD-1astocontrol cells andthat,asexpected, virusadsorption wasinhibitable
by heparin (48, 53).
Ability of HSV to initiate viral protein synthesis in trans-formed cells. Within 1 h ofaddition ofHSV to susceptible cells, cell protein synthesis is sharply inhibited because of
the action ofavirion-associated componentbroughtinto the cell along with the genome (8, 37, 42). This immediate inhibition of cellproteinsynthesisdoes notrequireviral gene
expression. Induction of viral protein synthesis normally
accompanies inhibition of cellprotein synthesis.
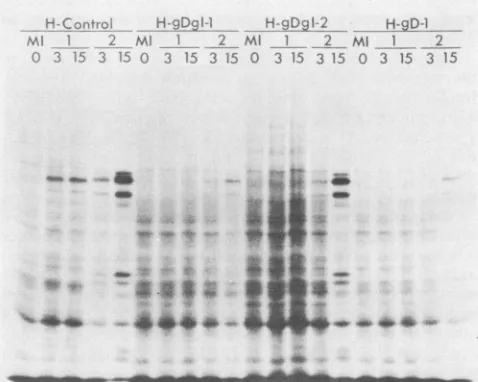
Experimentsweredonetodetermine whetherinhibition of cellprotein synthesisandinduction of viralprotein synthesis could be detected afterexposureof transformedcellstothe virus. For the experiments shown in Fig. 3 and 4, the transformed cells were exposed to HSV-1(KOS) or HSV-2(G) at two different input multiplicities and then pulse-labeledfor 30 min with
[35S]methionine
at 5 hafteraddition of the virus. Because HSV PFUsare notreadilyquantifiableon mouse Lcells, the multiplicities chosen for infection of the mousecell transformants with HSV-1 and HSV-2were thosethatinduced comparable ratesof viralprotein
synthe-sis at 5 h after infection of control cells. The lower
multi-plicity used(x in Fig. 3) corresponds to 1 PFU percell for
L-Control
*_D}
-
__ L-D91-2 L-D-1Ml __ _2 MI 1 2 Ml 2 Ml 1 _ 2
0 x 5x x 5x 0 x 5x x 5x 0 x 5xx 5x 0 x 5x x 5x
a a*
a aN--aa 40go :.. AD -W
4.;r:~a
W.
a
4di0
& A
FIG. 3. Polypeptides produced by mouse cell lines mock in-fected(MI) orinfectedwithlow(x) orhigh (5x) multiplicities of HSV-1(KOS) and HSV-2(G). Cells were labeled for 30 min with [35S]methionine at 5 h after infection, lysedin samplebuffer, and electrophoresed through anSDS-8.5% polyacrylamidegel.
HSV-1(KOS) and 1.5 PFU per cell for HSV-2(G), on the
basis oftiters obtained onHEp-2cells, oraboutone-third of
theconcentrationof the virustowhichtheHEp-2cellswere exposed (Fig. 4). The samevirus stocks had titers of6PFU per cell forHSV-1(KOS) and 3 PFU per cell for HSV-2(G)
when assayed on Vero cells, which we use routinely for plaque assays.
The results (Fig. 3 and 4) demonstrate that all of the
gD-i-expressing
clones (except L-gDgI-2) failed to respondnormallytochallengewithHSV-1(KOS) in that viral
protein
synthesis was diminished and cellprotein synthesiswasnot asefficiently shut off. In contrast, therewas onlypartial, ifC-4CfiDf
Hg.-Da
I-1 H-glA2 H-9D-1;;i1/ Ml 1 2=4 M} 2 Ml 1 2
15 35 0 3 15 3 15 0 3 15 3 15 0 3 15 3 15
_ _ .-a ^ " _
. : .A.9
[image:5.612.53.295.84.262.2]~~~~~e
FIG. 4. Polypeptides produced by human cell lines mock in-fected(MI)orinfected with low(3PFUpercell)andhigh(15PFU percell)multiplicitiesofHSV-1(KOS)andHSV-2(G)viruses.Cells werelabeledfor 30 min with
[35S]methionine
at 5hafterinfection,lysedinsample buffer, and electrophoresed throughanSDS-8.5%
polyacrylamide gel.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.315.554.473.664.2]824 JOHNSON AND SPEAR
L-Control L-Dgl-1 L-ODl-2 L-gD-1
Cd++ + Cd+ C+i Cd++
Ml 0 6 12 Ml 0 6 12 MI 0 6 12 Ml 0 6 12
Sao
.w..*2O am.
