0022-538X/92/020780-10$02.00/0
Copyright© 1992, American Society forMicrobiology
Second-Site Homologous
Recombination in
Epstein-Barr
Virus:
Insertion
of
Type
1
EBNA 3 Genes in Place of
Type
2
Has No
Effect
on
In Vitro Infection
BLAKE TOMKINSON* ANDELLIOTT KIEFF
Departments of Microbiologyand Molecular Genetics andof Medicine, Harvard University, 75FrancisStreet, Boston, Massachusetts 02115
Received 23 August1991/Accepted 17October 1991
Thisstudy was undertaken to develop ageneralstrategy fortheintroduction of mutations intospecificsites intheEpstein-Barr virus (EBV) genome. Previous approaches were limited by the need for physical linkage of thetransfected EBV DNA fragment to apositive selection marker. In ourexperiments, a positive selection marker was introduced into one site in the EBV genomeand adistant,nonlinked, markerwasintroduced into anothersite. Each marker was on alarge EBV DNAfragmentand wasinserted into thegenomebytransfection into cellscarrying a residentEBVgenome. The residentEBVgenomewassimultaneouslyinducedtoreplicate
by using a cotransfectedexpression plasmid for the EBVimmediate-early transactivator, Z (J. Countryman, H.Jenson, R. Seibl, H. Wolf, and G. Miller, J. Virol. 61:3672-3679, 1987; G. Miller, M. Rabson, and L. Heston, J. Virol. 50:174-182, 1984). Eleven percent of the resultant EBV genomes which incorporated the positive selection marker also incorporated the nonlinked marker. Both markers uniformly targeted the homologous EBV genome site. Inthis way novel EBV recombinantswereconstructed in which the EBVtype 1EBNA3A, EBV type 1EBNA3A and3B,or EBVtype 1 EBNA3A, 3B,and 3Cgeneswereintroduced into a largely type 2 EBV genome, replacing the corresponding type 2 gene(s). No difference was observed in primary B-lymphocyte growthtransformation, in latent EBV gene expression, orin spontaneouslytic EBV geneexpression. These new recombinants should be useful forongoing analysesof the typespecificityof the immuneresponse.
Moleculargenetic studies oftheEpstein-Barr virus (EBV)
genome have been limitedbythein vitrohost range restric-tion toprimateB lymphocytes and thepoorpermissivity of
Blymphocytes forvirusreplication.Mostmoleculargenetic approachesusedfor otherherpesviruses
require
largequan-tities of virus or of intact virus DNA as well as in vitro replication of progeny virus so that recombinants can be
selected, identified, or cloned. Two approaches have
re-cently been used to generate recombinant EBVs. Both approaches utilize and extend the principle of
high-fre-quencyhomologousrecombination betweenreplicating
her-pesvirus DNA and homologous transfected DNA (28). The
first strategy uses a transformation-defective (26), deleted (13) virus genome, resident in the P3HR-1 Burkitt's lym-phoma cell line (12, 26a), as one parent and a transfected
recombinant EBVDNAfragment which spans the deletion as the other parent (6, 7, 11). When virus replication is
induced in these transfected cells, largequantities of paren-tal P3HR-1 EBV are produced along with up to several
hundred homologously recombined EBV genomes (6). The recombinant genomes are usually restored for the deleted
DNA and are therefore uniquely able to cause growth
transformationorimmortalization of primary B lymphocytes
(6, 7, 11,31). Clonesof transformed B lymphocytes carrying asingle recombinantEBV genome canthereby be recovered
(6, 7, 11, 17, 18, 31, 32), characterized (6, 7, 11, 17, 18, 32), and in mostinstancespassaged to otherprimaryB lympho-cytes following induction of virus replication (17, 18, 32).
This strategy has been developed and exploited to create
recombinant EBVs with mutations in or around the EBV
*Corresponding author.
nuclearantigen2 (EBNA 2)orEBNA LPgenes, whichare
criticaltoinfectedB-lymphocyteoutgrowthandaredeleted
fromP3HR-1virus(6,7, 11,17). The strategy has also been used to introduce mutations into adjacent EBV genes,
ex-ploiting restoration of growth transformation as a linked positive selection marker (18, 32). The approach islimited to genes which can be physically linked to the deleted DNA segmentwhich is essentialtotherestoration oftheability of P3HR-1 to transform primary B lymphocytes. The second
strategy uses a cell line harboring a nondefective EBV
genome and transfection ofa recombinant EBV DNA
car-rying a linked positive selection marker (37). This latter strategywouldbeapplicabletoany EBV gene aslongasthe
neighborhood ofthegene wascompatible withafunctioning positive selection marker. In fact, mutations could, in the-ory, be made in essential transforming genes since the recombinant EBV genomes could be selected,
character-ized, and recovered from non-EBV-infected B-lymphoma cells (37). A weakness of this latter strategy is that the inserted positive selection marker not only must be
func-tional inthe neighborhood ofthe EBV gene, but also must notaffect EBVinfection.
We now demonstrate that an EBV genome which has
recombined withan EBVDNA fragment containing a posi-tive selection marker will also frequently recombine with a
second, nonlinkedcotransfected EBV DNA fragment. Res-toration of the P3HR-1 EBV-transforming ability was used as the nonlinked positive selection marker. The rate of
cotransfer ofacotransfected, nonlinked second marker was
investigated, and the resulting recombinant EBVs were characterized. The surprisingly high frequency with which second-site homologous recombination occurs remarkably 780
on November 10, 2019 by guest
http://jvi.asm.org/
extends the ease with which mutations can be made in the
EBVgenome.
The nonlinked second fragment which was investigated contains the EBNA 3A, 3B, and 3C genes. This fragment
waschosenbecauseEBNA 3A, 3B, and 3C and EBNA 2 are the genes which differ most extensively between the two naturally occurring andbiologically different EBV types (1,
9,29). Type 1 EBVstrains transform primary B lymphocytes
with a higher efficiency than type 2 strains, and the type 1-transformed lymphoblastoid cell lines (LCLs) are more abletosustaintheirgrowthatlow cell concentrations (7, 27). Otherwise isogeneic recombinant EBVs carrying type 1 EBNA2 in atype 2genome are similar in their effects on cell
growth to type 1 EBVs, indicating that EBNA 2 is the dominantdeterminantof thetype-specific growth effects (7). Although EBNA 2 is the dominant determinant of type-specific growth differences, the contribution of EBNA 3A,
3B, or 3C to thetype-specific growth properties could not be previously determined. Isogeneic recombinants carrying
type 1 or 2 EBNA 3A, 3B, or 3C would also be useful for dissecting the relative importance of type-specific
differ-ences in T-cell immune responses in EBV-infected people. Recent data indicate that EBNA 3A, 3C, and 2 contain important T-cell epitopes (4, 5, 23)and thatthe epitopes are frequently type specific (5, 23). These analyses would be facilitated by having identical cells infected with isogenic
EBVsdifferingonly in their EBNA 2 orEBNA 3 genes. MATERIALSANDMETHODS
Plasmid and cosmidconstruction. Plasmid pSVNaeI Z and
the
EcoRI
Acosmidhavebeendescribedpreviously (18, 32).The Sall E and Cfragmentsof the B95-8 EBVtype 1 DNA
werecloned from apartialSalIdigest intopDVcosA2 (14) by using an in vitro packaging extract and Escherichia coli PLK-A (Stratagene Corp.). pDVSalI-E/C (pDVSECWT) contains42 kb of EBV DNA frompositions 63201 to 105296
(3).
