Copyright © 1988,American
Society
forMicrobiologyExpression of
Herpes Simplex Virus
Type 1 DNA Polymerase in
Saccharomyces
cerevisiae
and
Detection of Virus-Specific
Enzyme Activity in Cell-Free
Lysates
MARY L. HAFFEY,1*JOHNT. STEVENS,' BRIANJ. TERRY,' DAVID I. DORSKY,2 CLYDE S. CRUMPACKER,2
STEVENM. WIETSTOCK,3 WILLIAM T. RUYECHAN,3 ANDA. KIRK FIELD'
Departmentof Microbial Biochemistryand Genetics, the SquibbInstitutefor Medical Research, Princeton, NewJersey08540-0130'; Division of InfectiousDiseases, Beth Israel Hospital, Boston, Massachusetts 022152;and
Department of Biochemistry, UniformedServices University of the HealthSciences, Bethesda, Maryland2081447993 Received 15 July 1988/Accepted 29 August 1988
The herpes simplex virus type 1 (HSV-1) (strain 17) DNA polymerase gene has been cloned into an Escherichiacoli-yeast shuttlevectorfusedtothegalactokinasegene(GAL-I)promoter.Genes controlled by the GAL-Ipromoterareinducedby galactose, uninduced byraffinose,andrepressed by glucose.Cellextractsfrom
astrain of Saccharomycescerevisiae harboring thisvector(Y-MH202,expressercells)growninthepresenceof
galactose andassayedin highsalt (100 mM ammonium sulfate) containedanovel DNApolymeraseactivity. No
significant high-salt DNA polymerase activity was detected in extracts from expresser cells grown in the
presence of raffinose orinextracts from control cells containing the E. coli-yeast shuttle vectorwithoutthe
HSV-1 DNA polymerasegenegrownin thepresenceof raffinoseorgalactose. Immunoblot analysisofthecell
extracts by usingapolyclonal rabbit antiserum prepared againsta highly purified HSV-1 DNA polymerase
preparation revealed the specific induction of the HSV-1 -140-kilodalton DNA polymerase polypeptide in expresser cellsgrown ingalactose. Extracts from thesamecells growninraffinose orcontrol cellsgrownin
eitherraffinoseorgalactose did notcontain this immunoreactive polypeptide. The high-saltDNA polymerase activity in the extracts from expresser cells grown in galactose was inhibited >90% by either acyclovir triphosphate oraphidicolin, asexpected for HSV-1 DNA polymerase. Inaddition, the high-salt polymerase enzymeactivity could bedepleted fromextractsbyimmunoprecipitationbyusing purified immunoglobulin G from thissamepolyclonalrabbit antiserum. These results demonstratethesuccessful expression of functional HSV-1 DNA polymeraseenzymeinS. cerevisiae.
Theherpes simplex virus type 1 (HSV-1) DNA polymer-ase (pol) has been identified as an approximately
140-kilodalton (kDa) polypeptide which is required for viral
DNAreplication (5,6,26). The viralenzymesharesspecific regions of amino acidsequencehomologywith other
herpes-virusDNApols andwithavarietyofeucaryoticDNA pols,
including the mammalian and yeast alpha pols in regions believed to correspond to substrate and pyrophosphate binding sites (13, 19, 25, 27, 30). Despitethese similaritiesto host cell enzymes, HSV pol activity is readily identified
because oftheionic strength optimum of the viral enzyme
and its much greater
sensitivity
toagents such asphospho-noacetate and acycloguanosine (acyclovir) triphosphate (ACVTP) (7, 12, 23). An improved understanding of the structureof the HSVpolcouldnotonlyaid indesigningnew
antiviral drugs but could also yield information about the
regulation of viral replication, DNA
synthesis,
and cellproliferation.
While mRNA
encoding
HSVpol
appears to be abundant in the infected cell, only low levels ofthepol
protein
are produced (16, 31). Several additionalpolypeptides
appeartoremain associated with the 140-kDa
pol
protein
through
standardpurification
procedures
(26);oneof these polypep-tides is the 65-kDaDNA-binding protein (24).
Therefore,
it isdifficult toobtainlarge
quantities
ofhomogeneous
140-kDaprotein from infected cells for
study.
Further mechanisticstudiesonthe HSV DNA
pol
wouldbegreatly
facilitatedby
*Corresponding author.
the availability of large quantities of soluble enzyme
ex-pressed in aheterologous system such as yeast orbacteria. Previously, Dorsky et al. (9) successfully expressed a
functional form ofthe HSV pol geneinCOS-1 cells fromthe
simian virus 40 early promoter. The low levels of active
enzymeexpressed in that systemweredetectedby comple-mentation of a temperature-sensitive HSV containing a
mutation in pol. Dorsky and Crumpacker (11) have also
reported the expression of HSV pol protein in an in vitro translation system. Inaddition, they reportedtheexpression
ofhighlevels of HSV 140-kDapol
protein
in Escherichiacoli(10);however,theexpressedproteininE. coliwasinsoluble and hadno readilydetectable
enzymatic
activity.Inthisstudy,theHSV pol genewasexpressedin the yeast
Saccharomycescerevisiae.Thisexpressionsystemwas cho-sen for several reasons. First, expression of another HSV enzyme (thymidine
kinase)
in S. cerevisiae had led totheproduction ofa
soluble,
functionalproduct (21).