L-Control
L-gDgl-2
4S &...
4
# ;,
** ..
LAL
.f.,nw.w p x
....---~ -M - -dbmMbdb -66
-66
-43
FIG. 5. Effect ofenhancing gD-1 expression before infection of
mousecell lines. (A) Polypeptide profilesofcelllinesinducedwith cadmiumfor0, 6,or12 h before infection withHSV-1(KOS). At 5 hafterinfection, cellswerelabeled for 30 min with [35S]methionine, lysed in sample buffer, andelectrophoresed through anSDS-8.5%
polyacrylamide gel. MI, mock infected. (B) Western blotofparallel samples showing the levels of gD-1 expressed at the time of infection.Molecular weight (in thousands) markersareshownatthe right.
L-gDl,1lV1
32
L-gDgl
16
0 10 20 0 10 20 0
20 10 20
Fluorescence Intensity
FIG. 6. Flowcytometric analysis ofmousecelllines before and after infection. Uninfected cells were stained with an anti-gD antibody(column 1) to show levels ofsurface expression of gD-1. Parallel cultures were infected with HSV-1(KOS) (column 2) or HSV-2(G) (column 3) and stained 9 h after infection with an antibody specific forgB. gB isexpressed at relatively high levels earlyafter infection. Thedottedprofilesin column 1 show thelack ofreactivityofanti-gBwithuninfected cells.Thedottedprofilesin the bottompanels ofcolumns 2 and 3 show thebackground staining with the second antibody(fluorescein isothiocyanate-coupled goat anti-mouseIgG)alone.
any, interference withHSV-2(G) infection. Interferencewith HSV-2(G) infection was most evident for H-gDgI-1 (the apparentinterference with HSV-2 infectionofH-gD-1 cells was probably due to an error in sample preparation or loading forthis particular experiment, since all of the other results obtained with these cells indicated susceptibility to HSV-2infection).
Ingeneral, thetransformed cloneswerenotasresistantto infection with HSV-2(G) asto infectionwith HSV-1(KOS). Similar results were obtained with HSV-2(333) and HSV-1(F), indicating that the interference phenomenon is
sero-type selective. Also,particularlyforHSV-2, high multiplic-ities of the challenge virus could overcome any resistance observed,asindicated bythetrendsseenatthemultiplicities tested.
Effects of enhanced gD-1 expression on resistance to HSV infection. Experiments were done to determine whether enhancement ofgD-1 expression could increase the
resis-tance toinfectionoftransformed cells exhibitingapartially resistant phenotype. Transformed mouse cells were incu-batedwith 2,uMcadmium chloride for0, 6,or12h andthen eitherharvestedforquantitation of gD-1 byWesternblotting orinfectedatamultiplicity of15 PFU percell (titeronVero
cells) with HSV-1(KOS). The infected cells were pulse-labeledwith [35S]methionine at5 hafter infection.
For two of the mouse transformants (L-gDgI-2 and
L-gD-1), cadmium induction increased the amountof accumu-latedgD-1 (Fig. SB) and renderedthecellsmore resistantto
HSV-1 infection, as judged by the rates of viral protein synthesisat5 h(Fig. SA). For thethird mousetransformant
(L-gDgI-1), full resistance to the dose of HSV-1 used was observedeven without cadmium induction.
A comparable experiment could not be done with the HEp-2 cell transformants, becausethey are all highly
resis-tant to HSV-1 infection.
Weconcludethat, forasinglecloned cell line, the level of gD-1 expression correlates with thedegree ofresistance to HSV-1 infection, whereas in comparisons ofdifferent cell lines, the degree of resistance to HSV infection may be influenced by as yet unrecognized differences between the clonesaswell asbythe levelofgD-1 expression.