Cells and cellculture. TheHH514-16 subclone of P3HR-1 (26a)(agift fromGeorgeMiller, YaleUniversity) containsa type 2 EBVgenomedeleted for the EBNA 2 gene and part of the EBNA LP gene (13, 26), rendering it nontransform-ing (26). BJA-B is an EBV-negative B-lymphoma cell line (20). Louckes is an EBV-negative Burkitt's lymphomacell
line (35). Human mononuclear cells used forestablishment of normal and mutant LCLs were obtained from normal
seronegative and seropositive donors. Enriched B-cell
pop-ulationswereobtainedbydepletingTcells with
S-(2-amino-ethyl)isothiuronium
bromide (Sigma)-treated sheeperythro-cytes as described previously (18). All cultures were
maintained in RPMI 1640 medium supplemented with 10% inactivated fetal bovine serum, glutamine, and 10
jig
ofgentamicin perml.
Cosmid transfection. P3HR-1 c16 cells were transfected with EBV cosmid cloned DNA after release of the EBV
DNA from vector DNA; pDVSECWT was digested with
Sall to liberate
SalI
E and C DNA fragments. pSVNaeIBamHI-Z was cotransfected to induce lytic replication (8, 21).P3HR-1 c16 cells (107 cells),harvestedduringlog-phase growth, were resuspended in 400 ,ul ofcomplete medium
containing 25 mg of pSVNaeI Z plasmid or 25 mg of
pSVNaeI
Z plasmid plus 10jig
of EcoRI-A or 25jig
ofpSVNaeI
Z plus 10,ugofEcoRI-Aplus 50pug
ofSaII-EIC.Resuspended cells were transferred to an electroporation
cuvette (0.4-cm gap;Bio-Rad) and, following a10-min
incu-bation at 25°C, pulsed with 200 V at 960-,uF capacitance.
Cells were immediately diluted into 20 ml of complete medium supplemented with 10
,ug
of gentamicin per ml. Lytic EBV replication in LCLs was activated for virus passage by transfection with 25jig
of pSVNaeI Z.Primary and secondary infections. Primary human B lym-phocytes were infected with
0.45-,uM
filteredsupernatants
from transfected P3HR-1 c16 cells obtained 3 days after transfection. Intracellular virus was released into the me-dium by three cycles of freezing and thawing. The superna-tant was filtered and used directly or first concentrated by centrifugation at 8,800 x g for 2 h. Virus was incubated with T-cell-depleted human mononuclear
cells
for 2 h at37°C.
Infected cells were resuspended in complete medium plus gentamicin to a concentration of 3.3 x
105
cells per ml, and150
,ul
(5 x 104 cells) was distributed into wells of a 96-microwell plate. The cultures were fed at 14 days post-plating with 100,ul
of medium. LCLs were macroscopically visible 3 to 5 weeks after plating. LCL recombinant virus was passaged by cocultivation with human T-cell-depleted mononuclear cells. Three days afterpSVNaeI
Ztransfec-tion, 5 x 104 irradiated (8,800 rads) LCLs were cocultivated
with 5 x 104 T-cell-depleted mononuclear cells in 150 pl of complete medium in a 96-microwell plate. Cultures were fed
at 14 days with 100
,ul
of medium. Transformed-cell out-growth was visible 3 to 5 weeks after plating. As controls, 5 x104
irradiated LCLs were plated without Blymphocytes.
PCRanalysis. (i) Primers. Oligonucleotide primers which distinguish among type 1 EBNA 3A (T1EBNA 3A), EBNA
3B (T1EBNA 3B), and type 2 EBNA 3A (T2EBNA 3A), EBNA 3B (T2EBNA 3B) have been described previously
(29). Aset of EBNA 3C (E3C-3') primers was synthesized to
a 3' region in EBNA 3C which would amplify a distinct fragment fortype 1 (158 bp) and type 2 (179 bp) EBV DNA
as aresult of a21-bp deletion in type 1 EBNA 3C DNA (29). E3C-3'-PF primer (CACATCTGCAATCGGAGACA) corre-sponds to nucleotides 100996 to 101015, and E3C-3'-PR primer
(GCTATCTTGTAACGGCATAG)
corresponds to nucleotides 101134 to 101153 (see Fig. 1 for location ofpolymerase chain reaction [PCR] fragments) (3).
(ii) Reaction. Genomic DNA was prepared from
approxi-mately 5 x 104 to 1 x
105
cells. Following centrifugation toremove medium, cells were resuspended in 0.2x phosphate-buffered saline, boiled for 10min, and mixed with 0.1 volume of 10-mg/ml proteinase K (Sigma), and the mixture was
incubated for 30min at 55°C. Proteinase K was inactivated
by incubationat 95°C for 20min.
PCRanalysis wasperformed by using Perkin-Elmer
ther-mal cycler with 10 to 20
pul
of DNA in a 50-pdl reaction. PCR-amplified DNA was analyzed by electrophoresis with 1%SeaKem ME-2% NuSieve agarose (FMC Corp.) gels andvisualized by staining with ethidium bromide.
Immunoblotting and Southern hybridization. Latent or lytic EBV protein expression was analyzed by immunoblot
following electrophoresis in 7% polyacrylamide gels with EBV immune human serum of previously characterized
specificities (2, 25, 32). LMP 1 was detected with the
monoclonal antibody S12 (16).
For Southern blot hybridization, 15 ,ugof cell DNA was digested with BclI restriction enzyme in a
200-pul
volume (15). Following complete digestion, DNA was precipitated and resuspended in 40 pLl of loading buffer. The DNA was size fractionated by electrophoresis in a 0.4% agarose gel and transferred to an activated nylon membrane (Gene-Screen Plus; New England Nuclear). The filter was hybrid-ized with a 32P-labeled EBNA 3A cDNA probe. Southern blot hybridization with BamHI was done similarly, excepton November 10, 2019 by guest
http://jvi.asm.org/
20 40 60 80 100
N
C
WWWWWWWWWWY H F QUPOaM
S L EIziRK
h el,2,3 g f
EBNA 2
B G DcbTXVdI A Nhet
EBNA 3A, 3B, 3C
P3HR-1 DELETION
Eco A (50-60kb)
(69,119)
Sal E/C (42.1Jcb
(105,296)
98 100 102kb
I
(63,201)
94 96
EBNA 3A -(E3A)
EBNA 3B
-(E3B)
EDNA 3C
(E3C-3 ')
--lxxxxxxxxx'-XXI
--FIG. 1. (A) Schematic representationof the EBV genome with the location of EBNA 2 and the EBNA 3genesandP3HR-1 deletion
indicated. Also shown are the cloned EBV DNAfragments which wereusedto constructthepDVEcoRI A and pDVSEC cosmids. (B)
Schematicrepresentationof the BamHI-E region indicatingtheexactlocationof the EBNA 3A, 3B,and 3Cgenesand theEBNA 3 PCR fragments.
that fragments were separated on a 0.8% agarose gel and hybridized with a32P-labeled BamHI-HDNA probe.
