Fate of Infecting Simian Virus 40-Deoxyribonucleic Acid in Nonpermissive Cells: Integration into Host Deoxyribonucleic Acid
8
0
0
Full text
Figure

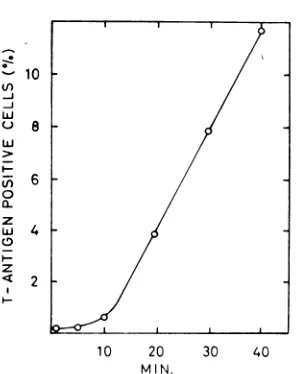

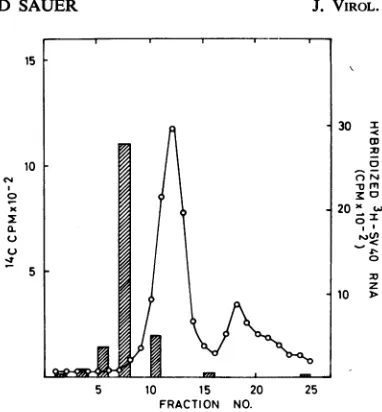
Related documents