0022-538X/88/030932-12$02.00/0
CopyrightC) 1988,American Society for Microbiology
A
Single
Point Mutation in the Envelope Gene
Is
Responsible
for
Replication and
XC
Fusion Deficiency of
the
Endogenous
Ecotropic
C3H/He Murine Leukemia Virus and for Its Repair in Culture
GUNAMANI SITHANANDAM ANDULF R. RAPP*
Laboratory of Viral Carcinogenesis, National Cancer Institute, Frederick, Maryland 21701 Received25June 1987/Accepted 2 November 1987
The molecular basis has been determined fordifferences in infectivity and XC phenotype of endogenous ecotropic murine leukemia virus of the low-leukemia mouse strainC3H/He,its relative in thehigh-leukemia mouse strain AKR, and highly infectious, XC-positive C3H virus variants selected in vitro. Endogenous ecotropic type C virus induced by iododeoxyuridine from the nontransformed C3H/1OT1/2 cell line is XC negative and replication deficient. In contrast, viruses produced late after iododeoxyuridine induction in chemically transformedC3H/10T1/2cells(MCA5)areXC positive and infectious. XC-negative viruses can be converted to XC-positive viruses by being grown in certain transformed cell lines. We have cloned the endogenous ecotropic provirus ofC3H/Hefrom MCA5 cells, which is XC negative and replication deficient,as well as two XC-positive C3H proviruses derived by in vitro conversion. Fragment exchange between the XC-negative molecular clone pllO and the XC-positive AKR virus clone p623 revealed that thedefect in pllO lies3'of the
Sail
site located in the pol region. Nucleotide sequencing established that the C3H pllO provirus wasintegrated within theR region ofanendogenous VL30 long terminal repeat (LTR)in reverse orientation and that thevirusdifferedfrom the infectiousAKRp623 provirus byapoint mutation, substituting Lys for Arg atthepotential precursor cleavage site forgp7Oandpl5E.Invitro-convertedXC-positive C3H proviralclones 3211 and4211are identicaltoXC-negative C3Hp110,except thattheyhaveArgatthis site and the normal cleavage site is thus regenerated inthese clones. The XC-negative C3H pllO was blocked in processing of Pr85env, whereas clones 3211 and 4211 had normal cleavage of the env precursor into gp7O. Both the XC-negative C3H provirus and the in vitro-converted XC-positive C3H proviruses had a single copy ofa 99-base-pairenhancer elementin theLTR, whereastwocopies of this sequencearepresentintheAKRproviralLTR. Substitution of Arg for Lys at the envelope precursor processing site of C3H pllO by site-directed mutagenesis is sufficient by itself to convert the virus to the XC-positive replication-competent phenotype. Thus,wehave established thatasingle point mutationat theprocessing site of the envelope precursor protein Pr85 is responsible for the difference in the infectivity and XC phenotype of endogenous ecotropic murine leukemia virusfromC3H/HeandAKRmice and that the basis for in vitro conversion isamutationatthissite. Inbredmouse strains differ in their incidence of leukemia
and the number and inducibility of endogenous ecotropic
murine leukemia virus (MuLV)proviruses(1, 15, 26, 30, 47, 53). Leukemia-negative strains lack
endcogenous
ecotropicMuLV, whereas low- and high-leukemic strains contain at least one copy of this provirus (30, 47). Initial studies on virus inducibility with halogenated pyrimidines (32)
indi-cated that viruses from low-leukemic strains were more
difficulttoinducethanvirusesfromhigh-leukemic strains (1,
32, 53, 54). These findings were interpreted to indicate
differences in cellular control ofexpression of endogenous ecotropic virus(21, 35,47,48)ordifferences intheir biolog-ical activities (1, 41, 53). Specifically, ecotropic virus recov-eredfrom induction of cellsfromhigh-leukemic strains such as AKR formed large XC plaques, whereas virus from the
low-leukemic BALB/c mouse cells formed small (1) or no (41, 45) plaques.
Inthe courseof studiesexamining the role of endogenous
C-type viruses in chemical carcinogenesis, we have ob-served that untransformed or transformed cells from low-leukemic C3H/He (C3H/1OT1/2 Cl 8) and BALB/c (BALB 3T3) mice produced XC-negative,replication-deficientvirus
early (phaseI)afteriododeoxyuridine(IdUrd) treatment (41, 44, 45). XC-positive virus did emerge late after IdUrd
*
Corresponding
author.induction ofchemically transformed C3H/1OT1/2 Cl 8 cells (phase III),presumably owingtogenetic changesthatwere selectedduring serialreinfection(44, 45).The lackof
infec-tivity and XC fusion activity of endogenous C3H/He and BALB/c ecotropic virus in phase I after induction was
interpretedtoreflectthedefectivenatureoftheendogenous proviruses, and their conversion to large-XC-plaque-in-ducing, infectious viruses similar to the AKR
ecotropic
provirus (41) was thought to result from mutation duringserial reinfection (41, 45). Moreover, the genotype ofthe
endogenous ecotropic provirus appeared to be a
major
determinant of leukemiaincidence,
since inoculation of NFS/N micewithXC-positiveAKRand C3H virus,butnot withXC-negative endogenousecotropicC3Hvirus, resultedin the development of leukemia (41). The existence of
XC-negative ecotropic BALB/c virus in the company of excess XC-positiveviruswasalsodescribedbyothers(18);
however, since this virus stock had been maintained in
chronically infected NIH 3T3 cells for some time before
analysis, no conclusion regarding the properties of the
endogenous BALB/cproviruscould be drawn.
Thepossible existence of cellularregulatory geneswhich control virusexpressioninBALB/cand otherlow-leukemic mice has since been furtherexplored (21, 34, 35).To date,
the nature of potential cellular control genes
determining
ecotropic
virus inducibility in BALB/c mice remains un-932on November 10, 2019 by guest
http://jvi.asm.org/
MuLV ENVELOPE GLYCOPROTEIN PROCESSING MUTANT 933
clear,
whereas structural defectsaffecting
viral infectivityhavebeen observed (22). Nevertheless, ithas been
demon-strated that in other systems,nonviralfactors suchasDNA
methylation
(7, 13, 17)andchromatinstructureofintegration sites affect theexpression
ofexogenousC-type virus (6, 13, 17, 25). Moreover, transregulation of C-type viral RNAexpression by
activation ofprotein
kinase C has beendemonstrated for the VL30
long
terminalrepeat(LTR) (46).Toestablishthemolecular basisforthe lackofinfectivity of
endogenous ecotropic
virusfrom C3H/Heand theinvitroconversion to an
infectious, XC-positive form,
we deter-mined thestructures of both viruses.MATERIALS AND METHODS
Cells. All cell lines were maintained in
Eagle
minimumessential medium with10% heat-inactivated fetal calfserum.
C3H/MCA5,
amethylcholanthrene-transformed
C3H/1OT1/2Cl 8 cell
line,
and the NIH R+Cl3 cell line(NIH
cellsinfected with
XC-positive
converted C3HR+C13virus)
wereused.The
origin
andcharacteristics of these cell lineshavebeendescribed
previously
(41, 44).Molecular cloning.
High-molecular-weight
DNA fromMCA5
and NIHR+Cl3celllineswasextracted anddigested
with the restriction enzyme EcoRI tocompletion
asrecom-mended
by
themanufacturer(New England BioLabs,
Inc.).The
digested
DNAs wereseparated
ina 10to40% sucrosegradient,
and MCA5DNAfractionscontaining
21-kilobase-pairs (kb) fragments
werepooled,
dialyzed,
andprecipitated.
Similarly, samples containing
9- to 11-kbfragments
fromNIH R+Cl3
gradients
werepooled
andprecipitated
(33).The 21-kb EcoRI
fragment
wasligated
togradient-purified
EcoRI-digested
Charon 35 arms(29).
Since BamHIcleaves the stufferfragment internally,
Charon 35 DNAwasdoubledigested
withEcoRIandBamHI,
and the smallerfragments
of stuffer wereeasily separated
from vector arms in thegradient.
NIH R+C13 EcoRIfragments
wereligated
to AWesB EcoRIarms
(28)
andpackaged
invitro(9),
and thetwolibraries were screened
by using
the Benton and Davismethod
(3)
witha32P-labeled p400 eco-specific
envfragment
(5).
Positive clones wereplaque purified
and subcloned inpBR322.
Gel
electrophoresis
and hybridization analysis. DNAsam-ples digested
with restriction endonucleases wereelectro-phoresed
in 0.8to0.9%agarosegels
withTris-acetate buffer(pH 7.8),
and the DNA was transferred to nitrocellulosepaper
by
theprocedure
of Southern(51).
The filters wereprehybridized
at60°C
foratleast2to3 handthenhybridized
overnight
at60°C
in thehybridization
buffercontaining
0.5 x 106 to 1 x 106cpm
of32P-labeled
probe per ml. Afterhybridization
the filters were washed twice with lx SSC(0.15
M NaClplus
0.015 M sodiumcitrate)-0.1%
sodiumdodecyl
sulfate(SDS)
at65°C
and thensubjected
to three30-minwashes at650Cwith0.lx SSC-0.1% SDS. Air-dried
filters were
exposed
to Kodak X-ray film at -70°C withintensifying
screens.Invitroconstruction of chimericviralgenomes. AKRp623
contains the entire AKR MuLVgenome cloned inpBR322at theHindlllandEcoRIsites.HindIllandEcoRIsites inp623
are located in 5' and 3'
flanking
cellular sequences,respec-tively (31).
C3HpllOcontains the C3H endogenousecotro-pic proviral
genome with 12-kb cellularflanking
sequences cloned into the EcoRI site ofpBR322.
Both p623 and pllOhave a
single
Sall restriction site within the ecotropicprovirus
in the polregion
and the otherSall site within thepBR
sequences.Fragments
3' of the Sall site in the polregion were switched between the two plasmids. AKR p623 and C3HpllO DNAs were digested with Sail, thefragments
were separated in a preparative gel, and the desired frag-mentswere isolated andligated with T4 DNA ligase (at14WC for 16h) and used to transform EscherichiacoliHB101 cells. Colonies were then screenedby the method of Grunstein and
Hogness (14), and the DNA from positive clones was mo-lecularly analyzed by using appropriate restriction enzymes to determine the orientation of theexchangedfragments.