Basisforpartial susceptibility of transformed cells to HSV infection. Experimentsweredonetoinvestigatewhether the transformed-cell populations were heterogeneous with
re-spect to gD-1 expression and whether the intermediate phenotype,withrespect toresistancetoHSVinfection, was duetoafraction of the cellsbecominginfectedortoreduced
ratesof viralprotein synthesis ineachinfected cell. Cells of four transformed mouse clones were divided into three samples.Onesampleofeachwassuspended, incubatedwith an anti-gD monoclonal antibody, stained witha fluorescein isothiocyanate-coupled second antibody, and then fixed. The other two samples were infected at a multiplicity of 5 PFU per cell (titer on Vero cells) with HSV-1(KOS) or HSV-2(G) and, at 9 h after infection, stained following reaction with an anti-gB monoclonal antibody. All samples wereanalyzedon a flow cytometer(EPIX).
The results (Fig. 6) show that the transformed clones constitutively expressed different amounts of gD-1 on cell surfacesand that thecells in eachpopulationwererelatively
A
B
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.319.559.67.315.2] [image:6.612.73.298.71.325.2]homogeneous with respect to the amount ofgD-1 expressed. (The background nonspecific level of staining with the anti-gD antibody was higher than that with the anti-gB antibody [upper left panel of Fig. 6], possibly because the anti-gD antibody was not purified from the ascites fluid, whereas the anti-gB antibody was purified.)
After exposure to HSV-1 or HSV-2, all of the L control cells became infected. They all expressed gB, as shown by the shift to a higher fluorescence intensity of virtually all of the cells. The lower mean fluorescence of HSV-2-infected L control cells than that of HSV-1-infected cells indicated that the antibody bound less avidly to gB-2 than to gB-1 or that less gB-2 than gB-i is expressed on infected cell surfaces.
Whereas the cells that expressed the highest constitutive levels ofgD-1 uniformly failed to express gB-1 after expo-sure to HSV-1 and therefore were probably not infected, L-gDgI-2 cells formed two distinct populations after expo-sure to HSV-1. Some of the cells expressed near-normal levels of gB-1 and were obviously infected, whereas the others apparently expressed little or nogB-1.
Similar results were obtained after exposure of the cells to HSV-2, except that for each of thegD-1-expressing clones a larger proportion of cells expressed gB after HSV-2infection than after HSV-1 infection. Also, the less intense staining of HSV-2-infected cells made it difficult to discern distinct subpopulations of cells that were either positive or negative for gB-2 expression.
These results indicate that the intermediatephenotype of L-gDgI-2 cells was due to the fact that a fraction of the cells was susceptible to HSV-1 infection, whereas the rest of the cells were resistant. Moreover, the results suggest that a certain threshold level of gD-1 expression is required for resistance to a given dose of a virus.
DISCUSSION
Hamster, mouse, or human cells expressing sufficient levels ofgD-1 can be resistant to infection by HSV (this study; 5). We show here that expression of gD-1 alone among HSV gene products is sufficient to render cells resistant to HSV infection. Moreover, in a given trans-formed cell line, the degree of resistance to HSV-1 infection is proportional to the amount ofgD-1 expressed. The resis-tance observed is not a generalized interference with viral infection but is specific for HSV among those viruses tested and is more pronounced for HSV-1 than for HSV-2. It remains to be determined whether cells expressing the HSV-2 form of the glycoprotein, gD-2, can also exhibit resistance to HSVinfection.
Although other cell lines expressing gD-1 have been isolated, resistance of these cells to HSVinfection has not been noted previously, in part because thestudies done had other aims and resistance was not looked for. In the casesof an L cell line (3) and aChinese hamster ovary cell line (2), both expressing gD-1, attempts to infect the cells were not reported; the latter cell line could not provide information about the role ofgD-1 in interference in any event, because Chinese hamster ovary cells are naturally resistant to HSV infection. With an L-cell line expressing gD-2, infection of the cells with HSV-1 was successfully performed to deter-mine whether the endogenous gD-2 was processed differ-ently in infected and uninfected cells (20). These results are not necessarily contradictory to those reported here and elsewhere (5), because gD-2 may differ from gD-1 in inter-fering activity, gD-2 may exhibit onlytype-specific interfer-ing activity, levels of gD-2 expressed by the particular
transformedcells used maynothave been
sufficiently
high
to induce complete resistance (partial resistance of the cellswas notruled outby theexperiments done),orthe variable expression of gD-2 on cell surfaces documented
by
the researchers may haveresulted inheterogeneity
of the trans-formed cellpopulationwith respect toresistance.Themechanism by which gD-1 renders cells resistantto HSVinfectionisnotknown,butitseems
likely,
assuggested
alsoby Campadelli-Fiumeetal. (5), thatgD-1
interactswith somecell surface componentrequired
forviralpenetration,
thereby preventing its interaction with
gD-1
in virions.Severalobservationsare consistentwith this
hypothesis.