In situlysing-gel analysis (10)wasperformed by loading2
x 106cells intoanin situlysing-gel wellcontainingagarose
with 1% sodium dodecyl sulfate andproteinase Kas
previ-ously described (37). In situ cell lysis was accomplished by
running the gel at 0.5 V/cm (15 V) for 4 h, and then the episomal and linear EBV DNAwasfractionatedby
increas-ing the voltageto 3.3 V/cm(100 V) for18 h. The DNAwas
transferredtoanactivated-nylon membrane and probed with
a32P-labeled EBVSall C DNA fragment.
RESULTS
Frequency of second-site recombination. To generate
sec-ond-siterecombinantscontaining thetype 1EBNA3genes,
P3HR-1cellswerecotransfected with the EBV DNA EcoRI
Afragment which spans the P3HR-1 deletion (Fig. 1), with
thetype 1 EBV DNA Sall C fragment which contains the
exonsencoding EBNA 3A, 3B, and 3C (Fig. 1), and with the
pSVNaeI Zexpression plasmidto induce replication of the endogenous P3HR-1 genome. In these experiments,
trans-fection with the Z expression plasmid resulted in EBV replication in approximately 1to10%of theP3HR-1 cells,as
measuredby expression of the major virion envelope glyco-protein, gp350 (data notshown). Theresultant virus stocks from 107 transfected P3HR-1 cells consisting of approxi-mately 108
(107
to 109) nontransforming parental P3HR-1 virus, based on a free-virus yield of 102 to103
EBV perpermissively infected cell (26a) and approximately 102
trans-forming P3HR-1 generatedas a consequence of
recombina-tion with EcoRI-A(6),wereusedtoinfect 1.5 x 107primary
human B lymphocytes so that transforming recombinants
could be isolated by their ability to cause long-term LCL
outgrowth (6, 7, 11). The primary B lymphocytes were
platedinto 300 wellssothatanaveragewellwouldhave less than1.0transforming recombinantvirus. At thismultiplicity ofinfection, theresulting LCLswouldprobablybe infected withasingle transforming recombinant virus, althoughthere
was a high probability that the LCLs would be initially
coinfected with parental, nonrecombinant P3HR-1 virus. The resulting LCLs were analyzed by PCR with primers which would detect whether the viralgenomesinthe
trans-formed cells had also undergone recombination with the transfected type 1 EBV DNA Sall C fragment and now
included type 1 EBNA 3A, 3B, or 3C genes. Since the parental P3HR-1genomeis type2 and thetransfectedtype 1 Sall C fragment is approximately 10% divergentin nucleo-tide sequence within the EBNA 3 genes, recombinationat
these loci is readily detected by type-specific PCR for each ofthe EBNA 3genes(29).
The results were remarkably consistent among
experi-mentsinwhichapproximately 89% of the transformed LCLs had only P3HR-1-derived type 2 EBNA 3 genes, whereas
approximately 11%wererecombinant and hadtype1EBNA 3A, 3B, or 3C (Table 1). Of the LCLs infected with virus carrying type 1 EBNA 3A, 3B, or3C, approximately 70%
were coinfected with virus carryingtype 2 EBNA 3 genes
(Table 1). Examples of the results showing evidence for infection withonly atype 1 EBNA 3A, 3B,or3C
recombi-nantvirus andnotwithaparental P3HR-1 virusaregiven in
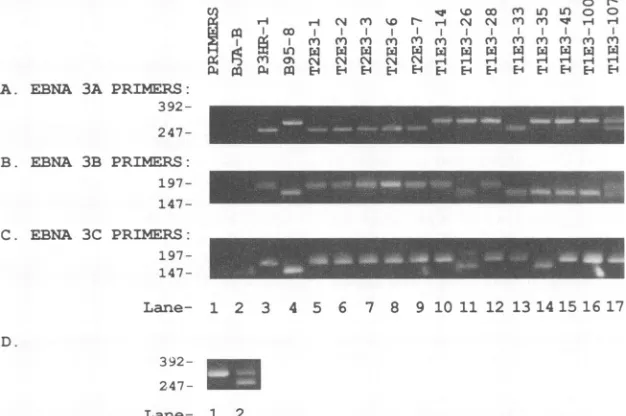
Fig. 2, lanes TlE3-14, T1E3-28, T1E3-33, T1E3-35, and TlE3-45. In eachof these examples, eithertype1ortype2 butnotbothgenesaredetectedatthe EBNA 3A, 3B,or3C
loci. An example ofa resultshowing evidence for
coinfec-tion with an EBNA 3 recombinant virus and a parental
P3HR-1 virus is given inFig. 2, lane TlE3-107. This latter 0
A.
L
120 140 160 kb
(7,315)
92
B.
Ion November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.61.551.59.328.2]TABLE 1. Frequency of second-site recombination
No.oftype1EBNA 3 recombinants
Expt (no. of P3HR-1 coinfected)/total no.
EBNA3A EBNA 3B EBNA3C
1 10(7)/107 1 (1)/25 1 (1)/25
2 3(1)/39 1/17 1/17
3 9(6)/68 9 (6)I68a 3(NT)b68a
4 11(10)/91 11 (10)/91C 1 (NT)/88C
Total 33(24)/305 20(16)/159 4(NT)/156
Total(% T1E3 11 12 3
recombinants)
Total(% P3HR-1 72 80 NT
coinfected)
aAllnine cloneswere type 1forEBNA 3A and 3B,andthree were alsotype
1for EBNA 3C.
bNT,nottested.
IAll 11cloneswere type 1for EBNA3A and 3B, and 1 was alsotype1for
EBNA3C.
LCL is coinfectedwith a type 1 EBNA 3A and 3B
recom-binant virus and with nonrecombinant P3HR-1 virus and
therefore hastype 1 EBNA 3A and 3B and type 2 EBNA 3C genes on therecombinantgenome and type 2 EBNA 3A, 3B,
and 3C genes on the parental P3HR-1 genome. Approxi-mately 11% of theLCL clones were infected with
recombi-nantvirus whichhad type 1 EBNA 3A or 3B, and almost all hadboth type 1 EBNA 3A and 3B (Table 1). Examples of
LCLs which wereinfectedwith an EBV that was
recombi-nantfor type1EBNA3A or 3B but not both aregiveninFig.
O4_4
A. EBNA 3A PRIMERS:
392-2,lanesTlE3-14, T1E3-28, and T1E3-33. Examples of LCLs whichwere infected with an EBV that wasrecombinantfor type 1 EBNA 3A and 3B are given in Fig. 2, lanes T1E3-26, T1E3-35, T1E3-45,TlE3-100, andTlE3-107. One clone had type 1EBNA 3A, 3B, and 3C (Table 1; Fig. 2, laneT1E3-35),
and none had EBNA3C only. The EBNA 3genotypesof the
recombinants are summarized in Table 2. The high fre-quency ofrecombination at the EBNA 3A and 3B loci and the low frequency ofrecombination atthe EBNA 3C locus may beattributed to thetermination ofthe EBNA 3Copen readingframe only 4 kb fromthe right end of the type 1 Sall
C fragment whichwasused in thesetransfections, whereas the EBNA 3A and 3B open reading frames are more cen-trally located withinSall C (Fig. 1).