Second,
it ispossible to construct a
high-copy-number,
regulatable expression vector to attempthigh-level
expression
of the gene. Finally, the yeast system is easy tomanipulate
and should readily allow introduction ofsite-specific
mutations into the pol gene. Such mutations could then be assessedas totheir effects on the enzymeactivity,
its association with other componentsof thereplication
complex,
and its intra-cellularlocalization.MATERIALS AND METHODS
Materials. T4 DNA
ligase,
E. coli DNApol
(Klenow fragment), and restriction endonucleases were purchased 4493on November 10, 2019 by guest
http://jvi.asm.org/
4494 HAFFEY ET AL.
from NewEngland BioLabs orBethesda Research
Labora-tories.
[3H]TTP
(25 Ci/mmol) was obtained fromDupont,
NEN Research Products. Unless otherwise noted,chemi-cals were
purchased
from SigmaChemical Co.Strains and media. The E. coli strains used for this
study
wereHB101(F- hsdS20 [r- m-] supE44 ara-14galK2lacYl
proA2
rpsL20 [Str']
xyl-5 leu mtl-l lambda- recA13)ob-tainedfrom BethesdaResearch Laboratories,andXL1-Blue
(endAl
hsdR17 [rk- Mk+] supE44 thi-J lambda- recAlgyrA96
relAl[Lac-]
[F' proAB lacIq ZAM15 TnWO(Tetr)])
supplied
by
Stratagene Cloning Systems. The yeast strainwas Y294 (MATa leu2-3 leu2-112 ura3-52 trpl Ahis3
Gal'
cir+),
obtainedfrom D. Kirsch (Squibb Institute for MedicalResearch,
Princeton, N.J.), and has been previouslyde-scribed
(2).
Bacteria were grown in Luria broth (22); yeast were grown in minimal media (0.7% yeast nitrogen base[Difco
Laboratories])containing required amino acids (20 to30
jig/ml)
and 2% raffinose or 2%galactose.Plasmids. The plasmids used in this study were YEp 352, a
high-copy-number
E. coli-yeast shuttle vector obtained from D. Kirsch (Squibb Institute for Medical Research,Princeton,
N.J.) which has been previously described (15). Theplasmid YCp125H
was obtained from J. Rine(Univer-sity
ofCalifornia, Berkeley); this plasmid was derived frompBM125
(18)
bytheadditionof aHindIII linker at the uniqueBamHI site. The HSV DNApol gene, derived from HSV-1 strain
17,
was obtainedfromplasmidpD702, a derivative ofpD7
inwhichthesequencesfor the short open reading frameupstream of thepol gene have been deleted (11).
Oligonucleotides.
Two50-basecomplementaryoligonucle-otides containing the CYC-J termination sequence were
synthesized
for this study on an Applied Biosystems model 380BDNAsynthesizer
byusing 3-cyanoethylphosphorami-dite
chemistry.
Theoligonucleotides
were purified bychro-matography
over a Sepharose G50 medium by previouslydescribed
procedures
(Applied Biosystems User Bulletin,issue
13,
p.
17, 1984).Approximately
15p,g
of each purifiedoligonucleotide
was annealed by heating to55°C
for 15min
and
cooling
atroom temperature for 30min before additiontothe
ligation
mixture.Recombinant DNAmethodology. Plasmid DNAs were
pu-rifiedandtransformation of E.coliwas achieved by standard
procedures
(20). Recombinant colonies were selected bycolony
hybridization.
Radiolabeled probes were prepared bynick translation oftheappropriateDNA fragment by using a
commercially
available kit (Bethesda ResearchLaborato-ries)
and 5'[a-32P]dCTP
(3,000 Ci/mmol; Dupont, NENResearchProducts).Yeast
transformation
was performed bythe
spheroplast
method (1). DNA fragments used tocon-struct the expression vectors were isolated from
low-melt-ing-temperature
agarose (Bethesda Research Laboratories)after
gel
electrophoresis. Where appropriate, DNA frag-mentscontainingoverhanging5'endswere filled in by using E. coli DNA pol (Klenow fragment). All ligations were carriedoutby using T4DNAligaseat 13°C overnight.Proteinanalysis. Therecombinantcultures were screened
forthe
production
ofthe140-kDaHSV DNA pol polypeptideby
analysis
of theenzyme extracts describedbelow on a 10%polyacrylamide
gel by the procedure of Heine et al. (14). After electrophoresis, the gels were stained by using acommercially available silver stain kit (Bio-Rad
Laborato-ries).
Proteindeterminations
were performed by using aCoomassie blueprotein assay kit (Bio-Rad).
Preparation
ofantiserum against HSV-1DNA pol. HSV-1 DNApol
was purifiedfromHeLa cells infectedwith strain mP aspreviously
described (26). Sodium dodecylsulfate-polyacrylamide
gel
electrophoresis
analysis
ofthepol
prep-aration used for immunization indicated the presence ofa
140-kDa band
corresponding
tothefull-sizepol
polypeptide,
as well as minorbandsat about 65kDa. Theantiserum was
raised in a New Zealand White rabbit
by
a series of five immunizations with 10,ug
ofpurified
pol
perdose;
the firstinjection
was withcomplete
Freundadjuvant,
and thesub-sequent
injections
were withincomplete
Freundadjuvant.