DNAsequencing. DNA wassequenced by the dideoxynu-cleotide chain termination method (2, 50) on fragments inserted in pUC18, pUC19, and single-stranded M13mpl8 andM13mpl9 vectors. Commercially available primers were used for priming, and primer extensions were carried out with the large fragment of E. coli. DNA polymerase I
(Klenow)and[35S]ATP (Amersham Corp.)wereused as the sourceof labeled triphosphate.
Site-directed mutagenesis. Theprocedureused for oligonu-cleotide site-directed mutagenesis was essentially that de-scribed by Zoller and Smith (57). A 520-base-pair (bp)
KpnI-to-XbaI fragmentwas cloned into the M13 vectorand was used as a template for site-directed mutagenesis. Two
oligonucleotide primers were used, a 26mer synthetic
ol-igonucleotide, 5'-GCCAAATATAAAAGAGAACCCGTC TC-3', and the universal sequencing primer (New England BioLabs). Preliminary tests were conducted by using the
mutagenic oligonuclotide as a sequencing primer in a stan-dard dideoxynucleotide sequencing proceduretodetermine the optimum conditions of primer-to-template ratio. Both
primers were simultaneously annealed to single-stranded
template DNA at 60°C for 5 min, extended with DNA
polymerase I (large fragment), and ligated at 15°C for 7 h. Diluted DNA wasthen used totransformE.coliJM103 cells. Theplaques were screened for mutants byfilter
hybridiza-tion with 5'-end-labeled mutagenic oligonucleotide as a
probe,andfilterswereprehybridizedat65°C for1 h and then
hybridized at 37°C overnightunderconditions of low strin-gency inwhich bothmutantandwild-typeDNAhybridized with oligonucleotide. The filters were then washed three times with 6x SSC at various temperatures. At 37°C both mutantandwild-typeDNAs weredetected,whereas at62°C only afewhybridizing plaques remained, which were puta-tive mutants. Strongly hybridizing plaques were purified, single-strandedDNA wasprepared,andportionswere spot-ted onto nitrocellulose and hybridized with the labeled
mutagenic oligonucleotide. Filters were again washed at
differenttemperaturesranging from37 to65°C.The
hybrid-izing
signals of wild-typeclones werelost aftera47°Cwash. Incontrast, hybridizing signalsfromputative mutantphageclones were stable even atveryhightemperatures.
Transfection of NIH 3T3 cells withproviral clones.
Trans-fection ofNIH 3T3cellswithproviralclones wasperformed by a modification ofthe method described by Graham and van der Eb (12). Linearized plasmid DNA was mixed with
pSV2neoDNA(ratio, 10:1) and carrier salmon sperm DNA to a DNA concentration of 20
pug/ml
and suspended intransfectionbuffer to afinal concentration of0.13 MCaCl2.
At 2dayslaterG418 was added to the medium at 400 ,ug/ml. Clones resistantto G418were selected and expanded.
Viralassays. Production of infectious virus particles was monitoredby XC andreversetranscriptaseassays. The XC test was aslight modification of thatdeveloped byRowe et al.(49). Target SC-1cellswereseededat105 cells per 60-mm dish. At 24 h later the cells were treated with Polybrene
(Sigma;
20,ug/ml)
for 1 hand then infected with 0.2 ml of virussuspension.At2 hafterinfection the virus suspensionVOL.62, 1988
on November 10, 2019 by guest
http://jvi.asm.org/
wasremoved and 5 ml of medium was added. At 4 days after
infectionthecells were lethallyirradiatedwith aUV
germi-cidallamp andwereoverlaid with 106ratXC cells. Depend-ingupon the confluency ofthe XC monolayer, the test was
terminated and the cells were fixed with methanol and
stainedwith Giemsa. Infectious-center assays were used as asensitiveassay to detectsingle-virus-producing cells.
neo-positive cells from transfections (test cells) were seeded at various concentrations and allowed to form small islands
duringgrowth for 2 days and then were lethally irradiated withUVandoverlaidwith XC cells. Plaques were scored 2 to3days later. Cocultivation assays with virus-free mouse
cells were used to determine the original phenotype of neomycin-positive clones in terms oftheir XC fusion
activ-ity. This test also determines whether there was a
conver-sion from the XC-negative to the XC-positive virus on
growth in different cells. neo-positive clones were mixed with uninfected SC-1 cells at various ratios (5:95, 10:90,
40:60, and 80:20), grown to confluency, and then lethally irradiatedand overlaid with XCindicatorcells. For reverse
transcriptaseassays, culturefluidsatearlyconfluency were
collected, cleared of cells and cell debris, and assayed for
reverse transcriptase activity essentially by the method
previously described (55).
Immunoprecipitation. Transfected cells were starved in leucine-free medium for2hbefore40,Ci of
[3H]leucine
wasadded. The cells werelabeled for 30 min andchased for0
and30min and 1, 2, 3, and4h. Labeled cellswere lysed in lysis buffer (0.05 M Tris [pH 7.2], 0.15 M NaCl, 0.5% Nonidet P-40,0.5% desoxycholate,
0.5%
SDS), and 1ml of the clarified lysates was incubated overnight with 10RI
of Rauchergp7O
antibody and100RI
of 10%protein A-agarose inlysis buffer at 4°C with gentle shaking.Immunoprecipi-tates were washed five times with lysis buffer, and pellets
weredissolvedin 50,ulofsample buffer,heatedfor2min at
100°C, and run on an SDS-10% polyacrylamide gel. After electrophoresis for 16 h at 35 V, the gel was soaked in
destainsolution for30 minand then foranadditional30min
in Amplify (Amersham Corp.). Thegel wasthen driedand
exposedto SB-5 film(Kodak). RESULTS
Cloning of the endogenous C3H ecotropic provirus from MCA5 cells. High-molecular-weight DNA from the MCA5
cell line was digested with various restriction enzymes, electrophoresed through agarose gels, and analyzed by
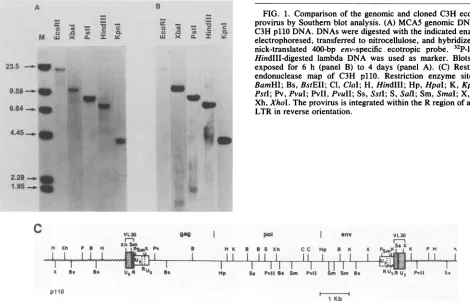
Southern blothybridization (51). UsinganecotropicMuLV env-specific probe, p400 (5), we detected a single 21-kb EcoRI fragment (Fig. 1A, lane 1). Since this enzyme does
notnormally cleave within thegenomes ofecotropic
provi-ruses(39), the 21-kb Eco-RI fragment contains the prototyp-ical 8.9-kb provirus and approximately 12 kb of flanking mousecellular DNA. Sucrosegradient-purified21-kbEcoRI
fragmentswerecloned intogradient-purified lambdaCharon
35EcoRI arms,and theresulting phage
library
wasscreened withthe envprobe p400(5).Afterscreeningapproximately5 x105
recombinant phages, we identified two positive plaques (clones C1109 and ClllO). Both clones had acom-plete proviral genome and identical flanking cellular se-quences. The results of Southern blot analysis of cloned
DNA confirmedthat the correct provirus was cloned(Fig.
1B). The 21-kb EcoRI insert ofClllO was subcloned into pBR322 and designated p110. To characterize the proviral
sequenceandtoidentify the unique cellularsequenceinthe
clonedDNA, we performed restriction enzymeanalysis of
the 21-kb fragment. The restriction mapping of the 8.9-kb provirus and approximately 2.1 kb of 5' and 2.5 kb of 3' flankingcellular DNA isshown inFig. 1C.
A
0 m
o a _ c
M us x n. T e
23.5 -_
9.59--o4
6.64-I4
4.45- i
2.29n 1.95 _
B
ccc
8 i (n . 0
w x
._.
Ki
FIG. 1. Comparison of thegenomic andcloned C3Hecotropic provirusby Southern blotanalysis. (A) MCA5genomic DNA; (B) C3H pllODNA. DNAs weredigestedwith the indicated enzymes, electrophoresed, transferredto nitrocellulose, andhybridizedtoa nick-translated 400-bp env-specific ecotropic probe. 32P-labeled HindIII-digested lambda DNA was used as marker. Blots were exposed for 6 h (panel B) to 4 days (panel A). (C) Restriction endonuclease map of C3H p110. Restriction enzyme sites: B, BamHI; Bs, BstEII;Cl, ClaI;H,Hindlll; Hp, HpaI; K, KpnI; P, PstI;Pv,PvuI; PvII, PvuII;Ss, SstI; S,Sall; Sm, SmaI; X,XbaI; Xh,XhoI.Theprovirus isintegratedwithin the RregionofaVL30 LTR inreverseorientation.
C VL30
r n 11 X.11 PB1 tt XhKSm
X s 5s Us R Uus as
gag I pol
a H K B XS Xh
Hp S. PullSs
pIO
env
CC Hp 8 K
11 I
Sm PW11 S Sm as
1Kb
VL30 r--l
I PSMKSs XK P H K
I ---I
T
---T-Pvli Ss
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.59.529.421.723.2]MuLV ENVELOPE GLYCOPROTEIN PROCESSING MUTANT 935 Biological activity of the C3H ecotropic proviral clonep110.
Totestthe biological activity of the proviral clonep110,NIH
3T3 cellswerecotransfected with pllO andpSV2neo,which
carries thegenefor neomycin resistance, ata10:1ratio (52).
Transfections with AKR p623 and pSV2neowerealso
per-formed as a positive control. Transfected cells were first selected for their ability to grow in the presence of the
neomycin analog G418, and individual colonieswereisolated
and assayed for XC fusion activity by theinfectious-center
assay at their first confluency. These conditions minimize the chance for secondary rounds of virus spread and
con-version in NIH 3T3 cells. Supernatant culture fluid was
tested forreleasedreversetranscriptase-containing particles
(55) and was also tested in XC assays (49). A total often neo-positive cloneswereassayed for each transfection. With
thecontrol p623 DNA, 10 of 10 G418-resistant clones were
positive forreverse transcriptase,indicating thepresenceof released viral particles. In thisway,producer cell lineswere
readily isolated. Incontrast,cellstransfectedwith C3H pllO DNA did not form fusions with XC cells, and release of
reverse transcriptase-positive particles was barely
detect-able(Fig. 2).