Stochastic and empiricalevidence indicates that the pres-enceofboth type 1andtype 2 EBNA3A, 3B, or 3C genes, as wasobserved in 72% of theEBNA3 recombinantLCLs, is almost always the result of coinfection with both a
parental, nonrecombinant P3HR-1 virus and a doubly recombinant virus whichistransformingandcarriesthetype 1 EBNA3 gene(s). In previous experimentswith mutations within EcoRI-A, parental, nonrecombinant P3HR-1 virus wasfrequently presentin such vast excess overrecombinant viruses that many or most primary B lymphocytes were initiallyinfectedwith theparental P3HR-1 virus in addition totransforming EcoRI-A recombinants (18, 32). This result was expected since the recombinant virus stocks are in P3HR-1 virus-infected cells and approximately 1 to 10%of the cells are induced to lytic virus infection, releasing approximately 108 parental P3HR-1 viruses and
approxi-O
r-v vo 0 m uU) 0O 0
r- N m kD r- H N cs M m v -4
-X X X X w D3 w w X X >3 X w
CN N cs N N ?- 4 H r- rA v- _ H
E h
247-B. EBNA 3B PRIMERS:
197- 147-C. FRBNA 3C PRIMERS:
197-
147-Lane- 1 2 3 4 5 6 7 8 9 1011 12 1314151617
D.
[image:4.612.59.302.85.212.2]392- 247-Lane- 1 2
FIG. 2. Analysisof theEBNA3genotypeofEBV-immortalized LCLscarrying wild-typetype2(T2E3)and recombinanttype 1(T1E3)
EBNA 3 genesby PCR. Panels A, B, and C illustrate the ethidium bromidestaining ofPCR-amplifiedfragments by using primers which distinguish betweentype 2andtype 1 EBNA3A,3B, and 3Cgenes,respectively.EBNA3Aprimers amplifya237-bpanda276-bpfragment fromtype2 andtype 1 DNA,respectively(29). EBNA3B primersamplifya218-bpanda183-bpfragmentfromtype2andtype 1 DNA, respectively (29).EBNA3C-3'primersamplifya179-bpanda158-bpfragmentfromtype2andtype1DNA, respectively. Lanes 1 and 2are negative controlamplificationswithprimersonly andEBV-negativeBJA-Bcell DNA. Lanes 1and 2 of EBNA 3C(panel C)show faint bands duetocontamination with B95-8 DNA.Lanes 3 and4arepositivecontrolamplificationsofP3HR-1(type2)and B95-8 (type1) cell DNA.
Lanes 5to9areamplifications of LCLDNAtransformedbyvirusestaken from P3HR-1 cellstransfected with theEcoRI-Acosmid and pSZNaeI Z plasmid only and therefore should carry wild-type type 2 EBNA 3 genes (LCLs designated T2E3). Lanes 10 to 17 are
amplificationsof LCL DNA transformedbyviruses taken fromP3HR-1 cells transfectedwith thepSVNaeIZplasmid,EcoRI-Acosmid, and Sal-Ccosmid andselected because of recombination in theEBNA 3genes(LCLs designatedT1E3).Panel D illustrates the ethidium bromide stainingofEBNA 3APCRfragmentfromDNAobtainedfromanearly(lane 2)andalater(lane 1)passageofTlE3-100demonstrating the lossofparental P3HR-1 virusovertime. Size markers(kilobases)ofRsaI fragmentsof4X174 DNAareshownonthe left.
-..l
...
on November 10, 2019 by guest
http://jvi.asm.org/
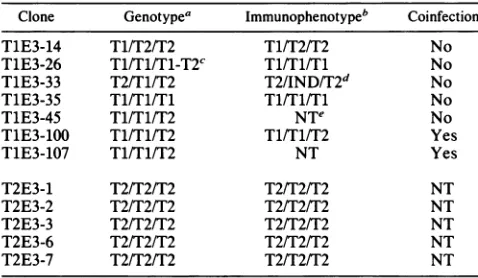
[image:4.612.154.467.394.602.2]TABLE 2. Genotypes andimmunophenotypesoftype2
EBNA3 recombinants
Clone Genotypea Immunophenotypeb Coinfection
TlE3-14 T1/T2/T2 T1/T2/T2 No
TiE3-26 Tl/Tl/Tl-T2C Tl/Tl/Tl No
TlE3-33 T2/T1/T2 T2/IND/T2d No
TlE3-35 Tl/Tl/Tl Tl/Tl/Tl No
T1E3-45 T1/T1/T2 NT" No
TlE3-iOO T1/T1/T2 T1/T1/T2 Yes
TlE3-107 T1/T1/T2 NT Yes
T2E3-1 T2/T2/T2 T2/T2/T2 NT
T2E3-2 T2/T2/T2 T2/T2/T2 NT
T2E3-3 T2/T2/T2 T2/T2/T2 NT
T2E3-6 T2/T2/T2 T2/T2/T2 NT
T2E3-7 T2/T2/T2 T2/T2/T2 NT
aType for EBNA3A/type for EBNA3B/typefor EBNA 3C.
bThe type 1 immunophenotype was detectedby atype 1-specificEBV
immune serum. The type2immunophenotypewas notdirectly tested,butwas
assumed to be type 2 since P3HR-1 virus isatype 2 EBV.
cT1-T2indicates the presence of both type1andtype2 EBNA3Cowing
to anonhomologousrecombination inEBNA3C.
dIND, indeterminate. The EBNA 3B of T1E3-33 didnot reactwiththetype
1-specific serum, despite the PCR amplification of a type 1 EBNA 3B
sequence.Type2EBNA 3Bexpressionwas notdirectly assayed. Therefore,
T1E3-33 encodesachimeric EBNA-3B withthetype1sequencelimitedto a
regionsurrounding the PCRprimers which doesnotencodedetectable type
1-specific epitopes;alternatively,therecombination hasdisruptedthe EBNA
3B openreading frame and noEBNA 3Bis expressed.
eNT, not tested.
mately
102
EcoRI-A recombinant, transforming P3HR-1 viruses. Independent recombination atthe EBNA 3 loci isexpected to be less efficient than within EcoRI-A since
EcoRI-Ais larger than Sall-C. Furthermore, EcoRI-A
con-tains30kb oflong internalrepeat DNAwhich ishomologous
totheparental P3HR-1genome, and EcoRI-A has latent and
lytic EBV DNA replication origins. The larger size, the repeats, and the replication of the transfected EcoRI A
fragment within transfected cells is expected to make
EcoRI-A more recombinogenic (24, 30, 33, 34). If the
fre-quency ofindependent Sall-C recombination is similar to
thatof EcoRI-A,therewould beapproximately
102
indepen-dent type 1 EBNA 3recombinants, and the chance that anEcoRI-A and a Sall-C recombinant would both infect the same primary B
lymphocyte
among 107 B lymphocytes which are used in one experiment would be very small.Direct evidence that the finding of both type 1 and type 2 EBNA 3A, 3B, or 3C genes in the same LCL is due to
coinfecting nonrecombinantparental P3HR-1 genomes came
from three observations. First, in all instances (except one
described below)whenan LCL was infected with an EBV
carrying
type 1 EBNA 3A or 3B, EBNA 3A and 3B, orEBNA3A, 3B, and 3C genes and also had thecorresponding
type 2 EBNA 3 gene(s), the cell had thefullcomplement of type 2 EBNA 3 genes, irrespective ofwhether one, two, or
threetype 1 EBNA 3 genes were present. Thus, these cells
infected with EBV genomes with an insertion of type 1 EBNA 3A, 3B, or 3C genes also had genomes in which
linkage was preserved through the entire type 2 EBNA 3
region.Thiscould occur on the same genome only if the type 1genes hadinserted intoa site 5' or 3' to the type 2genes.