Purified
immunoglobulins
wereprepared
by
affinity
chroma-tography onprotein A-Sepharose
(Beckman
Instruments,
Inc.).
Immunoblotting. Transfer of
proteins
tonitrocellulosewasdone as described
by
Towbin et al.(28).
Theprocessing
of filters was performed essentiallyby
manufacturer instruc-tionsby
using
the Immun-Blot assay kit(Bio-Rad),
exceptthat bovine serum albumin (BSA) was substituted for
gela-tin. Thedilution of
primary
antibody
was1:200,
and that ofalkaline phosphatase-conjugated anti-rabbit
secondary
anti-bodywas 1:1,000.Preparation of enzymeextracts. Yeast cellsweregrown in 100-ml
overnight
cultures and pelleted at4°C.
Celllysates
wereprepared as described by Johnson etal. (17). In
brief,
cellpelletswere
suspended
in 50 mMTrishydrochloride
(pH
8.0)-50
mMNaCl-1%
dimethyl
sulfoxide-10%glycerol-5
mM
,-mercaptoethanol-10
mM EDTA-2 mMbenzamidine-1 mM phenylmethylsulfonyl fluoride
(PMSF).
Lysis
was achievedbyvortexing
the cells 15to20times for1min
in the presence of an equal volume of acid-washed glass beads.Theextent oflysis wasmonitoredby
light
microscopy. The lysed extracts were transferred to1.5-ml
Microfuge
(Beckman) tubes and centrifuged for 10
min
at 12,000 x g. Extracts were stored at-80°C
in 15%glycerol until use. Thefinal protein concentration of each of the extracts was
between 5 and 6
mg/ml.
Infected cell extracts wereprepared as follows. HeLaS3 cells were infected withHSV-1 Schooleratamultiplicity of infection of10PFU per celland harvested at16 h
postinfec-tion. Cellpellets
(108
cells)were frozen overnightat-80°C,
thawed on ice, suspended in 20 ml of hypotonic solutionA (10 mMK2PO4
[pH 7.0], 10 mMKCI,
1 mM PMSF; 1 mM dithiothreitol[DTT]),
and incubated on ice for 30min
withoccasional vortexing. An equal volume of solution B (0.7 M
K2PO4
[pH 7.0], 28% glycerol, 6 mM DTT, 0.4% NonidetP-40, 1 mM PMSF) was added, and the mixture was
incu-bated on ice for an additional 30
min
with occasionalvortexing. The mixture was centrifuged for 1 h at 125,000 x
g, and Nonidet P-40 was added to a final concentration of 0.4%. Aliquots were stored frozen at
-80°C.
The finalprotein concentration ofthe extract was 5
mg/ml.
DNA
pol
assays. The assay for yeast DNA pol was performed essentially as described by Celniker andCamp-bell (3) but without the addition of ribonucleotide
triphos-phates. Reaction mixtures contained 5
,ul
of enzyme extract (25 to 30,ug
of protein) in 10 mM Tris hydrochloride (pH7.5)-10
mMMgCl2-10
mM(NH4)2SO4-0.1
mM DTT-100,uM
each of dATP, dCTP, and dGTP-100 p.M 3H[TTP] (10 cpm/pmol)-40 p.g of nicked calf thymus DNA per ml-2 mM spermidine in a final volume of 50p.l.
The assay for the HSV-1 DNA pol was performed as follows. The reaction mixture contained5,ul of enzyme extract (25 p.g of protein) 50 mM Tris hydrochloride (pH 8.0); 5mM
MgCl2; 100mM
(NH4)2SO4;
1 mM DTT; 5 p.M each of dATP, dCTP, anddGTP; 5 p.M
[3H]TTP
(200cpm/pmol);
30,ug
of nicked calf thymus DNA per ml; and 100,ug
of BSA per ml in a final volume of 50pl.
All DNA polymerase assays were per-formed by incubating the reactants for 15min
at37°C;
the J.VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
BamHiHindIII EcoRI BamH
EcoRI
G
I~ Ep352 Cp125H.
1. CutwithEcoRt &BamHt
2.Isolate810bpfragment
CutwithEcoRt /
& Barn Hti GAT/ BamHi HindIII
LIGATE EcoRI 5'-AGCTTAGTTAGTTAATTTTTATAGTTATGTTAGTATTAAGAACGTTATT CS
ATCAATCAATTAAAAATATCAATACAATCATAATTCTTGCAATAAATCGA URA3\;\
x ~ ~~ M I,--, I 8pMHIO0
Annealoligos 2
CutwithHind It
SSpI LIGATE
Hind
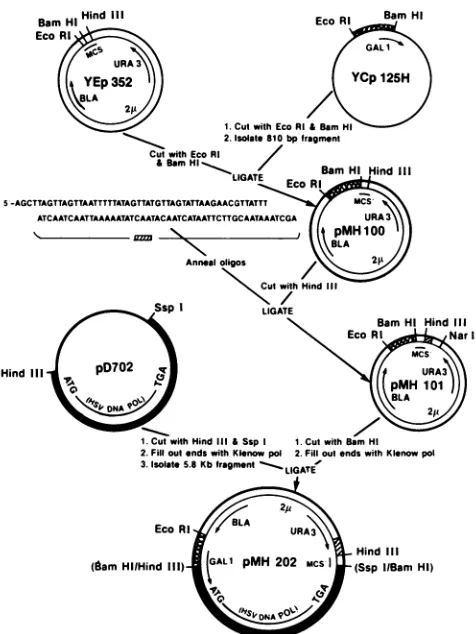
FIG. 1. Scheme for the construction
[image:3.612.63.301.67.384.2]vector, pMH101 and the HSV DNA pMH202.kb, Kilobasepairs.