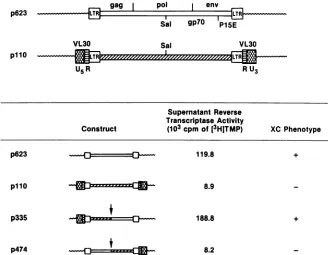
In vitro restriction fragment exchange and localization of thedefect in C311 p110.The fact that the XC-negative C3H
clone pllO and the XC-positive AKR infectious clone p623 share severalidentical restriction endonuclease sites allowed
ustomakeconstructs by switching fragments 3'tothe Sall site in thepolregion (Fig. 2). Chimeric plasmids were then
screened by colony hybridization (14), and plasmid DNA
waslinearized and transfected along with pSV2neoataratio
of10:1 onto NIH 3T3 cells. neo-positive single-cell clones
were tested for their reverse transcriptase activity and for
p623
p110
UDR
t
the presence of infectious virions as described previously
(49, 55). Switching the fragment 3' of the Sall site in p623 with that inpllO restored the biological activity of the pllO virus and generated infectious virus withreverse
transcrip-taseactivity (p335 in Fig. 2). Incontrast, 10 of 10 NIH 3T3 cells transfected with chimericplasmid p474 which has the 5' upstream Sall fragment from p623 and the 3' downstream SalI fragment from pllO didnotrescue the XC phenotype,
andparticle productionwasbarely detectable by thereverse
transcriptase assay. These results demonstrated that the
C3Hproviral clonepllOhadnodefectupstreamof theSalI site in thepolregion and that the defectmusttherefore lie somewhere downstream of this restriction site.
In vitro conversion of XC-negative to XC-positive pheno-type. We observed previously that poorly infectious, XC-negative ecotropic virus induced from C3H/1OT1/2 and BALB 3T3 cells convert to replication-competent, XC-positive viruswhengrown in certain transformed cell lines suchasSC-1, MCA5, and NIH 3T3 (41, 44). Totestwhether pllO could be converted, virus-negative target cells (SC-1)
were mixed in various ratios with neo-positive pllO-trans-fected clones and grown in the presence of Polybrene.
Different ratios ofvirus-positivetovirus-negative cellswere
used to allow forone or several cycles of infection in the
negative cells. With ratios of 5:95 and 10:90 of virus-positive pllO-transfected NIH 3T3 cells to virus-negative SC-1 cells, conversion from the XC-negative to the XC-positive phenotype was observed on continued coculture. No conversion was observed at a mixing ratio of 60:40or
80:20, in accordance with our previous observation with
biologically cloned viruses (44). These mixing ratios donot allow multiple rounds of infection in theconvertercells.
RU3
Supernatant Reverse Transcriptase Activity
Construct (103cpmof[3H]TMP) XC Phenotype
p623 119.8
p110
p335
p474
+
8.9
188.8 +
[image:4.612.145.473.405.660.2]8.2
FIG. 2. Infectivity of endogenous ecotropic proviral clones AKR p623, C3Hp110,andconstructsp335 and p474. Schematic
representa-tions ofthe proviral clones p623, p110, and the constructs p335 and p474 are shown. First-confluency supernatant culture fluid from
transfected celllineswasharvested 24 h after mediumchange. Mediumwasfreed of cells and cell debris by centrifugation and assayedas
previously described (45) forreversetranscriptase activity. Results areexpressed as103counts perminute of3[H]TMP incorporated ina
30-min incubation periodpermilliliter ofsupernatant; 105cpmrepresents1.2 pmol of TMP incorporated. Reverse transcriptase activities in thefigure arefor clones whichweretested bypulse-chase experiments. The XC phenotype wastested by the infectious-centerassay(45),
aswellasby the ability of thesupernatant toinduce syncytium formation in Rous sarcomavirus-transformedratXC cells.
gag
I
pol envSa gp7O
PIE
Sal 9P0 Pl5E
VL30 Sal VL30
TR LTR""2 z" "'z' "
VOL.62, 1988
f
on November 10, 2019 by guest
http://jvi.asm.org/
Ca)
-Z Yacc
tm
0E
uQB
U)
Q.
23.5
9.59
6.64
4.45
EcoRi Pstl
''-~~~~~~~~-''
v-- v- II v _ I
v-v- - C') v r- Ir-C' - v
CN CN r CD N r N N rT C' IK I C') t 1. _- M e t P.
8.3 -6.6
-am ,
sup 5.6
-awfl
40
cCT TGA
l K i
PvP
K P R +Ct3 3211 , IP Ss Sm
A 4 C13 4211
TACCITGA
RI
RI R+C13 7-1111
Ss
ga I
S HK a . 33
IL ~ I I I I I I
Ba I
II I
Hp So Pulls Sm
I env TCA CCTT
CC Hp BK X P K
Nil Sm Sm Sm TCAJTACC
PNll
c4I
;IIP x
R+C131013
Sm Sm RI
.0 ----.
_a <
_~~~S
[image:5.612.79.547.65.483.2]Kb
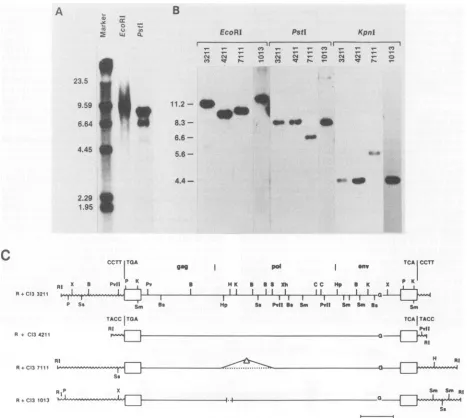
FIG. 3. Comparative Southern blot analysis of proviruses in NIHR+Cl3cellular DNA and C3Hecotropic provirusesisolated form NIH
R+Cl3 cells. (A) NIH R+C13 genomic DNA. (B) NIH R+C13 ecotropic proviral clones C13211, C14211, C17111, andC11013. DNAs were
digested with the indicatedenzymes,electrophoresed, transferredtonitrocellulose,andhybridizedtoanick-translated400-bp env-specific ecotropic probe. Blotswereexposed for 6 h (panel B)to4days (panel A). (C) Restriction endonucleasemapof C3H R+C13proviralclones
C13211, C14211, C17111, and C11013...Internal deletion. Cleavage sitesare denoted asfollows: B, BamHI; Bs,BstEII; Cl, ClaI; H,
Hindlll;Hp, HpaI;K, KpnI; P, PstI; Pv, PvuI; PvII, PvuII; Ss, SstI;S,SalI;Sm, SmaI;X, XbaI; Xh, XhoI; RI,EcoRI. The4-bpcellular
directrepeatattheintegrationsite is shown for C13211and C14211.
Molecular cloningof in vitro-converted replication-compe-tent C3H virus from NIH R+C13 cells. To compare the genetic structure of in vitro-converted XC-positive C3H
viruswith thatof the parentalXC-negative form,wechosea
biologicallycloned isolate for molecular analysis. R+C13 is
anXC-positive, replication-competent variantgeneratedby
growth ofIdUrd-induced XC-negative C3H/1OT1/2 virus in C3H/MCA5 cells. Thevirus wasclonedbyisolation from a
large XC plaque that developed in cultures infected at limiting dilution, and agar stabs from these plaques were
transferredtouninfectedNIH 3T3 cellcultures(40).
South-ern blot analysis with the eco-specific env probe (5) of
EcoRI-restricted NIH R+C13 genomic DNA detected a
stronghybridizing region of 9to10kb(Fig. 3A). Hybridiza-tion of PstI digested DNA with p400 probe detected two
bands of 8.2 and 6.8kb.ComparativeSouthern blotanalysis ofequal amounts ofPstI-digested MCA5 DNA and NIH R+C13 DNA indicated thepresence ofthree 8.2-kb
ecotro-picproviruses integrated in C3HR+C13-infected NIHcells (datanotshown).The 6.8-kbPstI fragmentwaspresumably
notderived fromendogenousxenotropicviruswhich carries
aninternal PstI site,sinceithybridizedwithaneco-specific env probe. Gradient-purified 8- to 11-kb EcoRI fragments
werecloned into XWES B(28)EcoRIarms,and theresulting phageswerescreenedwithp400. Fourpositive plaqueswere
identified after5 x 10i phagewerescreened(clones C13211, C14211, C17111, and C11013). To analyze the structure of ecotropic proviruses cloned form this cellline, wedigested cloned DNAs with the restriction enzymes PstI and KpnI, which should give internal viral fragments of defined size
A
Kpnl CD)
0-2.29 1:955
4.4
-#..m.
C
H RI I
11.2-
40
40,40
on November 10, 2019 by guest
http://jvi.asm.org/
MuLV ENVELOPE GLYCOPROTEIN PROCESSING MUTANT
when hybridized with eco-specific probe (Fig. 3B). C13211, C14211, and C11013 showed hybridizing fragments of 8.2 kb with PstI and 4.4 kb with KpnI. Incontrast, the hybridizing fragments from C17111 were 6.8 kb for PstI and 5.1 kb for
KpnI,indicatinganinternaldeletion ofa ca. 1.9-kb fragment
which includes one of the internal KpnI sites. C13211 and
C14211 had identical restriction maps within the provirus,
buttheir flankingsequences weredifferent. Nomajor struc-turaldifference could be detected for C11013 onthebasis of
Southern blots. However, restriction analysis indicated lack of the HindIlI site at nucleotide 2668 (nucleotides were
numbered by using AKR, p623 sequenceof Herr [16]), and
this clone had eitherapoint mutationor aminor deletion in
that region (Fig. 3C). All four clones were subcloned in
pBR322.