Southern
blot analysis of one putative coinfected and foursingly infected type 1 recombinant LCLDNAs proved that
both the type 1 and type 2 EBNA 3 genes were in the
expected context in EBV genomes (see below). Second,
although in initialPCR screening the P3HR-1-derivedtype2
EBNA 3 genes were frequently equally represented
(Fig.
2D, lane 2), subsequent PCR analyses of expanded LCL
cultures revealed that the amplified fragments from type 2 EBNA 3 genes were now less evident than the type 1 fragments (Fig. 2D, lane 1). This is consistent with the
expectation
that parental coinfecting P3HR-1 genomeswould be lost because they would be nontransforming ge-nomesand thereforedispensableforLCLoutgrowth.Third,
when viruswas passed fromtheputatively doublyinfected LCLs to primary B lymphocytes, the resultant LCLs had whichever type 1 EBNA 3 geneswerepresentin theoriginal
LCL (seebelow). The parental clone TlE3-100 in Fig. 2Ato
C has already lost most ofits coinfecting nonrecombinant P3HR-1, and the progeny virus-transformed LCL (seeFig.
6) has only type 1 EBNA 3B. Thus, the type 1 EBNA 3A gene cosegregates with the transforming virus genome and
thetype 2 EBNA3A gene does not.
Characterization of the type 1 EBNA3 recombinants. Five type 1EBNA 3recombinants(resultingfromrecombination with transfected SalI C and EcoRI A DNA fragments) and five type 2recombinants (resulting from recombination with EcoRI-A only) which had been derived from two experi-ments were further characterized by restriction endonucle-asedigestionand Southernblot, byin situlysis and Southern blot, by immunoblot for latent and lytic-cycle EBV gene
expression, and by growth rate at low cell concentrations. LCL DNA was cutwithBclI, which cleaves type 1 DNA at
positions 79894and 97110ortype2DNA at positions79894,
93135, and 97052 (Fig. 3A). Thus, type 2 DNA has a
distinctive BclI site near the center of EBNA 3A, which results in 13.3- and 3.9-kb fragments instead of the type 1 distinctive 17.2-kb fragment. Southern blots of LCL DNA were probed with EBNA 3A cDNA, which has two exons centered around the new BclI site characteristic of the type 2 EBNA 3 and an upstream exon from the BamHI U fragment (Fig. 1 and 3A). TheEBNA 3A openreadingframe probedetected a17.2-kbfragment in type 1 B95-8 DNA and 13.3- and3.9-kb fragments in type 2 P3HR-1 DNA (Fig. 3B). The upstream EBNA 3A BamHI-U exon also detected a 9.5-kb Bcll fragment in both DNAs (Fig. 3B). As expected, the clones which by PCR appeared to be type 2 nonrecom-binants at EBNA 3A had only the type 2BclI fragments (Fig. 3B,lanes T2E3-1,T2E3-2, andT2E3-3). The TlE3-33 clone, which by PCR analysis was singly infected with a type 1 recombinant restricted to the EBNA 3B locus, hadonly the type 2EBNA 3A Bcll siteand the expected 13.3- and 3.9-kb fragments (Fig. 3B). The TiE3-26, TiE3-28, and TlE3-35
clones, which were singly infected with type 1 EBNA 3A
recombinants by PCR analysis, had only the type 1 Bcll fragment (Fig. 3B). The absence of type 2Bcllfragments in these DNAs excludes the possibility that the type 1 EBNA 3Agenes hadinserted nonhomologously into another site in theEBV genome. The TlE3-100 Southern blot was particu-larly interesting in indicatingcoinfection withgenomes that had both the type 1 and type 2BclI fragments surrounding EBNA 3A (Fig. 3B; data not shown). This agrees with the
early-passage PCR analysis ofTlE3-100 shown in Fig. 2D, whereas PCR analysis of a later passage ofTlE3-100shown in Fig. 2 reveals that the coinfecting type 2 EBNA 3A gene has been lost, leaving a single virus genome which is recombinant for the type 1 EBNA 3A and 3B genes.
Southern blotanalysis of theBclland BamHI (see below)
digests of the LCL DNA probed with a SalI-C DNAprobe
demonstrated the expected restriction fragments, indicating a single Sall C fragment, further confirming the expected genome structures (data not shown). Thus, almost allPCR
on November 10, 2019 by guest
http://jvi.asm.org/
Bcll BclI BclI BclI
I I I _.i
9.5kb 17.2kb
3clI Bcll Bcll Bcll BclI
_I
C,
m CI L m
-4 -4 N 1 0 rDN1t m m OD
i g m I m m m m nmm
n' X X X W X X X X X X
F m os e' N N N '-4 '-4
m PE' -4Ed' E-4 Ep Ep E
TYPE 2 EBV
Z 4- 9. 5kb
PROBE
-- 1 kb
13.3kb 3.9kb /EBNA 3B
-EBNA 3C \EBNA 3A
116-
97-C
%D c m Ln 0
4 '-4 C'4 m CN CN ') ,-4 I co I
N m X
r-0 ow cs N N m w N w w
zw m Eq Ed E E-4 q Eq P
E-so1 4.wIIP -EBNA2
, _ _ _ 4om__ -EBNA 1
66-
23--w
9.9-6.
6-4
4-
2.3-Lane- 1 2 3 4 5 6 7 8 9 10
FIG. 3. Southern hybridization of genomic DNA from
repre-sentativeLCLs. GenomicDNAwasdigested withBcIlrestriction
endonuclease andhybridizedwith32P-radiolabeled B95-8EBNA 3A
openreading framecDNAprobe containinggenome sequence67649
to 67535 (from the splice site in BamHI-U) and 92238 to 95239
(EBNA 3Aopenreading frame) (panelA). (A) Aschematic
repre-sentation ofthetype1 andtype2EBNA 3Aregionandtheexpected
BclIfragments.Type 2 DNA(P3HR-1) hasanadditionalBcIlsite
(position 93135)in the EBNA 3Aopenreadingframe, resulting ina
prominent3.9-kb BcII fragment when hybridized withthe EBNA 3A
openreadingframeprobe. Incontrast, type1(B95-8) DNAhasno
EBNA 3ABclI site and thereforehas a17.2-kb BclI fragment. (B) Southernhybridizationresultsfromrepresentative LCLs. Digests of genomicDNAfrom P3HR-1 andB95-8(lanes1 and2), threeT2E3
LCLs(lanes 3to5),and fiveT1E3LCLs(lanes6to10)areshown.
Sizemarkers(kilobases) ofHindIll fragments of lambdaDNAare
shownontheleft.
and Southern blot results were consistent with insertion of
partof thetype 1Sall Cfragment into the P3HR-1genome
by homologous recombination. In addition, Southern blot analysis of the LCLgenomic DNA digested with BamHI and probed witha 32P-labeled cloned BamHI H DNA fragment
(Fig. 1) demonstrated proper replacement of the P3HR-1
deletionby recombination with the cloned type 1EcoRI A fragment(datanotshown). All the LCLs examined had the B95-8 (type 1) size BamHI H fragment, with no detectable
differencebetweenthe type2EBNA3 LCLsand the type 1 EBNA 3 LCLs(datanot shown).