of the yeast expression pol expression vector,
reactionswere spotted onto glassfiberfilters andquenched by the addition of 10 mM sodium pyrophosphate in 5% trichloroacetic acid. The filters were washed thoroughly
(twice with 1 N HCl and once with95%ethanol), air dried,
and quantitated by scintillation counting. All assays were
conductedin duplicate.
Immunoprecipitation of DNA polactivity. Protein-A bear-ing Staphylococcus aureus(Pansorbin; Behring Diagnostics) waspretreated with BSAtominimizenonspecificbindingby incubating 150
RI
of cell suspension with 0.3 mg ofBSA at 4°C overnight. The following day, cells were pelleted,rinsed, and suspendedin 125
RI
of 25 mMTrishydrochloride(pH 8.0) until ready foruse. Yeast cell enzyme extract (25
,ul)was incubatedwith 50
RI
ofdistilled watercontaining 0,1, or2,ugof purifiedimmunoglobulinG(IgG) for3honice.
Five microliters of the treated staphylococcus suspension wasadded, andincubationwascontinuedonicefor1h. The
mixturewascentrifugedat6,500rpminaMicrofuge, and the
supernatantwas assayed for DNA pol activity as described
above. The results of assays run either without IgG or
without both IgGandStaphylococcus aureuswereaveraged
and usedas acontrolvalue (100%). RESULTS
Construction of theexpressionvector. The construction of theHSV-1 DNApol expression vectorused in this studyis showninFig. 1.Plasmid YEp352is ahigh-copy-number, E.
coli-yeast shuttle vector used for the expression of genes which contain a functional yeast promoter (15). YEp 352 contains the 13-lactamase gene for selective growth in ampi-cillin-containing media in E. coli and the URA-3 gene for selectivegrowthinuracil-deficientmedia in S.cerevisiae.In addition, the vector contains DNA sequences for
autono-mous replicationin bacteria and in S. cerevisiae. We modi-fied thisvector toexpressforeign genes under the controlof
a regulatable yeast promoter, the galactokinase promoter
(GAL-I). The GAL-I promoterhas been describedindetail
elsewhere (18)and has been usedsuccessfully by others to express foreign proteins (18, 29). The use of a regulatable promoter would enable us to grow cells to high densities
before inducing foreign protein expression, thus avoiding
potentialdeleterious effects of foreign proteinaccumulation
oncellgrowth.Expression ofgenes controlledbytheGAL-I promoter isinducedbygalactose,repressedbyglucose,and uninduced by raffinose (2, 18).
YEp 352 DNA was digested with restriction endonu-cleases EcoRI and BamHI to remove part of the
multiple
cloningsite and togenerateoverhanging5'ends.An approx-imately 810-base-pair (bp) fragment containing the GAL-ipromotersequencewasisolatedfrom plasmid YCp125Hby digestion with EcoRI and BamHI. The two DNAs were ligated together toform the plasmid pMH 100.
While someinvestigators (2, 21) have reportedsuccessful expression of foreign proteins in S. cerevisiae without the addition of specific transcription termination sequences,
others (29) have used these sequences to improve
foreign
proteinexpression.Topotentially improvetheexpression offoreigngenes, ayeasttermination sequence was introduced
into thevector. The termination sequence wascomposed of
two synthetic oligonucleotides(Fig. 1)whichanneal toform
a fragment containing the sequence
required
for efficient transcription termination fromthe yeastiso-1-cytochrome
c (CYC-I) gene (32) and translation stop codons in the three possible reading frames. The double-stranded oligonucleo-tide was designed to be directly inserted into theHindIlI
restriction site in pMH100. When inserted in the correct
orientation,theoligonucleotidewould retain theHindIII site
only at the end upstream of the three stop codons. The
proper orientation ofthe insert was established
by
specific
differences in the patterns generated
by
digestion
of recom-binant plasmids with appropriate restriction enzymes(data
notshown).pMH101, the new construction
containing
thecorrectly
oriented
oligonucleotide,
wasdigested
withBamHI,
and theoverhanging 5' ends were filled in with E. coli DNA
pol
(Klenow fragment) to createblunt ends which could accepta DNAfragment
containing
the HSV-1 polgene. The HSVpol gene was mobilized from
pD702,
aplasmid
in which aHindIlI linker was inserted 79
bp
upstream of the3,705-bp
pol openreading frame
following
BAL 31mutagenesis.
This vector contains a deletion of the short upstream openreading frame, 124 bp proximal of the
assigned
translation initiator (13, 27).PlasmidpD702 was
digested
withrestrictionenzymesSspI
andHindIII, and the
overhanging
5' endsweremade bluntasdescribed above. The
5.8-kbp
HindIII-SspI
fragment
con-taining theentireHSV-1polgenewasthen blunt end
ligated
into the modified BamHI site of
pMH101.