Infectious properties ofprovirus clones from NIH R+C13 cells. NIH 3T3 cells were cotransfected with linearized
plasmid DNA of C13211, C14211, C17111, or C11013 and
pSV2neo, and G418 (52)-resistant clonesweretested for XC
fusion activity by the infectious center assay. Supernatant
culture fluid from these cells was tested for the release of
reverse transcriptase-containing particles andwasalso used
for the XCassay. Cellstransfected with C13211 and C14211 DNAwere XC positive and released reverse
transcriptase-containing particles, whereas cells transfected with C17111 and C11013werenegative for XC fusion activity and released no reverse transcriptase-containing particles into the
super-natant medium (Table 1). For comparison, results are also
shown for cells transfected withendogenous C3H viral DNA
p110,testedattheir firstconfluency(phase I) and again after five subculture generations (phase III), aswellaswith AKR
p623 viral DNA. C3HpllOclonesreleasedbarely detectable levels of reverse transcriptase-containing particles early
after transfection, butwere highly positive afterpassage in
culture, atwhich time theywerestill XC negative.
Sequence analysis of XC-negative C3H pllO and in vitro-convertedXC-positive C3H R+C13 proviral clones: molecular basis forthe XCphenotype. Toidentify the defect in the C3H proviral clone p110,the clonewas sequenced from the Sall
siteatnucleotide 3720tothe3' end of theprovirusandwas
compared with the XC-positive infectious AKR provirus p623sequencepublished by Herr (16). pllO fragmentswere
subcloned in pUC18, pUC19, and M13mpl8 and M13mpl9 vectors and were sequenced by dideoxy chain termination
(2, 50). Of the 4,660 bp sequenced, there were 21 base
substitutions: 10wereinthe3'pol region, 8wereintheenv
region, and 3 were in the LTR region. Of these, only 6
changes ledtoamino acidsubstitutions(2inthe3'pol region
and4in theenvregion), correspondingto99.5% amino acid homology between AKR p623 and C3H p110. The most interesting amino acid substitution occurred atthe proteo-lytic cleavage site of Pr85, wherea 1-bp change of GtoAat position 7191 substituted lysine for arginine (Fig. 4). A similar 1-bp change has been observed ina BALB/c
endog-enous ecotropic provirus clone (22), presumably because germlineinfection occurredpriorto divergence of BALB/c and C3H/He mice from a common ancestor. Nucleotide
sequenceanalysis of the converted C3H R+Cl3 virus clones
showed that all four coded for arginine at this potential cleavage site. Comparison ofa ca.1,500-bpsequenceof C3H R+C13 clones C13211 andC14211 shows thatthey differ from thatof C3HpllO attheproteolytic cleavage site by coding for Argatposition 7191 (Fig. 4). No othersequencechanges
between C3H R+Cl3 clones C13211 and C14211 relative to C3H pllO were detected in this 1,500-bp segment of the envelopegene sequence(Fig. 4). However, it is possible that
the XC-positive, replication-competent variant clones have additional changes in the remainder of the envelope gene, for example, at either or both of the other two positions outside
this segment in which the AKR env sequence differs from thatofC3HpllO(Fig.4). Wedidnot explore this possibility further, but instead concentrated on evaluating the effect of the Lys-to-Arg mutation on the XC and replication pheno-type, because of its likely consequences for envelope pre-cursorprocessing.
Comparison of LTR sequence and integrationsitesof endog-enous C3H XC-negative and converted XC-positive provi-ruses. Analysis of flanking sequences of the C3H
XC-negative clone pllO and the in vitro-converted XC-positive
C3H R+C13 clones showed that the pllOprovirus is inte-grated within the R region of a VL30 LTR in reverse
orientation (Fig. 5) (23, 24). A similar integration site has
also been observed for the BALB/c endogenous provirus
(20, 22). Flanking sequence analysis of C13211 and C14211 show that they are different from each other and are not integrated within the VL30 LTR, but within other unique sequences.Figure5BshowstheflankingsequenceofC14211
with the integration site, and Fig. 3C schematically shows the 4-bp direct repeat of cellular DNA at theintegrationsite
for C13211. The LTR from both endogenous (p110) and
converted (C13211 and C14211) C3H MuLV had a single
enhancer element in the U3 region oftheir LTRs (Fig. 4), whereas in the AKR provirus there are two copies ofthe enhancer sequences (16). In addition, the pllO LTR se-quence shows three nucleotide differences (two in the U3
region andoneintheUSregion) relativetop623,all of which
are identical to those changes observed for the BALB/c ecotropic viral LTR (22). Apart from sharing these three
nucleotide changes, proviral clones from NIH R+Cl3 cells differ from the LTR sequence ofpllO at onenucleotide at
position7889. Sinceall theecotropic provirusesclonedfrom
NIHR+Cl3cells sharethis 1-bpchange,they were
presum-ablyderived from asingle converted virus.
Site-directed mutation of C3Hp110.Todetermine whether the difference observed in infectivity between the C3H
XC-negativeand invitro-convertedC3H XC-positive clones isduesolelyto thepoint mutationattheproteolytic cleavage site or to otherfactors such as integration site orthe 1-bp change observed in the LTR, we performed a Lys-to-Arg
substitution in C3H pllO by site-directed mutagenesis. A
[image:6.612.317.559.585.678.2]520-bp KpnI-XbaI fragment which includes the gp7O/pl5E junction of the env region of pllO was subcloned into M13mpl9and used as atemplatefor site-directed mutagen-esis(seeMaterialsandMethods). Sequence analysis of these
TABLE 1. Infectivity of C3H endogenous ecotropic proviralclonesa
Supernatant
XCClone RTactivityb phenotype
R+Cl3 3211 129.2 +
R+C134211 162.5 +
R+C137111 1.4
R+C13 1013 1.8
pllOphaseI 8.9
pllOphaseIII 125.1
AKRp623 191.9 +
aCulture fluids fromNIH 3T3-transfected cellsat their first confluency
wereharvested24hafter mediumchange.Infectivitywasassayed byreverse
transcriptaseandXCassay asdescribedin thelegendtoFig.2.
b Reversetranscriptase (RT) activityisexpressedas103countsper minute of[3H]TMPincorporatedpermilliliter ofsupernatantculturefluid.
VOL.62, 1988 937
on November 10, 2019 by guest
http://jvi.asm.org/
ulmalns,.al vrklsLvsevvelrlntvI Iavt^r^"rb^^Ii I^rt I.t_t__;IL-a^ S_"_-A
C3H GTcG _ 3839
T
LouhrgM-tValAlaAalsiAlaValLsurhrLssAsal*GlYLYsfuThrMotGlyGlnPro. uValIllL*uAiProHislaValGrluAlaiLuValLysGlnProProAs
C3H CCGTGGTAGCAGCCATTGCCGTcTACAAAGA 3959
AKR
ArgTrpLuSrAsnAM ArqMetThH GArValGlnPholYProValVAlaLouAsnProAlaThrLuL5 uProLuProGluGlu
C3H CGCTGCTACCAACGCCCGCATGACCCACTACCGCATGCTCCTAG^iCACTGACCGAGTTCAGTTGACCAGTGGTGGCCCTAACCTGCCACCTTACTCCCTCTCCCGGAAGhM 4079
AKR
GlYAlaProHisAspCysL*uGluXleLouAlaGluThrHisGlvrThrAr ProAspL6uThrAspGlnProIlloProAspJuAspHis*ThrTrpTErThrAsDG1YSirS*rPh*LAu
C3H GGAGCCCCCCA7rGCTGACAGCCCATCCCAGACGCCGAGCCACTCACT AACC TG;ACArC5T0 4199
AKR
GlnGluGlyGlnArqLvsAlaGlvAlaAlaValThrThrGluThrGluValIllTrMl ArgAlaL*uProAlaGlyThrSerAlaGlnArgAlaGluL*uIleA1aLuThrGInAla
C3H _CCACCGCCATACACTC CC 4319
AKR A
L*uLYsMotlaGlu1LYLsAFrqIuAsnValTYrThrAspS*rArgTrAlaPheAlaThrAlaHisIlHisGl1yGuIleTyrAr&Ar rFG1yL*uL*uThrS*rG1uG1YAr
C3H TXAT 4439
AKR G
GluIleLysAsnLysSrImI1L*u/laLouLouLysAlaL*uPhoLFuPrOLvsAr L*uS.rSlleZl.HisCysLouGlyHisGnLsGlvAs *rAlaGluAlaArqg1n
C3H GAMTCAAAACAGACCGAGAC CAAAGACTCAGTATAATT C AGC C 4559
AM
ArqLouAl AspGlnAlaAiaArgGluAI&lall*TysThrPcroProAspThrSorThrL uL*uIl-&lUAsSSrThrProTyrThrProAlaT
rPhqoHisTyrThirGluThrAsp
C3H CGCACAGACCCAGCGGCCCGC;GAGGCTGCCTTACTACGCCT 4679
AKR
L.uLysLysLauArqGluL.uGlyAlsThrTyrMrAnGlnS-rLysGiyTyrrpalPhoGlnGl LysProValM*tProA- uAsS*rLeuHisAr C3H CTATAACAAACCAGAGAGCTTCvGGCCACCTATAACTGAGTTTACTCCAGCCGGGTGGTGCCCG heVaPTA GTTAA LCATCAC 4799 AKR
Lu~ThrHis'LuGlyTYrGiDnLYsM tLysAla*)uLuAspArgGlyGluS*rProTyrTyrM tLouAsnArgUDLyeThrLuGlnTyrValJilaAs S-r YsThirValCysAla C3H CTChCCCACCTCGG,TACCACACTCCTTGAICCCTCCGgXCCCGCCTAACCCCGGACAACCCTCCAATATGTGA CCTGCjCoGTshTGCC 4919
AK
G1nValAsnAlaS*rLys sIlGlyAlaGlyValArgValArqGlyHisArqProG r isTrGluIl1Ph*hrGluValLysProGlvLsuTvrG1yTyrLysTY
C3H CACGTACTGCCCGCCTGCATG:TAGGGGATCGATCNAACAGT3CACGCAGCTGTA AT.TAC 5039 AKR
L.uLeuValPh-7ValAspThrPhSruaPh*ProThryhslTh lValSrsL LruLouGluGlullPheProArPhOGl tPro
C3H CTC CCTCCAAC 5159
AKR
GlnValLsuGlySerAsDpsnGlyProAlaPheThrSerGlnValSerGlnSerValAlaAspL;uL.uGl IleAspTr LsLI*uHisCysAlaTyrArgProGlnSFrSrGlyGln
C3H CAGGTATTGGGATCTGAACGGGCCTGCCTTCACCTCCCAGGTAAGTCAGTCGGTGGCCGAACCCAGAGTCTCAG 5279
VlGluArqM
tAsnArFThrIlsLysGluThrLouThrLysLeuThrLeuAlaAlaGlyThrArgAspTrpValLeuL*uLeuProLeuAlaLeuTyrArgAlaArFAsnThrProGly
C3H C539CATCA9CTCTAACTAA9TTACGCTTGCAGCTGGCAC GTACTCCTACTCCCCTTAGCTCTCTACCGAGCCCGGAACACTCCGGGC 5399
AKR T
ProHisGlYLeuThrProTYrGluIleL.uTyrGlyAlaProProProLeuValAsnPheHisAspProAspMetSerGluILuThrAsnSerProSerL.uGlnAlaHisL*uGlnAla
C3H CCCCATGGATTGACTCCGTATGAAATCTTGTACGGGGCGCCCCCGCCCCTTGTCAACTTCCATGACCCCGACATGTCAGAATTACTAATAGCCCATCTCTCCAAGCTCACTTACAGGCC 5519
AKR C T
LouGlnThrValGlnArqGlull*TrpLysProLouAlaGluAlaTyrArq As uAspG1nProVa1I1ProFHisProPheArFIlGlvAs S*rValTr ValAr r His
C3H CTCCA5ACGG73GCAGGAATTGGCCACTGGCCGAGGCCTACCGGTGATACCACACCCCTTC9;lG RCCGTr3MWTGCGC98CAC 5639
Al~
In
A~
AG1nThrLysAsnLeuG1uProArFTr LYsGl ProTyrThrValLsuLsuThF 'rOThrAlaeuLysValAs GlyIl*SrAlaTr IlHisAlaAlaHisValLysAlaAla
C3H CAGACCAAAAACTTAGAACCTCGTGGAG ACCCTACACCGTCCTACTGACCACCCCACCGCTCTCAAGGTAGACGGCATCTCTGCAGGATACACGCCGCCCACGTCGCG 5759
AKR.