(i)
gD-1 is essential for virion
infectivity;
it isrequired
forpenetration, not adsorption (26).
(ii)
Penetration occursby
fusion of the virion envelope with the
plasma
membrane (12), and anti-gD monoclonal antibodies can neutralizein-fectivity by blocking this fusion without
inhibiting
adsorp-tion (12, 16). (iii) Virions bind
normally
to resistantgD-1-expressingcellsbutareunabletopenetrate
(this
study; 5).
Expression ofgD-1
by
cells could somehow tie up all oftheunidentified cell surface component with which
gD-1
in virions is proposed to interact.Alternatively,
expression
ofgD-1 could result in
sequestering
inside the cell of the putative cell surface component,thereby
rendering
the cellsurfacedevoidofareceptorneededforHSV
penetration.
A precedentfor this latterhypothesis
exists in part. There isareduction inamount of CD4presentonthe surfacesofcells
producing HIV or
expressing
the HIVglycoprotein,
as mentioned above.It seems likely that an
orderly
cascade of interactions between virion and cell surface components isrequired
for induction offusion between the virion and the cell(P.
G. Spear, M. Wittels, A. 0.Fuller,
D.WuDunn,
and R.Johnson, in R.
Compans,
A.Helenius,
and M. B. A. Old-stone, ed., CellBiology
of
VirusEntry,
Replication,
andPathogenesis, in press). The first interaction is
binding
to virions tocell surfaceheparan
sulfate(53).
Wepropose that this interaction is followedby
otherspecific
interactions,
including one between
gD
and some other cell surfacecomponent.
Some important
biological
consequences ofgD-mediated
interference can be envisioned. The presence of
gD
in membranes of infected cells mayensurethat progenyvirionsdo not superinfect the cells that
produced
them. Because HSV acquires itsenvelope
at the inner nuclear membrane and is transported out of the cell in membrane-boundedvesicles and cisternae of the
Golgi
(21),
a mechanism to prevent superinfection may beespecially
important
to per-mit escapeof progeny virions from infected cells.The consequences ofthis
gD-mediated
interference haveprobably already been seen in studies of HSV-induced
cell-cellfusion. It is known that fusion occurs more
readily
at low
multiplicities
ofinfection, <1,
than athigher
multi-plicities (17, 39).
Mixing
an infected cellpopulation
with a noninfected cellpopulation
gives
muchhigher
ratesof fusionthan
mixing
two infected cellpopulations (23, 24).
To theextent that virus-induced cell-cell fusion resembles virion-cellfusion, thedecreased
ability
of infected cellstofuse with one another mayparallel
the decreasedability
of virions tofuse with
gD-expressing
cells.While this report was under
review,
another kind of interference with HSV infection wasreported
(10).
It was found thattransformed L cellsexpressing
a truncated form of the HSVregulatory
protein
VP16(also
known asax-TIF)
did notsupport HSVreplication
aswell as did control cells. Afterinfection, the transformed cellsproduced
about 1/12 ofon November 10, 2019 by guest
http://jvi.asm.org/
826 JOHNSON AND SPEAR
the normal levels of an immediate-early viral mRNA, the
synthesis of which is influenced by VP16. Whether
resis-tance of cells to HSV infection or replication is caused by
expression of truncatedVP16orwild-type gD,both phenom-ena belong to the general category of viral interference mediated by homologous viral gene products. The two
phenomenamay differ in that the interfering VP16 must be mutated,whereas theinterferinggDmayhavetoretainmost
orall wild-type functions. Thetruncatedformof VP16 used, which itselflacks normal regulatoryactivity, ispostulated to
occupy sites that wild-type VP16 must interact with to
initiate viral gene expression (10). As discussed above,
wild-typegDisproposedto have different roles, depending
onthemembraneinwhichit isfound-interference activity when present in cell membranes and an essential role in
infectivity when found in the virion envelope.