Recombinant isolates were selectedby
colony
hybridization,
and the proper orientation of the inserted gene was determinedby
digestion with
appropriate
restriction endonucleases(data
notshown).The final construction
(pMH202)
isdesigned
togenerate
aon November 10, 2019 by guest
http://jvi.asm.org/
4496 HAFFEY ET AL.
transcript,
underinducing conditions,
which contains about 140bases of5'noncoding
sequence before the natural ATGinitiation codon of the HSV-1 pol gene and about 2.1
kilobasesdownstreamof the natural TGA termination codon
before
reaching
theoligonucleotide
sequence fortranscrip-tion terminatranscrip-tion.
Therefore,
the vector should encode anRNAwhich can be translated tothe
complete
HSV-1 140-kDa DNApol polypeptide
aspredicted
from the DNA sequence(13, 27).
Expression
of HSV DNApol
inS. cerevisiae. To determine whether the 140-kDa HSVpol
protein
wasexpressed
and whetherHSV-specific
DNApol
activity
could bedetected in yeast cellscontaining pMH202,
thefollowing
experiments
were
performed.
Yeast cells(Y-294)
weretransformedwithpMH101 (control)
orpMH202 (expresser)
DNA and selectedfor uracil
prototrophy by growth
on minimal media supple-mentedwithleucine, histidine, tryptophan,
andglucose
butlacking
uracil. Onecolony
of each type(designated
Y-MH101 orY-MH202,
respectively)
was selected at randomfor further
analysis. Overnight
cultures were grown from these two strains in the presence of 2% raffinose. On thefollowing day,
cultureswere dividedand cellswerepelleted,
suspended
in fresh mediacontaining
either2% raffinose or 2%galactose,
andagain
grownovernight.
On thefollowing
day,
the fourcultures werepelleted
andlysates
werepre-pared
as described in Materials and Methods. Thelysates
were run on sodium
dodecyl sulfate-polyacrylamide gels,
and theseparated proteins
weretransferredtonitrocellulose forimmunoblotting
with antiserum which weprepared
against
ahighly purified
HSV-1 DNApol
preparation.
The results are shown inFig.
2,
lanes 3 to 6. For comparison, lane 1 contains HSV-1 DNApol
from thehighly purified
preparation
used asimmunogen
to prepare our antiserum and lane 2 contains a crude HSV-1pol
preparation
frominfectedHeLacells
prepared
as acontrol for thepol
enzyme assays.Analysis
of the yeastlysates clearly
shows thespecific
induction of an immunoreactivepolypeptide
ofapproximately
140 kDaonly
in Y-MH202 cells grown underinducing
conditions(lane 6).
Inaddition,
theyeast-expressed
polypeptide
exhibitsthesameelectrophoretic
mobility
asthe HSV-1pol
from crude infected-cell extracts. It should benoted that the
pol polypeptide
in lane 1(derived
fromstrainHSV-1
mP)
has aslightly
slowerelectrophoretic mobility
than thepol
in lane 2(derived
from HSV-1Schooler).
Thisdifferencein
electrophoretic
mobility
may be dueto incon-sistencies betweenthetwovirusstrains with respecttoDNA sequence ortranscriptional
orposttranslational
modifica-tion. Inaddition,
it should be notedthat ourantiserum alsoreacts with
HSV-specific protein(s)
of about 65 kDa inmolecular mass. In the yeast cell extracts, this 65-kDa
protein
mostlikely
represents adegradation product
of the140-kDaHSV DNA
pol polypeptide.
Thegeneration
of such adegradation
product during purification
of pol fromin-fectedcells has been noted
previously
(W.T. Ruyechanand C. S.Crumpacker,
unpublished observations).
Alterna-tively,
it ispossible
that this protein may result fromalterations in the
transcription
or translation of mRNAencoding
the HSV pol gene. The 65-kDa protein band inlanes1 and
2,
however,
may also contain the 65-kDa HSVDNA-binding
protein
which is often foundtightlyassociatedwith 140-kDa DNA
pol
polypeptide through purificationprocedures
(24).
The
lysates
were alsoanalyzed
for DNA pol enzymeactivity.
Each of the four yeast cellextractswasassayedfor DNApol activity
undertwo sets ofconditions. The yeast DNApol
assayconditionsweredesignedtodetect thealphaMW
YEAST HSV-1 iNF. CONTROL HELA CELLS RAF GAL
1 2 3 4
YEAST EXPRESSOR
RAF GAL
5 6
200.0
97.5
66.2
45.0
FIG. 2. Immunoblot of the HSV DNA pol expressed in S.
cerevisiae as described in Materials and Methods. Molecular weights (MW; 103)arereported alongthe leftmargin.Lanes 1and2 containHSV DNApolfrom infected HeLa cells: -9jigofpurified
enzyme preparation preparedfrom HSV-1 (mP)-infectedcellsand -60
jig
of a crude enzyme preparation from HSV-1 (Schooler)-infectedcells,respectively.Lanes 3 and 4containyeastcelllysates preparedfrom control cells(Y-MH101)growninraffinose(RAF)or galactose (GAL), respectively. Lanes 5 and 6 contain extracts prepared from the expresserstrain (Y-MH202) grown in RAFor GAL, respectively.Lanes3to6contain 50to60 ,ugeach.<,-140 kDaHSVpol; 4, -65kDaprotein(s).polwhich should be present in all
lysates.