The single evidence for nonhomologous recombination
among 33 clones which were analyzed was the T1E3-26 clone which byPCR and Southern analysis hadonlytype 1 EBNA 3A and 3B (Fig. 2 and 3). The absence of type 2 EBNA 3A and 3Bindicates that this LCLwasnotcoinfected withanother EBVgenomeand that thetype1EBNA 3Aand
3B genes had replaced the corresponding type 2 genes
throughsite-specific integrationintothe EBVgenome.
How-ever,theT1E3-26genomeisdiploidfor EBNA 3C(Fig. 2C). Thetriplet bands notedinthePCRanalysis areduetotype 1, type 2, andheterohybridEBNA 3C bands. Thissuggests that recombination at the right side of SalI-C for this
440Om N _ EBRNA LP
Lane- 1 2 3 4 5 6 7 8 9 10 11 12 13
FIG. 4. Immunoblot analysis oftotal cellular proteins from
rep-resentative LCLs. Protein blots were incubated with a serum
specificfortype1 EBVlatentproteinstoexaminerecombinationat
theprotein level (25).Type 1 EBNA 3A, 3B,and 3Care145-, 165-,
and 155-kDa proteins, respectively (lla, 25, 27a). Protein lysates
from5 x 105 cells were loaded and separatedon a7%
polyacryl-amide gel, transferred to nitrocellulose, incubated with a 1/10
dilutionof thetype 1 specificimmune serum(previously absorbed
with EBV-negative Louckes cells), and radiolabeled with 1251_ protein A. A negative control from the EBV-negative BJA-B is
shown in lane 1. Proteinlysatesfrom P3HR-1 and B95-8 cellsare
shown in lanes 2 and 3.Thespecificityofthetype1-specificimmune
serumis shownbythestrongreactivitytotheB95-8type1 EBNA 3proteins (lane 3)andthe lackofreactivity totheP3HR-1type 2 EBNA3proteins (lane 2).Proteinlysatesfrom fiveT2E3(lanes4to
8)andfive T1E3(lanes9to13)LCLsareshown.Thelocation of the EBV latentproteins is shownontheright. Theproteindetected in
thetype2 LCLs(lanes2 and 4to8)andthe T1E3-33LCL(lane 12) whichismigrating slightly fasterthan EBNA 3A,isacross-reacting
band and is not to be confused with the type 1 EBNA 3A band detected in lanes3, 9to 11, and 13. Size markers (kilodaltons) of
proteinmolecularmassstandardsareshownonthe left.
recombinant was nonhomologous and that the
recombina-tion was 3' to the type 1 EBNA 3C and 5' to the type 2 EBNA 3C site. PCR analysis of this recombinant with an
additional set of EBNA 3C primers (29) located approxi-mately 900 bp 5' tothefirstsetof EBNA 3Cprimers (Fig. 1) also demonstrated amplification of both type 1 and type 2 EBNA 3C-specific fragments, further indicating that the EBNA3C recombinationwasnonhomologous.Theanalysis
with the secondset ofprimers indicates thattype 1 EBNA 3C ispositioned5' to mostorall of type 2 EBNA 3C in the recombinant genome (PCR data not shown). With virus replication in the T1E3-26 clone, progeny transforming
vi-ruses further recombined to delete the type 1 EBNA 3C
gene, resulting in apparently fully homologously
recombi-nantgenomescarryingtype 1 EBNA 3Aand 3B genesand thetype 2 EBNA 3Cgene(see below).
Immunoblot analyses confirmed the genotypic analyses andindicated that therewas no effect oftype 1 EBNA3A, 3B, or3C substitution on the expression of other latent or
lytic-cycle EBV genes. A largely type 1-specific human
antiserumdidnotdetecttype1 EBNA 3sinthefully type2
cell linesT2E3-1-, T2E3-2-, T2E3-3-, T2E3-6-, and
T2E3-7-infected LCLs (Fig. 4). The antiserum did detect type 1 EBNA 3A, 3B,or3C in the TlE3-14-,T1E3-28-, T1E3-35-, T1E3-33-, and TlE3-100-infected LCLs (Fig. 4). EBNA 2
A.
TYPE 1 EBVi
B.
200
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.79.281.73.313.2] [image:6.612.325.553.75.248.2]CnS1 m14e1
XNC1i3(
NS NN
H .H
0
er cc ur en C
r H N~ e) en H
I
NS H H H H H
H HHEHHHPE
-EA
B.
H H N enwk
m1 m0
Ct 1¢ n M
,en cNN NN
P1pP4 H H HH
0
t en en o
r, - N m m 14
N enH ('1 NI)
N H1
Hq Hq
-LUP1
-DlLHP1 *
6 7 8 9 10 1112 13
FIG. 5. Expression of lytic (A) and lI.MP I (B) proteins in
representative LCLs. Polyacrylamide gels (7%c) were loaddc with
protein Iysaite samplcs. and separated pi-oteins weretransftrred to
nitrocellulose. (A) l xtic-antigen expression wasexamiined by
inCL-batioin with /10)dilution Of SPhumanseillmand radiolabelingwith
25I-protein A. Ihe location of the lvtic antigens (carlys antigens
IEAI) isshown ontheright. SPalso hats reactswith thetspc 1 Iatent
antigens. (B) LMIP 1 expression was cxamined bs incLbaitingwith
the S12 LMP 1-specific monoclonal antibody (16). a ribbit
anti-mouse secondai aintibody andi '2`1-protein A. IMP 1 and the
lytic-cycle prodiuct of the LMP 1 gene. DILM1I1. arc indicated. Bothareexpressedin1395-8cells. EBVintectioninthe i-ccombinaint LCLsx unlike B95-8 cells, is primarily latent and DIL MP is not
detected. Aproteinlysate fromEBV-negative BJA-B cellsis shown
in lane 1. Protein lvsates from induced P3HR-1 (lane 2) induced
B95-8 (lane3).and fiveT2E3(lanes4to8)andfivc TIlE3 (lancs9to
13) LCLsare shown. Sizemarkcrs (kilodaltons)of piotein
moleCLi-larmass standards are shownon the let.
and EBNA LP and LMP 1 expression was simila am-ong dtl
of the celllines, includingT1E3-35. which wastype 1 att the EBNA 3A. 38.3 and 3C loci (Fig. 4 and SB). Most LCI.
clones exhibited little evidence of spontaneouLs lytic-cycle EBV gene expression irrespective of whether they were
recombinant or nonrecombinant at an EBNA 3 lCouS (Fig. 5A). In situ lysis a-ind Southern blot analysis of five LCLs
infected withtype 1EBNA 3A and 3BorEBNA3A. 3B.and
3Crecombinants and of fourLCLs infected with fullytype2
EBNA 3 genomes reveailed no diflerence in EBV episome
copy number or in spontanCous EBV replication as cvi-denced by linear EBV genomes (dattanot shown).
Replication and passage of the recombinant virus. Virus
could be passatged from LCLs infected: with recombinant
virus whether ornot the recombinant virus had also
incor-porated one ormore type 1 EBNA 3 loci. Asexpected. the
resultant infected LCLs had the EBNA 3 genotypc and immunophenotype characteristics of the parental virus (Fig.
6 to8; data not shown). The time to oLutgrowth and
subse-quentpassage of primary B lymphocytesinfected withtype 1 EBNA3Aand 3Brecombinants was no different from that ofcells infected inparallelwith an otherwise isogenic type 2 control.