LtGluSG1 rThrThrL.uS.rLysProPh.LysAsnGlnValAsnProTrpGlYProLsuI1*ValLsuLouIl-LsuGlyGlyVaiAsnProVa
ThrThrProProIlLysProSrTrpAr ValGlnArqsorGl AsnProLsuLysIllArgLvuThrArqGlyAlaProEnd
C3H ACCACACCCCCGATAAAACATCATGACAACTCTCAAAACCC7'TAAAAALTCAGGTTAACCCGTGGGGCCCCCTAATTGTCCTTCTGATTCTCGGAGGGGTCAACCCG 5879
AlR G
1LuGlyAsnSFrProHisGlnValPheAsnL.uThrTrPGluValThrAsnGlYAspArgGluThrValTrpAlaIleThrGlyAsnHisProLuTrpThrTrpTrpProAspLou C3H 6119ACAGCCCCCACCAGGTTTTCCTCACCTGGGAGTGACTATGGAGACCGAGAAACGGTGTGGGCAATACCGGCAATCACCCTCTGACTTCGTGACCA 5999 AKA
roAsprouCsA AltAouAaLuisF GlyProSoT
rGlyuGlPhuTyrCrgMaProPhGiyCyrProProPrGlyArorCsCy
sSYrG
cy
rS.rASrThrProGC3H CSSAACTCTTTTTGCTCACGGGCCGTCCTTTCGCCTAGAATATCGGGCTCC-Trrl7lTCCTCCTCCCCCGGGOCCCCCCTECTETTCAGGAAGtGCGACTCCACGCCAG 6119 AKR
lyCysS-rArgAspCvsGluGluProLouThrSrTyrThrProArqCysAsnThrAl aTrpAsnArgLouLysLoeuSrLysValThr}iisAlaHisA.n G1 lyhTyrValCysP
C3H GCTGCCAAATGGGGAGCCCCTGACTTCATATACTCCCCGGTGCAATACGGCCTGG;ACAGACTTAAGTTAT=AAAAGTG CACAAGC GG G CTATGTCTG;CC 623 9
AKl A A
roGlyPoHisArProAr TrAAgSCyGlyGlGyProluS-rPheTyrCvysAlaSorTr GiCsluThrThrGlyArgAlaesrTrEw rSSrTApT
C3H CCGGGCCACATCG CCC GCCTCG CCAGAATCCTTTGCCT CTGCGAAACCACAGGCCGAGCATC CCATCCTCGTCCTCGGACT 6359
AKR A
vrIlThrValSerAsnAsnLeuThrS-rAspGlnAlaThrProValCYsLysGlyAsnGluTrpCysAsnSerLouThrIleArqPheThrSerPheGlyLysGlnAlaThrSerTr V
C3H ACATCACAGTAAGCAACAATCTAACCTCAGA67CAGGCAA9CCCAGTATGAGTGGTGCAACTCCTTAACTATCCGGTTCACGAGCT97GAAAACAGGCCACCTC6479
AlR
alThrGlyHisTrDTrDG1 LouArgLo uTyrVal1SrGlyHi sAspProGlyLouIl1 PhoGlyIl1eArgL uLyslIl*ThrAspSe rGlyProArgVal1ProIl*GlyProAsnP roV
C3H TAAGC
W
1GCCTTCTT7GAAGCCGGTA GACGCTAATCGTCGGCCGTCATGGCACCG6599AKR
alLeuSerAspArgArgProProSerArqProArgProThrArgSerProProProSerAsnSerThrProThrGluTh'rProLeuThrL.uProGluProProProAalyValGluA
C3H TCTGTCAGACCGACGACCACC7TCCCGGCCTAGACCCACCAGATCTCCCCCGCCTTCAAACTCCACCCCAACCGAGACACcccTcAcccTcCCCGAACCCCCGCCAGCGGGAGTCGAAA6719
AKR
6nArgLeuLsuAsnLeuValLysGlyAlaTyrGlnAlaLeuAsnLeuThrSerProAspLysThrGlnGluCysTrpLeuCysLeuValSerGlyProProTyrTyrGluGlyValAlaV
CM AC 6839
C3H ACCGATTGTTAAATCTAGTAAMGACCTACCAAGCCCTCAACCTAACCAGTCCTGATAAAACCCAAGAGTG;CTGG;TTATGCCTAGTATCGGGACCCCCATACTACGAGGGGGTTG CG 683
AKR C
R+CL3 - R+CL3
alLeuGlyThrTyrSrAsnHisThrSrAlaProAlaAsnCysS rValAlaSrGlnHisLysLouThrLuSrGluValThrGlyGlnGlyL*uCysI lGlyAlaVa1ProLYsT C3H TCCTAGGTACCTACTCCAACCATACTTCTGCCCCAGCTAACTGCTCTG9GGCCTCTCAACACAAATACCTGTCCGAAGTGACC oGGAcTCTGCATAGGCGGTcCCTALbA 6959
AM
[image:7.612.64.562.59.648.2]R+CL3
FIG. 4. Envelope gene sequenceof C3Hp110. The sequence ofC3HpllO from theSall siteat
position
3719ofthepolregion
tothe virus/cellularjunction (top row)iscomparedwith thatofAKRp623provirus (16).C3HR+Cl3clonesweresequenced
from nucleotide6835 tothevirus/cellularjunction. Clones C13211andC14211 hadidenticalsequencesinthis region. Different aminoacids encodedatthepointmutations areboxed. - - - -,Absenceofone copyofthe99-bpenhancer element in the U3
region
of C3HpllOandC3HR+C13clones. clones showed(Fig. 6)that thecorrectA-to-Gtransversion from thereplicative
form of the mutantphage.