The HSV DNApol assay conditions were chosen to detect HSV-specific DNA pol
activity
which is enhancedby high
salt(100 mM ammoniumsulfate) (23).
In contrast, the yeastalpha DNApol is inhibited under these salt conditions (3). Table 1 reveals thatonly Y-MH202, HSV DNApol expressercells, grownunder
inducing
conditions(galactose) produce signif-icant amounts ofhigh-salt
DNA pol activity. In addition,Table 2 reveals that this novel high-salt activity in the Y-MH202 yeast cell extracts is inhibited by ACVTP and
aphidicolin,
both potent andwell-characterized inhibitorsof the HSVDNApolenzyme(8, 16).Table 2 also reveals that thelow-saltDNApolactivity in all of theextractstested is sensitivetoaphidicolin,
which ischaracteristic of alphapots(3, 17)
and isnot sensitive to ACVTPat the concentration tested.Amore
complete comparison
of the inhibitionofACVTP of HSV polproduced
in S. cerevisiae versus HSV polproduced
in HeLa cells is shown in Table 3. These resultsJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.319.557.74.398.2]EXPRESSION OF HSV POL IN S. CEREVISIAE 4497
TABLE 1. DNApolactivity
Result of DNA pol assaya Extract andgrowth condition HSV S. cerevisiae
Ib Ilb I II
HSV-1-infectedHeLa cells 6.90 5.78 129.1 118.6 Y-MH101 yeast control cells
Raffinose 0.26 0.18 139.6 113.2
Galactose 0.11 0.87 95.6 125.4
Y-MH202yeastexpresser cells
Raffinose 0c 0.40 129.1 139.6
Galactose 6.69 5.53 132.4 129.6
a Pol activity is expressed as picomoles of [3H]TTP incorporated per reaction; assay conditions are as described in Materials and Methods. All values are the average of duplicate assays.
bI andII areseparate experiments.
cValue belowbackground.
reveal that the HSV polenzyme from both sources behaves in asimilarfashion; the 50% inhibitory dose is at a
concen-tration of between 1.15 and 2.3 ,uM ACVTP.
To further demonstrate the HSV-specific nature of this
high-salt DNA pol activity, we incubated the cell extracts
with purified IgG from our rabbit anti-pol serum or with
control IgG from a nonimmunized rabbit. Immune
com-plexeswereremovedwith proteinA-Staphylococcus aureus
by centrifugation, and then residualDNApolactivity from
the supernatants was assayed. Two yeast extracts were
chosen forcomparison; Y-MH101 (galactose-induced)
con-trol cell extract was assayed under yeast DNA pol assay
conditions to monitor effects of IgG on alpha DNA pol activity, and Y-MH202 (galactose-induced) expresser ex-tractwas assayed under HSVDNApolconditionsto
mon-itor effects ofthe IgGonthehigh-salt pol activity. Figure 3 reveals that the high-salt activity in galactose-induced
Y-MH202 expresser cells can be reduced >50% by
immuno-precipitation by using IgG derived from our antiserum to HSV-1 DNA pol, whilethe alpha pol activity in the galac-tose-induced Y-MH101 control cell extract is only
slightly
reducedby reactionwith the immuneIgG.DNApolactivity in bothextractsisonlyslightly affected by reactionwith the
control(normalrabbit) IgG.
These results indicate the successful
expression
of anactive form of the HSV-1 DNA
pol
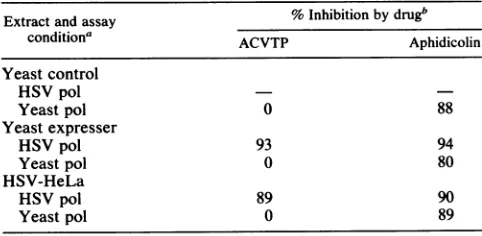
inS. cerevisiae.TABLE 2. Inhibition ofDNApolactivityby ACVTP andaphidicolin
Extractandassay % Inhibition by drugb
conditiona ACVTP Aphidicolin
Yeastcontrol HSVpol
Yeastpol 0 88
Yeast expresser
HSVpol 93 94
Yeastpol 0 80
HSV-HeLa
HSVpol 89 90
Yeastpol 0 89
aExtracts wereprepared asdescribed in Materials and Methods. Yeast
control(Y-MH101) andexpresser(Y-MH202) cell extracts were prepared
from culturesgrown in thepresence ofgalactose. Assayconditionswere as
described in Materials and Methods.
bACVTP andaphidicolin were present in reactions at 23and 5.3 FtM, respectively. 0, Relative activity was -1.0; -, value is not statistically
[image:5.612.64.308.76.197.2]significantbecauseoflowpolactivity.
TABLE 3. Inhibitionof HSVpolby increasing amountsofACVTP
Concn (>M) of %HSV pol activity inextracta
ACVTP HeLa Yeast
0 100 100
0.023 78 93
0.115 91 90
0.23 82 81
1.15 60 59
2.3 36 38
11.5 17 6
23.0 6 10
a HSV assay conditions were as described in Materials and Methods. All values are the average of duplicate assays. Extracts were prepared as described in Materials and Methods. Yeast extracts were prepared from cultures of expresser (Y-MH202) cell extracts grown in the presence of galactose. HeLa extracts were prepared from HSV-1-infected cells.