Passage of virus was useful in separating coinfecting genomes and in resolving the single putatively partially homologous recombinant. The T1E3-100 LCL was pre-sumed to be coinfected with parental P3HR-1 and a trans-forming recombinant which also had type 1 EBNA 3A and 3Bgenes. When progeny virus from TlE3-100was used to infect primary B lymphocytes. the resultant transformants had type 1 EBNA 3A and 3B by genotype and immunophe-notype (compare the parental TlE3-100and progeny
T1E3-10)A. T1E3-100G. and T1E3-100J in Fig. 6, 7. and8). Tlhe cosegregation oftransformation and type 1 EBNA 3A and 3B in the progeny transformed LCLs is strong evidence for physical linkage between these genetic markers in a single genome in the parental LCL. Otherwise, progeny transfor-mants would be expected to be as frequently or more
frequentlytype 2 atthe EBNA 3A and 3B loci.TheTIE3-26 LCL was infected withanEBV whichhadtype I EBNA 3A and 3B and type 1 and type 2 EBNA 3C. Ihis LCL was
presu-med to be infected with a single virus which had incorporated type I EBNA 3A. 3B, and 3C, displacing the type 2 EBNA 3Aand 3B genes, hut witha nonhomologous recombination 3' to thetype 1 EBNA3C gene, insertingthe type 1 EBNA 3C 5' to the type 2 EBNA 3C gene. Thetwo
progeny LCLsthat arosefollowing infection with viruis from
T1E3-26 were infected withgenomes which had deletedthe r-edundant type 1 EBNA 3Cgene and weretype 1 for EBNA 3Aand 3B and tvpe 2 ftor EBNA 3C (Fig. 6 and 7).
I)ISCUSSION
'Ihese experimcnts demonstrate important principles which will be useful for subsequeint EBV recombinant
mo-leculatr gcnetic experiments. First, a cotransfected EBV DNA fragment can be incorporated into replicating EBV genomes which had also recombined with a nonlinked pos-itive selection marker. TIhus, a replicating EBV DNA mole-cule which pairticipates in one recombination event can
participate in at second recombination event. Second, the recombination of the nonlinked EBV DNA fragments into the EBV genome isalmost always fully homologous.
Previ-ous studies with transtected large EBV DNA fragments
which included the DNA deleted from P3HR-1 and which used restoration of transformation as a positive selection revealed that the transformation-competent recombinants had incorporated the previously deleted DNA into the
'correct' site(6, 7, 11, 17, 18, 31, 32). Ourdata indicate that thenonlinked cotransfectedEBV DNAis also almost
invari-ably targeted to the correct site, probably by reciprocal
homologous recombination. PCR analysis of eight of nine LCLs singly infected with independently derived type I EBNA 3 reconmbinantsdemonstrated that the type 1 EBNA 3gene(s) had replaced the type 2gene(s). Most oftheinitial
type1 EBNA 3 recombinant-infected LCLs were coinfected with P3HR-1. complicating the interpretation of their initial
PCR
results.
Even in those instances, passage of virusresulted insegregationofthetransformation-competenttype 1 EBNA 3 recombinant genomes from the nonrecombinant
P3HR-1 genome.
The
single exception to complete replace-ment was agenome inwhichthe three type 1 EBNA 3 genes hadreplacedthe type2 EBNA3A and 3B genes but hadnot replaced the type 2 EBNA 3C gene. Passage of virLsS fromthis LCIL to primary B lymphocytes resulted in an EBV
A.
m co
1,
m P4 m E-4
200-
116-
97-
66-
45-Lane- 1 2 3 4 5 6 7 8 9 10 11 12 13
97-
66-Lane- 1 2 3 4 5
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.66.293.62.348.2]X m n1 w d e 0 0 0 o)
H U: w w N N N HHv H H
0
m MM MmXm m M m mm
m EdN N
A. EBNA 3A
PRIMERS:
392-247-B. EBNA 3B PRIMERS:
197-
147-C. EBNA 3C PRIMERS:
197-
147-Lane- 1 2 3 4 5 6 7 8 9 10 11 12 13
FIG. 6. PCR analysis of T2E3 andT1E3progeny clones. Parental clones T2E3-6,T1E3-26, and TlE3-100 were transfected with pSVNaeI Ztoinduce virus replication, lethallyirradiated (8,800 rads), and cocultured with human B cells. The PCRanalysis of three progeny T2E3-6 LCLs(lanes4to 6)derived from the parental T2E3-6 LCL (lane 3), three progeny TlE3-100 LCLs (lanes 11 to 13) derived from the parental TlE3-100 LCL (lane 10), and two progenyT1E3-26LCLs(lanes 8 and 9) derived from the parentalT1E3-26 LCL (lane 7) analyzed with the EBNA3A, 3B, and 3C-3' primers is shown. The PCR analysis of P3HR-1 (lane 1) andB95-8 (lane 2) is also shown. Size markers (kilobases) of RsaI fragments of4X174are shown on theleft.
genomewhich deleted thepreviously duplicated EBNA 3C
gene, presumably resulting- ina fullyhomologously
recom-bined type 1 EBNA 3A and 3B and type 2 EBNA 3C
genome. Third, the frequency ofincorporation of the
non-linked EBVDNA fragmentwas 11%despite the absence of
repeatDNAand latentorlytic-cyclereplication origins. The
lack ofdependence on repeat DNA and on latent or lytic
.4
0 0 0
w x'0 0 0 .-4 %D %' -4 .-4 4 a: 0a
(In (Im enm m rn
, a' CN (N N .-4
m m H El h
0 -1 (N4
m
H4
.4 aQ
(N N M c'
H4
200-
116-
97-/FBNA 3B v -EBNA 3C
\EBNA 3A
66- _ =
[image:8.612.155.463.71.264.2]
45-Lane- 1 2 3 4 5 6 7 8 9 10 11 12 13
FIG. 7. AnalysisofprogenyLCLsforexpressionof latent
pro-teins. ProteinanalysisofthreeprogenyT2E3-6 LCLs(lanes4to6)
derived from the parental T2E3-6 LCL, three progeny TlE3-100 LCLs(lanes8to10)derived fromtheparentalTlE3-100LCL(lane 7),andtwoprogenyT1E3-26 LCLs(lanes12and13)derived from
theparentalT1E3-26 LCL(lane 11)areshown. Also shownarethe protein analysesfromBJA-B (anegativecontrol[lane1]), P3HR-1 (lane 2), and B95-8 (lane 3). Protein lysates were separated and analyzedwiththetype1specificimmuneserum asdiscussed inthe
legendtoFig. 4.
origins should make it possible to extend this approach to
specifically introduce mutations into any site in the EBV genome.
The relatively high efficiency with which second-site
ho-mologousrecombination occurredintheseexperiments sug-gests that second-site homologous
recombination
may begenerally applicable
to otherherpesvirus
orother virus orhost cellmoleculargenetic experiments. Suchanapproach is particularly useful because it simplifies the strategy neces-sary to make mutations in any specific site in a genome
which is linked toanother site that is subject to apositive selection strategy. Indeed, the fact that successfully
trans-fectedcellstakeup andincorporate largequantities ofDNA hasbeenwidelyuseful andisanimportant componentofthis procedure (28, 38). A high frequency of recombination
among DNAs which
participate
in a single recombinationevent hasalsobeen noted in many systems(19, 22, 36).