Itistemptingtospeculate,asdidFriedmanetal. (10), that these observations will lead to the development of novel
antiviral agents, particularly inasmuch as expression of
truncated VP16 or gD is not toxic to cultured cells. It remains to be seen, however, what effects these viral
pro-teins ortheiranalogs mighthaveinnormalcellsof develop-ing and adulttissues in intactanimals.
ACKNOWLEDGMENTS
WethankI. Halliburton forthe rabbit antiserumused, E. Kieff
and D. WuDunn for plasmids, N. Soltys for assistance with cell culture, andJ.Hartley for assistance withcytofluorometric analysis. R.M.J. isanMSTP trainee supported by Public Health Service
traininggrant5T32GM07281 fromtheNationalInstitutes ofHealth. This workwassupported bygrantsCA19264 and CA21776fromthe National Institutes of Healthand byaMarietta Klinmanmemorial
grantforcancerresearch fromthe AmericanCancerSociety. LITERATURE CITED
1. Ackermann, M., R. Longnecker, B. Roizman, and L. Pereira. 1986. Identification, properties, and gene location ofa novel glycoprotein specified by herpes simplex virus 1. Virology 150:207-220.
2. Berman, P.W., D. Dowbenko,L. A.Lasky,and C.C.Simonsen.
1983. Detection of antibodies to herpes simplex virus with a
continuous cellline expressing cloned glycoprotein D. Science 222:524-527.
3. Blacklaws,B. A., A. A. Nash,andG. Darby.1987.Specificityof the immuneresponseofmicetoherpessimplex virus
glycopro-teinsB and Dconstitutivelyexpressedon L celllines. J. Gen.
Virol.68:1103-1114.
4. Cai,W.,S. Person,S. C. Werner,J.H. Zhou, andN. A.DeLuca.
1987. Linker-insertion nonsense and restriction site deletion mutations of the gB glycoproteingeneof herpes simplex virus
type 1. J. Virol. 61:714-721.
5. Campadelli-Fiume, G., M. Arsenakis, F. Farabegoli, and B.
Roizman. 1988. Entry ofherpes simplex virus 1 in BJcellsthat
constitutively express viral glycoprotein D is by endocytosis and resultsin degradation ofthe virus.J. Virol.62:159-167.
6. Desai, P.J., P. A. Schaffer,and A. C. Minson. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a
temperature-sensitive mutant of herpes simplex virus type 1:
evidencethat gH is essentialfor virion infectivity.J.Gen.Virol.
69:1147-1156.
7. Everett, R. D. 1983. DNA sequence elements required for
regulated expression of the HSV-1 glycoprotein D gene lie
within 83 bp of the RNA cap sites. Nucleic Acids Res. 11:
6647-6666.
8. Fenwick,M.L., andJ.Clark. 1982. Early and delayed shutoffof
host protein synthesis in cells infected with herpes simplex
virus.J. Gen. Virol. 61:121-125.
9. Frame, M. C.,H.S. Marsden,andD.J.McGeoch. 1986. Novel
herpes simplex virustype 1 glycoproteins identified by antise-rumagainstasyntheticoligopeptidefrom thepredictedproduct
of gene US4. J. Gen. Virol. 67:745-751.
10. Friedman, A. D., S. J. Triezenberg, and S. L. McKnight. 1988. Expression of a truncated viral trans-activator selectively im-pedes lytic infection by its cognate virus. Nature (London) 335:452-454.
11. Fuller, A.O., and P. G. Spear. 1985. Specificities of monoclonal antibodies that inhibit adsorption of herpes simplex virus to cells and lack of inhibition by potent neutralizingantibodies. J. Virol. 55:475-482.
12. Fuller, A. O., and P. G. Spear. 1987. Anti-glycoprotein D antibodies that permit adsorption but blockinfection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc. Natl. Acad. Sci. USA84:5454-5458.