TheBstEII-wasinsertedinto themutant.Noother
base-pair change
was to-XbaIfragment
from a 1.8-kb BamHI-SstI subclone of observed in this region. C3H pllO was substitutedby
thecorresponding
fragmentFor reconstruction of C3H p110-R7191, the
KpnI-XbaI
from themutagenized
subclone.Subsequently,
theBamHI-fragment containing the
gp7O/pl5E
junction was recovered SstIfragment
was substituted into the HindIII-SallpBR
on November 10, 2019 by guest
http://jvi.asm.org/
MuLV ENVELOPE GLYCOPROTEIN PROCESSING MUTANT 939
C3H
R+CL3 C3H R+CL3 C3H
ARM
R+CL3
hrHisGlnValLeuCYsAsnThrThrGlnLysThrSerAspGlySerTyrTyrLAuAlaAlaProThrGlyThrThrTrpAlaCysSerThrGlyL*uThrProCysIleSerThrThrI
CCCATCCTCTACTAATACCACCCcTGc TATTTGGCCCTCCCACAGGAACTACCTGGGCTrOTAGTACTOGACTCACTCCCTGTATCTCAACCACCA 770 79 T
1e*L.uAsp euThrThrAls pTyrCysVa LUValGluL*uTrpProArqValThrTyrHisSrProSrTyrValTyrHisGGlnPh*lGuAr Ar A1 sr 1uProV
TACTA7GACCTCACCACCGATTACTGTGTCCTGGTCGAiCMCCAAGGGTGACCCATCCCCCACCAATTAAAGACGAGCCATwIAACCCG 7199
alS*rL.uThrL.uAlaLeuLuLJulGl 1 L.uThrM.tGlyGlyIlOAlaAlaGlyValGlyrGlThrThrA1aLeuVa1A1aThrGlnGlnPh.GlnGlnLu'±lnAlaA1aM
TCTCACTAACTCTGGCCCTACTATTGCTCACTATGGGCGCCCTAGGGCCACTCAGCAGTTCCAACAiCTCCAGGCTGC-ACCG
*tHi sAsDAsoL uLy5GluValGluLysS-rIlThrAsnL*uGluLysS rLeuThrS-rLouS-_GluVal1Val1LouGlnAsnArArqglyLuAs2LeuLeuPheLeuLysG1uG
C3H TGCACGA GAtCT A AGTCCATCA aAATTCCCTCCGTCCGAAGTAGTGTTACAGAATCGTAGAGGCCTAGATCTACTATCCTMGAGG
AICR
R+CL3
lvcflvLAuCvsAiaAjaauLvsG1uG1uCv3CVSPh.TyrA5pHihThrGyLUValArqASSrM@tAlaLysL4uArqGluArgL.uSerGlnArqGlnLysLeuPheGluS
C3H
T?GGTACGGACATGGCCAAAC7TAGAGAAAGAGTCAGAGACAAAAGCTCTrGAPAT
AKR
R+CL3
C3H AMm R+CL3 C3H R+CL3 C3H AM R+CL3 C3H
AM
R+CL3 C3H R+CL3 C3N R+CL3 C3H R+CL3
C3H
R+CL3
*grOlGniYTr hiGuGlL*uPhoAJnLYsS*rProTrDPhbThrThrLeuI
CTGTITAATAXGTCCC
lSeSrThrIlul*MtGlyProL*uI lolIl*L*uLouLosul 1*L*uLouPhoGlyProCYsIl eLC CCTGTATCCACCATCATGGGTCCCCTGATAATCCTCTTGTTAAT
TTACrCTTTGGGCCTATIC
*uAsnAr L[uVaIGlnPheIl*LvsAspArgIllSrValValGlnAlaLuValL4uThrGlnGlnTyrHisGlnu sThrIl spCy sLVsS.rArgGluEnd
TCAATCGCCTCGTCCAGTTTATCAMGACAGGATTTCGGTAGTGCAGGCCCTGGTTCTGACAACAATTCATCAACACAT TTGTAACACGTGAATAAAAGATTT
I-) U3
TA,TCAGTTACAGAAAGAGGGGGAT*AAACCCCTTC ATAAGGCTTAGCCAGCTAAC TGCAGTAACGCCATCCTGCAAGGCATGGGAAAATACCAGAGCTGATG1TTCTCAGAAAAA
T 0
A
CA8;AACAAGGAAGTA
CCGGGACTACGGGCCAAACAGGATATCTGTGGTCAAGCACTAGGGCCCCGGCCCAGGGCCAAGAACAGATGGTCCCCAGAAA---CAGAG
TAGCTAAAACAACAACAGTTTCAAGA
AGOCTOGAAAGTACCGGGACTAGGGCCAACAGGATATCTGGWTCAAGCACTAGGGCCCCGGCCCAGGGCCAAGAACAGAATGGTCCCCAGAAA
LACCCAACT rCCCCC AGTACsCGATCTACCCA TCTT TCTCTCGC TCTTC TGTACCCGC GCTTATTGCTGCCCCTAT
- ) R 82651-> US
AAAGGGTAAACCCCACACTCCGCGCGCCAGTCCTCCGATAGACTGAGTCGCCCGGGTACCCGT'GTATCCAATAAAGCC --- CTGBTTGCATCGAATCGTGGTCTCGCTGATCC'TT G
GGGAGGGTCTCCTCCAGAGTGATTGACTGCCCAGCl iWGGGGTCTrrCA
FIG. 4-Continued.
7319
7439
7559
7679
7799
7919
8034
8060
810
25
74
subclone. Finally, the 5' part of the viral genome with cellular flank was ligated to this construct to generate the provirus C3H pllO-R7191.
InfectivityassayofC3H
pllO-R7191.
DNA from the muta-genized clone C3HpllO-R7191wastransfectedontoNIH 3T3 cells with pSV2neo as described above. Ten neo-positivecolonies were selected, and the infectivity of these clones was determined by release of reverse
transcriptase-con-taining particles and assay of their XC fusion ability. In contrast to cells transfected with C3H pllO DNA, cells transfected with the mutagenized C3H
pllO-R7191
DNA released reverse transcriptase-containing particles andscoredpositiveinthe XC assay(Table 2).
Processing of Pr85to gp7O and pl5E. During the biosyn-thesis of the murine leukemia virus envgene products, the
precursorpolypeptide Pr85 isproteolyticallycleavedtogp70 andpl5E. It has been shown that the cleavage recognition
sequences forthe envgeneproducts lieatthe Arg(Lys)-X-Lys-Arg for a number of retroviruses, as well as for the
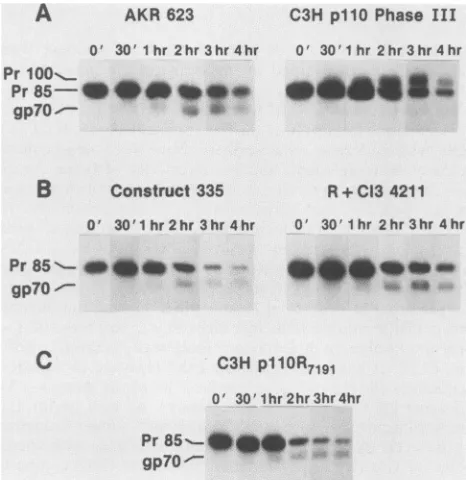
hemagglutinin glycoprotein of avian and human influenza viruses(11, 38, 56). Eucaryotic endopeptidases with speci-ficity forLys-Arg have been identified in theGolgi compart-ment(27). To examine whether the point mutationobserved inC3HpllOfunctionally blocks the cleavage of Pr85togp7O andpl5E,wecompared therateofprocessing of Pr85env in NIH3T3 cells transfected withdifferentproviral DNAs. The cells were pulse-labeled with [3H]leucine for 30 min and
chased for0 and 30min and 1, 2, 3, and 4 h. The labeled proteinswere immunoprecipitated with ananti-gp70 antise-rumand separated byelectrophoresis inan
SDS-polyacryl-amide gel. In cells transfected with AKR p623 DNA, the
precursor protein Pr85 was present at high levels after a
30-min pulse and cleavage was observed after a 1-h chase (Fig. 7A). Similarly,cellstransfectedwith the DNA of the in
vitro-converted infectious C3HR+Cl3 virus (clone C14211) or with the AKR p623-C3H pllO construct 335, which containthe 5' halfof C3HpllO and the 3'half ofAKRp623,
showed cleavage ofPr85 (Fig. 7B). Alternatively, in cells
transfected with C3HpllO(phaseIII) there was no cleavage
ofPr85.Instead, aftera1-hchaseahigher-molecular-weight formmigratingas aPrlOO wasobserved whichwasstablefor
theremainder ofthechaseperiod(Fig.7A). Incontrast,Pr85 wascleavedinto
gp70
with identicalkinetics to those of the AKR virus control in cells transfected with DNA of themutagenized clone C3H
p110-R7191
(Fig.7C).
DISCUSSION
Wehave previously described, on the basis of work with
biologically cloned viruses, that the endogenous ecotropic
virus in at least two low-leukemic mouse strains, C3H/He
and BALB/c, is XC negative and replication defective. XC-negative, replication deficient virus could, however, be
convertedto aninfectious XC-positive form whengrownin certain transformed mouse cells in culture. The change in phenotype involved mutation and selection as judged from
conversionexperimentsatlimitingdilutions (41, 44, 45) and under conditions which restricted successive rounds of infection (41). The converted virus was comparable with virus fromhigh-leukemic AKR mice inits ability to spread and form large XC plaques. The nonpathogenicity of the
XC-negativevirus insusceptible NFS/N mice,incontrast to the similarity of the converted, XC-positive infectious
C3H/Hevirus and ecotropic AKRvirus(41) in induction of VOL.62, 1988
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.66.560.59.386.2]A
TGGATCTCTGAGTTTGAGGCCAGTCCATCTACACAGTGAGTTCTAGGACATTCAGGGCTACACAGAGAAA 70
cellular flank VL30 US
CCTTGTCTTGTAAAACAAATCAAAACAAAAAATTACCCTGAAAGACCCTCGAGGGGAGACCCTCACTCAG 140
VL30 R
ACACTCAAGTCCCGGGACAGCCGCGTACCCAATGAAGACACTGAGACCATACATAAAATGTAGAAGGCAA 210
GATTTAATAAGGCAGCAACATGAAAGC1I- C3H pllO provirus I-gIAGACCAGAGCTC 257
VL30 R
TGGGGTCGAAATAAAGCAGCAACATGGAAGCACACAGAGCTCTGGGGTCGAAACTTCATACACCTTAGCA 327
CAGGGTAGAGGAGTCTCGACGGTCAGCCAGAATTTTTCACAGGCTTATATAGTAAAACTCAAAGGGGGAG
VL30 U3
AACTGGGCAGGGAAAGTACAAGTTTACATCACTAGGGAGTTCTGCCAAGGGACAAGGGGTTCTGCCAAAG
GATTCTACGTAACTAAGGAGTCATGTCCTATCAAGGAACCTACGTAACTAAGGAGTACCTGGTTCATTTT GAGGTTGTTCCAGGAGGCCTTTATCTCA
397
A5,7
TABLE 2. Comparisonof infectivity of C3Hp110-R7191
andR+Cl34211provirusesa
Plasmid Supematant XC
RTactivity' phenotype
p110 10.1
-p110R7191 111.4 +
R+Cl3 4211 183.7 +
a NIH 3T3 cells weretransfected with plasmid C3Hp110-R719,and
R+C13
537 4211. Culturefluid from transfected NIH 3T3 cells was tested for reverse
565 transcriptase and XC fusion activity as described in the legend to Fig. 2.
b Reverse transcriptase (RT) activity is expressed as103counts per minute
of[3H]TMPincorporated per milliliter of supernatant culture fluid. B
TCTTGCCTCTGTTAAATACAGCCCCAAAACTATCTGGATGGGACTTCATACTGAAATTACCATGTCTACC 70
TAAACAAAAAACTTAGCCTGGTAGGATCTCCTACCATCACCACTCCCAAATATCTTTATCTCCTTCCTTA 210
GTATCATCCTGACAAGGTGGCTCACAGCACAGCTACTTTTGTTCCTTTAGATTCGGGAAGCCACACATAT 280
AATTCATACTCCATCCTTATTTTTTTGAGAAATTACATATTTTTGAACCCTGGTTTTAGAGTCCTTTCCA 350
GAGCCTGAAGGTCCCAGCATT|A---4211 PROVIRUS--- ATTGCTGAG 388
ACATAGCACACCTTTGTGTTTCTGATTATCATGGAATTCATTGGCATTAACAG 441
FIG. 5. Flanking cellular sequences of XC-negative and XC-positive C3H proviruses. (A) Flanking cellular sequence ofC3H p110. C3H pllO is integrated within a VL30 LTR in reverse orientation. The directrepeatsequences that occur at the junction of the VL30LTRand the ecotropicprovirus are boxed. The U3, R, andU5 regions of VL30 LTR are indicated. (B) Flanking cellular sequence of the C3H R+C13 4211 provirus. The direct cellular repeat attheintegration site of the provirus is boxed.
leukemia, led us to conclude that the infectivity of the
endogenous ecotropic virus was a major determinant of
leukemia incidence. The properties ofthe molecular clones
of endogenous and in vitro-converted ecotropic C3H/He
viruses presented here provide definitive evidence for our
previous conclusions and furthermore precisely locate the
lesion in theendogenous C3H/He virus.