DISCUSSION
Thisreport detailsthe successful expression of an active
eucaryoticDNApol in a heterologous host. Inthis study,the HSV pol open reading frame was inserted into a modified yeastexpressionplasmidsuch that the HSV DNApolcould
beinducibly expressedinyeast cells under thecontrol ofthe
GAL-I promoter. The enzyme expressed in S. cerevisiae
possesses the saltresistanceandinhibitor sensitivityprofiles ofthe authentic pol enzyme isolated from HSV-1-infected cells.Furthermore, theexpressed productpossesses HSV-1
polantigenic determinantsasshownby immunoblotting and byimmunoprecipitation of pol enzymatic activity fromyeast
cell extracts.
Expression of
the
HSV-1 DNA pol enzyme in quantity from a heterologous system will allow further mechanistic studiesaswellascharacterization ofcomplexesbetween theHSV-1 DNA pol and the other components of the DNA
replication machinery of HSV (4).Eventhoughthe amounts
of HSV-1 DNA pol obtained from infected cells and the yeast cells appear to be essentially the same when this constructisused,theabilitytogrow
large
quantities
ofyeast-1 00 - ci pol
\
~~~~~~~~~HSV
pol80 _apol
60
0
U 40 ~* HSV pol
|
-A control IgG
20
~A
* immune IgG 02
IgG
(Qsg)
FIG. 3. DepletionofHSV DNApol activityfromyeast
lysates
byimmunoprecipitationwithpurified immunoglobulinsfroma rab-bit antiserum against HSV-1 DNA pol (immune IgG) or normal rabbitserum(controlIgG).Yeastextractsfrom Y-MH202grown in galactosewereused fortheHSVpolenzyme assay. Extracts from Y-MH101 grown ingalactosewere used forthe alpha polenzyme assay. Allvaluesarethe averages ofduplicateassays.
VOL.62,1988
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.321.561.95.202.2] [image:5.612.329.564.500.659.2] [image:5.612.65.306.553.671.2]4498 HAFFEY ET AL.
canpotentially result in the availability of large
quantities
of enzyme. Becausetheproductisexpressedfromaplasmid, it should bepossibletoengineeravarietyof mutations into thegene to test their effects on such enzyme properties as substrate specificity and sensitivity to inhibitors. Further refinements ofthis expression system should also allow the bulkpurification ofahomogeneous product
leading
tophys-ical-chemical studies designed to elucidate the functional domains and three dimensional structure of the HSV pol
enzyme.
ACKNOWLEDGMENTS
We thank Donald Kirsch, Margaret Lai, and Rose Kelly for invaluableadvice and instructionin themolecular biologyofyeast. Weacknowledgetheexcellenttechnicalassistance ofJeanLynchin thepreparation ofpurifiedimmunoglobulins and KathrynMazinain the preparation of synthetic oligonucleotides. For their careful preparation of the manuscript and for the illustrations, we thank
Doris GardnerandJoseAlcantara, respectively.
Part ofthe work done by W.T.R. wassupported by PublicHealth
Servicegrant A122468 from the National Institute of Allergy and Infectious Disease.
LITERATURE CITED
1. Beggs, J. D. 1978. Transformation of yeast by a replicating
hybrid plasmid.Nature (London) 275:104-109.
2. Brugge, J. S., G. Jarosik, J. Andersen, A. Queral-Lustig, M.
Fedor-Chaiken, andJ.R. Broach. 1987. Expression of Rous
sarcomavirus transforming proteinpp6Ov-src in Saccharomyces
cerevisiae cells. Mol.Cell. Biol. 7:2180-2187.
3. Celniker, S. E.,and J. L. Campbell. 1982. Yeast DNA replica-tion in vitro: initiation and elongation events mimic in vivo
processes. Cell 31:201-213.
4. Challberg,M. D. 1986. A methodfor identifying theviralgenes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA83:9094-9098.
5. Chartrand, P., C. S. Crumpacker, P. A. Schaffer, and N. M.
Wilkie. 1980. Physical and genetic analysis of the herpes sim-plexDNA polymerase locus. Virology 103:311-326.
6. Coen,D. M.,D. P. Aschman, P. T. Gelep, M. J. Retondo, S. K. Weller,and P. A. Schaffer. 1984. Fine mapping and molecular cloningof mutationsonthe herpessimplex virusDNA
polymer-aselocus.J. Virol. 49:236-247.
7. Derse, D.,K. F. Bastow,andY.-C. Cheng. 1982. Characteriza-tionofthe DNA polymerases induced by a group of herpes
simplextype 1 variants selected for growth in the presence of
phosphonoformicacid. J. Biol. Chem. 257:10251-10260. 8. Dicioccio, R. A., K. Chadha, and B. I. S. Srivastava. 1980.
Inhibition of herpes simplex virus-induced DNA polymerase, cellularDNA polymerasea, and virus production by
aphidico-lin. Biochem. Biophys. Acta609:224-231.