13. Gatti, R. A., P.Concannon, and W. Salser. 1984. Multiple use of Southern blots. Biotechniques May/June:148-155.
14. Gompels, U., and A. C. Minson. 1986. The properties and sequence of glycoprotein H of herpes simplex virus type 1. Virology 153:230-247.
15. Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640-651.
16. Highlander, S. L., S. L. Sutherland, P. J. Gage, D. C. Johnson, M. Levine, and J. C. Glorioso. 1987. Neutralizing monoclonal antibodies specific for herpes simplex virus glycoprotein D inhibit virus penetration. J. Virol. 61:3356-3364.
17. Hoggan, M. D., and B. Roizman. 1959. The isolation and properties of a variant of herpes simplex producing multinucle-ated giant cells in monolayer cultures in the presence of anti-body.Am. J. Hyg. 70:208-219.
18. Hoxie, J. A., J. D. Alpers, J. L. Rackowski, K. Huebner, B. S. Haggarty, A. J. Cedarbaum, and J. C. Reed. 1986. Alterations in T4 (CD4) protein and mRNA synthesis in cells infected with HIV. Science 234:1123-1127.
19. Ikura, K.,J. L. Betz, Z. R. Sadler, and L.I.Pizer. 1983. RNAs transcribed from a 3.6-kilobase SmaI fragment of the short unique region of the herpes simplex virus type 1 genome. J. Virol.48:460-471.
20. Johnson,D.C., and J. R. Smiley. 1985. Intracellular transport of herpessimplex virus gD occurs more rapidly in uninfected cells than in infected cells. J. Virol. 54:682-689.
21. Johnson, D. C., and P. G. Spear. 1982. Monensin inhibits the processing of herpes simplex virus glycoproteins, their trans-port to the cell surface, and the egress of virions from infected cells. J. Virol. 43:1102-1112.
22. Karin, M., and R. I. Richards. 1982. Human metallothionein genes-primary structure of the metallothionein II gene and relatedprocessed gene. Nature (London)299:797-802.
23. Keller, J. M.1976. The expression of the syn- genes of herpes simplexvirus type 1. I. Morphology of infected cells. Virology 69:490-499.
24. Lee, G. T. Y., and P. G. Spear. 1980. Viral and cellular factors that influence cell fusion induced by herpes simplex virus. Virology 107:402-414.
25. Lewis, J. A., and A. B. di Girolamno. 1987. Activation of metallothionein expression is potentiated by DNA sequences present in the herpes simplex virus thymidine kinase gene. FEBS Lett. 217:292-296.
26. Ligas, M. W., and D.C. Johnson. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by
P-galactosidase
sequences binds to but is unable to penetrateintocells. J. Virol. 62:1486-1494.
27. Longnecker, R., S. Chatterjee, R. J. Whitley, and B. Roizman. 1987. Identification of a herpes simplex virus 1 glycoprotein gene within a gene cluster dispensible for growth in cell culture. Proc. Natl. Acad. Sci. USA 84:4303-4307.
28. Longnecker, R., and B. Roizman. 1987. Clustering of genes dispensible for growth in culture in the S component of the HSV-1 genome. Science 236:573-576.
29. Luytjes,W., R. Vlasak, P. Palese, and W. T. M. Spaan. 1988. A receptordestroyingenzyme (esterase) is associated with bovine coronavirus. J.Cell. Biochem. Suppl. 12C(J110):19.
30. Malissen, B., M. P. Price, J. M. Goverman, M. McMilan, J. White,J. Kappler, P. Marrack, A. Pierres, M. Pierres, and L. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
Hood. 1984. Gene transfer of H-2 class II genes: antigen presentation by mouse fibroblast and hamster B cell lines. Cell 36:319-327.
31. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold SpringHarbor Laboratory, ColdSpring Harbor, N.Y.
32. McGeoch, D. J., A. Dolan, S. Donald, and F. J. Rixon. 1985. Sequence determination and genetic content of the short unique region of the genome of herpes simplex virus type 1. J. Mol. Biol. 181:1-13.
33. Neidhardt, H., C. H.Schroder, and H. C. Kaerner. 1987. Herpes simplex virus type1glycoproteinEisnotindispensible for viral infectivity. J. Virol. 61:600-603.