The sourcefor molecular cloning of endogenous ecotropic
C3H MuLV consisted of C3H/MCA5 cells, a chemically
transformed derivativeofC3H/1OT1/2Cl 8 cells, from which
XC-positive virus could be obtained late (phase III) after
induction withIdUrd (41, 44, 45). The fact that the proviral DNA from these cells encoded a XC-negative replication
deficient virus for which these cells were homozygous
(unpublisheddata) established that chemical transformation
A
had not mutated the proviral DNA to yield the infectious virus form, confirming ourbiological observations (41, 45). The nature of the defect in infectivity and XC fusion activity of endogenous C3H/He virus is a point mutation at
the precursor cleavage site of Pr85 as judged by sequence
comparisonand DNAfragment exchange with the ecotropic AKRprovirus. An identical mutation has been observed in theendogenous BALB/c ecotropic virus (22). In contrast to our findings, the biological activity determinations with
cloned BALB/c ecotropic viral DNA were interpreted to
indicate levels of infectivity comparable to those of AKR virus clone p623 (22). However, the assay conditions used were different from ours in that they allowed for multiple
cycles of infection and thus selection (22). Moreover, the target cells were of the converter category (see below), according to ourexperience, and the time of reverse
tran-A
AKR 623O' 30 1hr 2hr 3hr 4hr
C3H p110 Phase III
O' 30'1hr2hr3hr4hr Pr100_
gp7l _ r
B Construct 335 O' 30'1 hr2hr3hr4hr
R+C134211
0' 30'1hr 2hr 3 hr4hr
Pr851
4000V
***004
amgp70
-,-*B
A T G C A T G C
-.NO_
A. C3HpI1O 5' TTT GAA AGA CGA GCC AAA TATAAAAA GAA CCC 3' Phe Glu Arg Arg Ala Lys Tyr LysILysiGIu Pro B. R7191 5' TTT GAA AGA CGA GCC AAA TAT AAAIAGAIGAA CCC 3
Phe Glu Arg Arg Ala Lys Tyr Lys ArgJGlu Pro
FIG. 6. Site-directed mutagenesis of C3H p110 (Lys-7191) to C3H p11O-R7191 (Arg-7191). The M13 dideoxynucleotide sequence ofp110 and R7191 clones in the region of mutation is shown. The position of the A-to-G transition is indicated by an arrow. Amino acids encoded by the region shown in the sequenceare indicated below. TheLys-to-Argtransition is boxed.
C
C3H p11OR7191O' 30'lhr2hr3hr4hr
Pr85
-*@*w-gp70"O
FIG. 7. Pulse-chase analysis of the env gene product Pr85 ex-pressedfrom cells transfectedwith (A) AKR p623 andC3H pllO phase III, (B) construct 335 and C3HR+Cl3 4211, and (C) C3H pllO-R7191.Transfected cells were labeledfor30min with 40,Ciof
[3H]leucine,chasedfor0 and 30 min and 1, 2, 3, and 4 h, and then lysed. Lysates were immunoprecipitated withanti-gp7Oantiserum. Immunoprecipitates were analyzed on SDS-10% polyacrylamide, and the gels were exposed for 3 weeks. Sizes of proteins were determined from theirmigration relative to a 14C-labeled marker protein.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.314.551.89.147.2] [image:9.612.318.551.386.626.2] [image:9.612.61.300.533.663.2]MuLV ENVELOPE GLYCOPROTEIN PROCESSING MUTANT 941
scriptase assay waslate aftertransfection, presumably
cor-responding to what we have termed phase II/III of virus spread (44,45).
Thesequenceofthecleavagesite intheXC-negativevirus is Arg-Ala-Lys-Tyr-Lys-L-Glu-Pro and Arg-Ala-Lys-Tyr-Lys-Arg-Glu-Pro in the AKR virus (the Lys-to-Arg
substi-tution is underlined).ThisArg(Lys)-X-Lys-Argcleavage site has been conserved in a number of other retroviruses, including humanimmunodeficiency virusand some
myxovi-ruses (11, 38, 56). The Arg(Lys)-X-Arg-Arg sequence at the
cleavage site ispresent in human T-cell lymphotropic virus type I,bovine leukemia virus, and feline leukemia virus (56). Fromexperiments with surface-labeled IdUrd-induced C3H/ 1OT1/2 Cl 8 phase III cells, wehadprevious evidence fora
defect in envelope glycoprotein processing (N. Famulari, and U. Rapp, unpublished data). Pulse-chase experiments withcellstransfected with C3H/HeproviralDNAclonepllO
confirmtheseearly observations,aspresentedin thisreport,
since proteolytic cleavage of Pr85 into gp7O and pl5E is
blocked (Fig. 7) and leads to the accumulation ofa
unpro-cessed precursor. Processing of precursor polyprotein to
functional gene products plays a fundamental role in the
infectivity of viralparticles. Forexample, ithasbeen shown for avian influenza virus that the
proteolytic cleavage
of hemagglutinin glycoproteinto HAl andHA2isrequired
for full infectivity but not for full assembly (4). We haveobserved, by electron-microscopic examination of virus produced by
C3H/1OT1/2
Cl 8 cells acutely infected with C3H/10T1/2 phase III virus, that the budding particlesap-peared bald relativetoecotropic AKRvirus, indicative ofa lackof mature envelope glycoprotein (unpublished data).
Conversion of
XC-negative
toXC-positive
infectious virus in cultureby growthintransformedmousecells restores the Pr85 cleavage site by a point mutation (A-to-G transition, resulting in a Lys-to-Arg substitution) as demonstrated forthe molecular clones C13211 and C14211 derived from C3H R+Cl3 grown in NIH 3T3 cells. Recombination with
non-ecotropic
endogenousenvelope
sequences as a source forthe amino acid substitution is
virtually
ruled out, since noadditional sequence alterations relative to clone pllO were
observed in this
region.
Both C3H R+C13 clones and theinfectious AKR ecotropic virus have an identical sequence atthecleavage
site, indicating
that otheramino acidsubsti-tutions that could be achieved by
single-point
mutation of this region may not becompatible
with Pr85 cleavage. We have examined the consequence of this mutation forenve-lope glycoprotein precursor processing by pulse-chase
ex-periments andfound it to restorecleavage.
Why does conversionoccur
preferentially
incertaintrans-formed cells? One
possibility
isthattransformedcells which behave as converter cells haveanincreased mutation rate.However, several lines of evidenceargue
against
this.First,
the endogenous C3H/He virus in chemically transformed C3H/MCA5 cells was not mutated, as shown above. Simi-larly, infection of converter cells under conditions that
minimize subsequent virus spread (as achieved by using
high-percentage
virus donor cells andby
the absence of Polybrene in the cocultivation) eliminate conversion.Fi-nally, long-termpassage ofC3H/10T1/2 Cl 8 phase III cells foratleast 18months didnotyield convertedvirus(40, 41, 44) as might be expected iftheir potentially lower rate of mutation during DNAsynthesis was the limiting factor for conversion. Therefore, we conclude that mutation occurs
duringreversetranscription. Transformed cells tendtogrow virus to ahighertiter than normal cellsdo,and this would be
expected to facilitate subsequent rounds of
infection,
evenfor
poorly
infectiousparticles.
Once ahighly
infectiousmutantemerged, it would
quickly
beamplified by selection,
provided that a sufficient reservoir of virus-free cells was available. The basis forbetter virusgrowth
by
transformedcells is not known and may include differences in DNA
methylation activityand enhancerutilization.
The LTR ofthe
ecotropic
C3H/Heprovirus
hasonly
asingle
enhancerelement,
raising
thepossibility
thatsomeofthe differences in
infectivity
relative toecotropic
AKR MuLVmight
be LTRmediated.However,
virusproduction
levels,
as determinedby
reversetranscriptase
assays, areonly
<5-fold lower in chronicproducer
cultures of C3H/10T1/2 Cl 8phase
III than in AKRphase
IIIcell lines(41, 44),
indicating comparable
transcription
ratesfrom theproviral
LTRs. Asingle-nucleotide change
has beenob-served at
position
7889 between theproviral
LTRs oftheendogenous
and convertedlarge
XCplaque-derived
C3H virus cloneR+C13, suggesting
that LTRmutationsmay alsocontribute to the increased
infectivity
of this converted virus. However, these mutations were not necessary for conversion oftheXCphenotype,
as shown inthisreportby
DNA
fragment exchange
betweenendogenous
C3H/Heand AKRproviral
DNA.The step in the
replication cycle
at whichXC-negative
ecotropic
C3H/He virus is blocked in acute infections is at thelevel of virusrelease(41).
Thus,
thespecific infectivity,
as measured
by p30
antigen production
inacutely
infectedcells,
wascomparable
betweenXC-negative
C3H/He,
XC-positive
convertedC3H/He,
andXC-positive
AKR viruses(44), whereas an assayfor release ofreverse
transcriptase-containing particles by
infected cells showed a 100- to1,000-fold
lowerinfectivity
for theXC-negative
virus(41,
44). Theinitially
nonproductive
infectioneventually
be-comesproductive
oncontinued subculture of infected non-convertercells,
without anydetectablechange
in the stableproperties
oftheproduced
virus. Themechanismunderlying
this processisunknown. One
possibility
isviral gene ampli-ficationviarepeated
cycles
of infectionas aresultofinitially
incomplete
levels ofinterference. C3H/10T1/2Cl8phase
III cellschronically producing
XC-negative
C3H/He virushaveglycoprotein
ontheirsurfaces,
areGlX cell surfaceantigen
positive,
and arecompletely
resistant tosuperinfection by
XC-positive
virus(P.