9. Dorsky, D., P. Chatis, and C. Crumpacker. 1987. Functional
expression of a cloned herpes simplex virus type 1 DNA polymerasegene. J. Virol. 61:1703-1707.
10. Dorsky, D. I., and C. S. Crumpacker. 1987. Cloning and
expressionof theherpes simplex virustype1 DNA polymerase inbacteria. AntiviralRes. 9:92.
11. Dorsky, D. I., and C. S. Crumpacker. 1988. Expression of
herpessimplex virus type 1 DNA polymerase gene by in vitro
translationand effectsofgenedeletionsonactivity. J. Virol.62:
3224-3232.
12. Furman,P.A., M.H. St. Clair,J. A. Fyfe, J. L. Rideout, P. M.
Keller,and G. B.Elion. 1979. Inhibition of herpes simplex virus
inducedDNA polymeraseactivityand viral DNA replication by
9-(2-hydroxyethoxymethyl)guanine andits triphosphate. J.
Vi-rol. 32:72-77.
13. Gibbs, J. S., H. C. Chiou, J. D. Hall, D. W. Mount, M. J.
Retondo, S. K. Weller, and D. M. Coen. 1985. Sequence and mappinganalysis of the herpes simplex virus DNApolymerase genepredictaC-terminal substratebindingdomain. Proc.Natl. Acad. Sci. USA 82:7969-7973.
14. Heine, J. W., R. W. Honess,E.
Cassai,
and B. Roizman. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptidesof type 1 strains. J. Virol.14:640-651.
15. Hill, J. E., A. M. Myers, T.J. Koerner, and A. Tzagoloff. 1986. Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 2:163-167.
16. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C. Glorioso, and M. Levine. 1984. Transcriptional and genetic
analyses of the herpes simplex virus type1genome: coordinates 0.29 to 0.45. J. Virol. 49:947-959.
17. Johnson, L. M., M. Snyder,L. M. S. Chang,R. W. Davis, and J. L. Campbell. 1985.Isolation of the geneencodingyeastDNA polymerase
I.
Cell 43:369-377.18. Johnston, M., andR. W. Davis. 1984. Sequences that regulate the divergent
GAL1-GALIO
promoter in Saccharomyces cere-visiae. Mol. Cell. Biol. 4:1440-1448.19. Larder, B. A., S. D. Kemp, and G. Darby. 1987. Related functional domains in virus DNA polymerases.EMBOJ. 6:169-175.
20. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold SpringHarbor Laboratory, Cold SpringHarbor, N.Y.
21. McNeil, J. B., and J. D. Friesen. 1981. Expression of the herpes simplex virus thymidine kinase gene inSaccharomyces cerevi-siae. Mol. Gen. Genet. 184:386-393.
22. Miller, J. H. 1972. Experiments in molecular genetics, p. 433. Cold Spring Harbor Laboratory, Cold SpringHarbor, N.Y. 23. Ostrander, M., and Y.-C. Cheng. 1980. Properties of herpes
simplex virus type 1 and type 2 DNA polymerase. Biochem. Biophys. Acta 609:232-245.
24. Parris, D. S., A. Cross, L. Haarr, A. Orr, M. C. Frame, M. Murphy, D. J. McGeoch, and H. S. Marsden. 1988. Identifica-tion of the gene encoding the 65-kilodalton DNA-binding protein ofherpes simplex virus type 1. J. Virol. 62:818-825.
25. Pizzagalli,A., P. Valsasnini, P. Plevani, and G. Lucchini. 1988. DNA polymerase I gene of Saccharomyces cerevisiae: nucleo-tide sequence, mapping of a temperature sensitive mutation, andproteinhomology with other DNA
polymerases.
Proc. Natl. Acad. Sci. USA 85:3772-3776.26. Powell, K. L., and D. J. M. Purifoy. 1977. Nonstructural proteins of herpes simplex virus. I. Purification of the induced DNA polymerase. J. Virol. 24:618-626.
27. Quinn, J. P., and D. McGeoch. 1985. DNA sequence of the region in the genome of herpes simplex virus type 1 containing genes for DNA polymerase and the major DNA binding protein.
NucleicAcids Res. 13:8143-8163.
28. Towbin, H., T. Staehelin, and J.
Gordon.
1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA76:4350-4354.29. Wen, D., and M. J. Schlesinger. 1986. Regulated expression of Sindbisandvesticular stomatitis virus glycoproteins in Saccha-romyces cerevisiae. Proc. Natl. Acad. Sci. USA 83:3639-3643. 30. Wong, S. W., A. F. Wahl, P.-M. Yuan, N.Arai,B. E. Pearson, K.-I. Arai, D. Korn, M. W. Hunkapiller, and T. S.-F. Wang. 1988. HumanDNA polymerase a gene expression iscell prolif-eration dependent and its primary structure is similar to both procaryotic and eucaryotic replicative polymerases. EMBO J. 7:37-47.
31. Yager, D. R., and D. M. Coen. 1988. Analysis of the transcript of the herpes simplex virus DNA polymerase gene provides evidence that polymerase expression is inefficient at the level of translation. J. Virol. 62:2007-2015.
32. Zaret, K. S., and F. Sherman. 1982. DNA sequence required for efficient transcription termination in
yeast.
Cell 28:563-573.J. VIROL.