34. Noble,A.G., G.T. Y.Lee,R. Sprague,M.L. Parish, and P. G. Spear. 1983. Anti-gD monoclonalantibodies inhibit cell fusion induced by herpes simplex type 1. Virology 129:218-224. 35. Palese, P., K. Tobita, and M. Ueda. 1974. Characterization of
temperature sensitive influenzamutantsdefective in neuramin-idase. Virology 61:397-410.
36. Para,M. F., M. L. Parish, A. G. Noble, and P. G. Spear. 1985. Potentneutralizing activity associated with anti-glycoprotein D specificity among monoclonal antibodies selected for binding to herpes simplex virions. J. Virol. 55:483-488.
37. Read, G. S., and N. Frenkel. 1983. Herpes simplex virus mutantsdefective in the virion-associated shutoff of host poly-peptide synthesis and exhibiting abnormal synthesis of a (im-mediate early) viral polypeptides. J. Virol. 46:498-512. 38. Richman, D. D., A. Buckmaster, S. Bell, C. Hodgeman, and
A.C. Minson. 1986. Identification of a new glycoprotein of herpessimplex virus type1andgenetic mapping of the genethat codesfor it. J. Virol. 57:647-655.
39. Roizman,B. 1961. Polykaryocytosis induced by viruses. Proc. Natl. Acad.Sci. USA48:228-233.
40. Roizman, B.,B. Norrild, C. Chan,and L.Pereira. 1984. Iden-tification and preliminary mapping with monoclonal antibodies ofaherpessimplex virus type2glycoprotein lackingaknown type1 counterpart. Virology133:242-247.
41. Sarmiento, M., M. Haffey, and P. G.Spear. 1978. Membrane proteins specified by herpes simplex viruses. III. Role of glycoprotein VP7(B2) in viiion infectivity. J. Virol.
29:1149-1158.
42. Schek, N., and S. L. Bachenheimer. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J. Virol. 55: 601-610.
43. Spandidos, D. A., and N. M. Wilkie. 1984. Expression of exogenousDNA inmammalian cells, p. 1-48. InB. D. Hames and S. J. Higgins (ed.), Transcription and translation. IRL Press, Cambridge.
44. Spear, P. G. 1985. Glycoproteins specified by herpes simplex viruses, p. 315-356. In B. Roizman (ed.), The herpesviruses, vol. 3. Plenum Publishing Corp., New York.
45. Spear,P.G.,and B.Roizman. 1972.Proteinsspecified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J. Virol. 9:143-159.
46. Stevenson, M., C. Meier, A. M. Mann, N. Chapman, and A. Wasiak. 1988. Envelopeglycoprotein of HIV induces interfer-ence and cytolysis resistance in CD4+ cells: mechanism for persistence in AIDS. Cell53:483-496.
47. Stevenson, M.,X.Zhang,and D.J.Volsky.1987. Downregula-tion of cell surface moleculesduring noncytopathic infection of T cells with human immunodeficiency virus. J. Virol. 61: 3741-3748.
48. Vaheri,A. 1964.Heparinand relatedpolyanionicsubstances as viral inhibitors. Acta Pathol. Microbiol. Scand. Suppl. 171:7-97. 49. Vogt, P. K. 1965. Avian tumor viruses. Adv. Virus Res.
11:293-385.
50. Watson,R.J., J.H. Weis, J. S.Salstrom, and L. W. Enquist. 1982. Herpessimplex virus type-1glycoproteinDgene: nucle-otide sequence and expression in Escherichia coli. Science 218:381-383.
51. Weber, P. C., M. Levine, and J. C. Glorioso. 1987. Rapid identification of nonessential genes of herpes simplexvirustype 1 by Tn5mutagenesis.Science 263:576-579.
52. White, J.,M.Kielian,andA. Helenius. 1983.Membrane fusion proteins of enveloped animal viruses. Quart. Rev. Biophys. 16:151-195.
53. WuDunn, D.,and P.G.Spear.1988. Initialinteraction ofherpes simplexvirus with cells is bindingto heparin sulfate. J. Virol. 63:52-58.