0. Donnell and U. R.Rapp,
unpub-lisheddata).
In contrast, cellsacutely
infected withXC-negative
C3H/He virusexpressp30,
butonly
very low levelsof
gp70,
are GlXantigen negative,
and areonly
partially
resistanttosuperinfection.
The lackofGlXantigen
may be due to the low level ofenvelope
glycoprotein expressed
at the cell surface or may be a consequence ofthe structuralmutation
(19).
The
inability
ofXC-negative
C3H/He virus torapidly
establish aproductive
infection ispresumably
the basis forits lack of XCfusion
activity.
Fromexperiments
inwhichratXC cell infection was facilitated
(use
ofPolybrene)
orblocked
(addition
ofvirus-neutralizing antibody)
afterover-lay
ofXC-positive
virusproducer
cultures,
it appears that suchaninfection isarequirement
for effective fusion(U.
R.Rapp,unpublished data). These
findings
do notexplain
the basis for the lack ofXC fusionactivity
of other hostrange classes ofXC-negative
type Cretroviruses,
nor dothey
establish that all
XC-negative ecotropic
murine type C viruses must be defective atthe same site.Clearly,
thecell surfacereceptor-binding specificity
of viralgp70
for theecotropic
viralreceptor(8, 43)
isarequirement.
Theecotro-pic
viral receptor is apotential
fusion receptor, whereas otherxenotropic
or mink cellfocus-forming
viralreceptors VOL.62, 1988on November 10, 2019 by guest
http://jvi.asm.org/
are not.Thus, introduction of a different receptorspecificity by recombination between ecotropic and endogenous am-photropic minkcellfocus-forming-class murine type C virus (43) destroys XC fusion activity of the parental ecotropic
virus. Our experiments establish that cell surface
presenta-tion of ecotropic Pr85 with an intactreceptor-binding siteis not sufficientto induce XC plaque formation. We ascribe the
basisfor this inabilitytothe associatedreplicationdeficiency
of the virus, which is not effective in establishing a rapid productiveinfection of XC cells.
Multiple mechanisms contributetothe controlofleukemia
in low-leukemic mouse strains. In BALB/c mice, virus
spread is suppressed by the restricting allele of a host resistancegene, Fv-J (36, 37), and, additionally,the
endog-enous ecotropic virus has a structural defect (22). For C3H/He mice it appears that the structural defect of the
endogenous virus is by itself sufficient to contain virus spread, since this strain carries apermissive Fv-J allelefor
its endogenous ecotropic virus (36, 37). With the molecular clones described in this report, we are now inapositionto test the impact of a single proviral point mutation on leukemia incidence in C3H/He mice. Mutantviruses,
gener-ated by site-directed mutagenesis, have nowbeenprepared and are being tested for their ability to increase leukemia
incidence in C3H/He mice.
After submission of this manuscript, inhibition of
enve-lope precursor processing by an Arg-to-Lys substitution in AKR MuLV was reported (10).
ACKNOWLEDGMENTS
WeacknowledgePatriciaLloyd and Robert Nalewaikfor excel-lent technical assistance. Wethank Steve Oroszlan for providing gp7O antibodyand JamesIhleand John Cleveland for criticalreading of themanuscript.
G. S. is a Ph.D. student in thegraduateprogram of Geneticsat George WashingtonUniversity, Washington,D.C.
LITERATURE CITED
1. Aaronson, S. A., and C. Y. Dunn. 1974. Endogenous C-type viruses ofBALB/c cells:frequenciesof spontaneous and chem-ical induction. J. Virol. 13:181-185.
2. Beggin, M. D., T. J. Gibson, and G. F. Hong. 1983. Buffer gradient gels and35S labelas an aid to rapid DNA sequence determination. Proc. Natl. Acad. Sci. USA 80:3963-3965. 3. Benton,W.D.,and R. W. Davis. 1977. Screeningof gt
recom-binant clonesbyhybridizationtosingle plaqueinsitu. Science 196:180-182.
4. Bosch, F. X., W. Garten, H. D. Klenk, and R. Rott. 1981. Proteolytic cleavageofinfluenza virushemagglutinins: primary structure of the connecting peptide between HAl and HA2 determines proteolytically cleavability and pathogenecity of avianinfluenza viruses. Virology113:725-735.
5. Chattopadhyay,S.K.,M. R.Lander,E.Rands,and D. R.Lowy. 1980. The structure ofendogenousmurineleukemiavirus DNA inmousegenomes. Proc.Natl. Acad. Sci. USA77:5774-5778. 6. Chumakov, I.,H.Stuhiman,K.Harbers,and R.Jaenisch. 1982. Cloningoftwogeneticallytransmitted Moloneyleukemia pro-viral genomes: correlation between biological activity of the clonedDNAandviral genome activationin theanimal.J.Virol. 42:1088-1098.
7. Copeland, N. G., H. G. Bedigian, C. Y. Thomas, and N. A. Jenkins. 1984. DNAs oftwo molecularly cloned endogenous ecotropic provirusesarepoorlyinfectious inDNAtransfection assays.J. Virol.49:437-444.
8. DeLarco, J.,U. R.Rapp,andG.J.Todaro. 1978. Cellsurface receptorsforecotropicMuLV: detection of tissue distribution of free receptors in vivo. Int. J. Cancer21:356-360.
9. Enquist, L., and N.Sternberg. 1980. Invitro packaginglamda
dam vectors and their use in cloning DNA fragments. 1980.
MethodsEnzymol.68:281-298.
10. Freed,E.O.,and R. Risser.1987.Role ofenvelopeglycoprotein processing in murine leukemia virus infection. J. Virol. 61: 2852-2856.
11. Gething,M.J., J.Bye, J. J. Kethel,and M. D.Waterfield.1980. Cloning and DNA sequence of double stranded copies of hemagglutinin genes from H2 and H3 strains elucidates anti-genicdrift andshift in human influenza virus. Nature(London)
287:301-306.
12. Graham,F.C.,and A.J.vanderEb.1973.Anewtechniquefor the assayofinfectivityof human adenovirus5 DNA. Virology 52:456-467.
13. Groudine,M.,R.Eisenman,and H. Weintraub.1981.
Chroma-tin structure ofendogenous retroviral genes and activationby
inhibitor ofDNAmethylation. Nature(London)292:311-317. 14. Grunstein, M., and D. Hogness. 1975. Colonyhybridization:a
methodfor the isolation of cloned DNAs that containaspecific
gene. Proc.Natl. Acad. Sci. USA 72:3961-3965.
15. Hartley, J. W., W. P.Rowe,W. I.Capps,and R.J. Huebner. 1969. Isolation of naturally occurring viruses of the murine leukemia virus group in tissue culture. J. Virol. 3:126-132. 16. Herr,W. 1984. Nucleotide sequence of AKV murine leukemia
virus. J. Virol. 49:471-478.
17. Hoffmann,J. W.,D.Steffen, J. Gusella, C.Tabin,S. Bird,D. Cowing,and R. A. Weinberg.1982. DNAmethylation affecting
expressionof murine leukemiaproviruses.J.Virol. 44:144-157. 18. Hopkins, N., and P. Jolicoeur. 1975. Variants of N-tropic
leukemia virus derived fromBALB/c mice. J. Virol. 16:991-999. 19. Hopkins, N.,H. D.Keller,J.Rommelaere,and R. W.Ellis.1980. Nucleotidesequencesassociated with differences in
electropho-retic mobility of envelope glycoprotein gp7O and with GIX antigen phenotype of certain murine leukemia viruses. Proc. Natl. Acad. Sci. USA 77:1642-1645.
20. Horowitz,J.M.,G.D.Holland,S. R.King,andR. Risser.1987. Germ line integration of a murine leukemia provirus into a
retroviruslike sequence. J. Virol. 61:701-707.
21. Horowitz, J. M.,andR. Risser.1982.Alocus that enhances the induction ofendogenous ecotropic murineleukemia viruses is distinct fromgenome-lengthecotropic proviruses. J. Virol.44: 950-957.
22. Horowitz, J. M., andR. Risser. 1985. Molecular andbiological
characterization of the endogenous ecotropic provirus of BALB/c mice. J. Virol. 56:798-806.
23. Itin, A.,andE. Keshet. 1983. Nucleotide sequenceanalysisof thelongterminal repeat of murinevirus-likeDNA(VL30)and itsadjacentsequences:resemblancetoretrovirusproviruses.J. Virol. 47:656-659.
24. Itin, A.,and E. Keshet. 1986. Diverselongterminal repeatsare
associated with murine retroviruslike (VL30) elements. Mol. Cell. Biol. 6:1276-1282.
25. Jaenisch, R., D. Jahner, P. Nobis, I. Simon, J. Lohler, K. Harbers, and D. Grotkopp. 1981. Chromosomal position and activation of retroviral genomes inserted intothe germline of mice. Cell 24:519-529.
26. Jenkins,N. A., N. G.Copeland, B. A.Taylor,and B. K. Lee. 1982. Organization, distribution, and stability of endogenous ecotropic murine leukemia virus DNA sequences in
chromo-somesofMusmusculus. J. Virol.43:26-36.
27. Julius, D., A. Brake, L. Blair, R. Kunisawa, and J. Thorner. 1984. Isolation of the putative structural gene for the
lysine-arginine-cleaving endopeptidase required for processing of yeastprepro-a-factor.Cell 37:1075-1089.
28. Leder,P.,D.Tiemeier,and L.Enquist.1977.EK2derivatives of bacteriophagelambda usefulin thecloningofDNAfromhigher organisms:theAgtWESsystem. Science 196:175-177. 29. Loenen, W.,and F. R. Blattner.1983. Lambda charonvectors
(Ch32,33, 34,and35)adaptedforDNAcloningin recombinant hosts.Gene 26:171-179.
30. Lowy, D. R.,S. K. Chattopadhyay, N. M. Teich,W. P.Rowe,
and A. S. Levine. 1974. AKR murine leukemia virus genome:
frequency of sequences in DNA in high, low, and non-virus yielding mouse strains. Proc. Natl. Acad. Sci. USA