Theseexperiments useda30-kbEBV DNAfragment, and
the large fragment size probably contributed to the
high
efficiency of targeted homologous recombination. Ina
pre-viousexperiment with apositive selection markerin a3-kb
EBV DNA fragment,
nonhomologous
insertion was noted (37). The highfrequency of insertion of bothtype 1 EBNA3A and 3B and the low frequency of insertion of type 1 EBNA 3Care compatiblewiththe
working
hypothesis
that mostofthe30-kbtransfectedDNAis insertedinto the EBV genome andthat theefficiency
decreases within4kboftheends.This occurred in these
experiments despite
type 1andtype 2EBNA3C havingasimilar levelof
homology
tothat between type 1 and type 2 EBNA 3A and transfection of Sall-C as a free, linear DNAfragment.
Although
homolo-gous recombination in other systems is facilitatedby
freeDNAends
(5a),
free ends maynotbemorerecombinogenic
inthis system.
These
experiments
demonstrated no difference between EBV recombinants with type 1ortype 2 EBNA 3 genes intheir
ability
to transformprimary
Blymphocytes
or in them .M
M-IV',
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.63.295.446.627.2]
A-m
m 1fi _ El
200- 116-
97-
66-45-.
Lane- 1 2 3 4 B.
11 1.D k0
m Co
e a aN (a a4
-0
a: D H
mn .e m m N N S
H H EH E
.4
h~-o 0 o 0 0
- -I
-F- B
m
N1
h~
*a:-:
10 11 12 13 14
0 0 0 0) C 0 0 o HD H H H H
f" f: -1 tr
--El Eq E El El
(4 (Q CH
In M. m
W X ;S
Eq1
97
66--1mp1
-D1LIPI
45-Lane-- 1 2 3 4 5 6 7 8 9 ln ll 1'2 13 14
FIG. 8. Analysis ofprogenyLCLsforexpressionof lytic (A) and
LMP1 (B) proteins. Three progeny T2E3-6 LCLs (lanes 5 to 7) derived from the parental T2E3-6 LCL (lane 4), three progeny
TlE3-100LCLs (lanes9to 11)derived from the parental TlE3-100
LCL (lane 8), and two progeny T1E3-26 LCLs (lanes 13 and 14)
derived from theparental T1E3-26 LCL (lane 12)are shown. Also
shownaretheprotein lysates fromBJA-B (anegativecontrol [lane
1]), P3HR-1 (lane 2), and B95-8 (lane 3). Protein lysates were
separatedandanalyzed asdiscussed inthelegend toFig.5.
growth of the resultant LCLs. Furthermore, there was no
difference in latent or lytic-cycle EBV gene expression.
These results are consistent with previous results which indicated that type-specific differences in EBNA 2 are
pri-marilyresponsible for the different effectsoftype 1ortype
2 EBVin primary B-lymphocytegrowth transformation (7, 27). Although no differences were observed in vitro, it is possible that EBV encodingtype1ortype2 EBNA 3differs in some aspect which may only be apparent in vivo. The functions of the EBNA3proteinsarenotknown,exceptfor the observation that EBNA 3C expression in Burkitt's lymphoma cells results in increased expression of CD21
(36a). Whethertype 1 andtype 2 EBNA 3C havethe same
effectonCD21 expressionhas notbeen examined.
Further-more,wehavenotexaminedorcomparedthe level of CD21
oranyother antigen expressioninourEBNA3recombinant
LCLs.
The type 1 EBNA 3A, 3A and 3B, and 3A, 3B, and 3C
recombinants derived inthese experiments will be useful in
subsequent studies of the effect of type-specificdifferences
on cellgrowth or immune T-cell cytotoxic killing of
EBV-infected B lymphocytes. Forthesepurposes,twoadditional
type 1 EBNA 3A, 3B, and 3Crecombinant EBVs have been derived (data not shown). Previous analyses of immune T-cell cytotoxic recognition ofEBV-transformed B lympho-cytes indicated that EBNA 3A has an epitope which is recognized in the context of HLA-B8 andthat this epitope differs between type 1 and type 2 viruses (5, 23).
ACKNOWLEDGMENTS
Wonkeun Lee and Jennifer Leecontributed valuable assistance, and George Miller generously provided the HH514-16 clone of
P3HR-1 (26A).
Thisresearchwassupported by grantCA00449 fromtheNational
Cancer Institute, Public Health Service. B.T. was supported by postdoctoral research awards from the American Cancer Society
(grant PF-3561) and the BaxterFoundation. REFERENCES
1. Adidinger, H., H. Delius, U. Freese, J. Clarke, and G.
Bornkamm. 1985. A putative transforming gene ofthe Jijoye virusdiffers from that ofEpstein-Barr virus prototypes. Virol-ogy141:221-234.
2. Alfieri, C., M. Birkenbach, and E. Kieff. 1991. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology
181:595-608.
3. Baer, R., A. Bankier, M. Biggin, P. Deininger, P. Farrell, T. Gibson, G. Hatfull, G. Hudson, S. Satchwell, C. Sequin, P. Tuggnell, and B. Barrell. 1984. DNAsequence andexpression ofthe B95-8 Epstein-Barrgenome. Nature (London)
310:207-211.
4. Burrows, S.,I.Misko, T. B. Sculley, C. Schmidt, and D. J. Moss. 1990. An Epstein-Barr virus-specific cytotoxic T-cell epitope
present on A and B type transformants. J. Virol. 64:3974-3976.
5. Burrows, S., T. Sculley,S. Misko, C.Schmidt,and D. J. Moss. 1990. AnEpstein-Barr virus-specific cytotoxicT cellepitope in
EBV nuclear antigen3(EBNA 3). J. Exp. Med. 171:345-349. 5a.Carroll, D., S. H. Wright, R. K. Wolff,E.Grzesiuk, and E. B.
Maryon. 1986. Efficient homologous recombination of linear DNA substrates after injection into Xenopus laevis oocytes.
Mol. Cell. Biol. 6:2053-2061.
6. Cohen, J. I., F. Wang, and E. Kieff. 1991. Epstein-Barr virus nuclear protein 2 mutations define essential domains for
trans-formation and transactivation. J. Virol. 65:2545-2554.
7. Cohen, J. I., F. Wang, J. Mannick, and E. Kieff. 1989. Epstein-Barrvirus nuclear protein 2 is a keydeterminantoflymphocyte transformation. Proc. Natl. Acad. Sci. USA86:9558-9562.
8. Countryman, J., H.Jenson, R. Seibl, H. Wolf, and G. Miller. 1987. Polymorphicproteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J. Virol. 61:3672-3679.
9. Dambaugh, T., K. Hennessy, L. Chamnankit, and E.Kieff.1984. U2regionof Epstein-BarrvirusDNA may encodeEpstein-Barr
virusnuclear antigen 2. Proc. Natl. Acad. Sci. USA
81:7632-7636.
10. Gardella,T., P.Medveczky, T.Sairenji, and C.Mulder. 1984.
Detection ofcircular and linearherpesvirusDNAmoleculesin
mammaliancells bygelelectrophoresis. J. Virol. 50:248-254.
11. Hammerschmidt,W., and B.Sugden.1989. Genetic analysis of
immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature (London) 340:393-397.
11a.Hennessy, K., F. Wang, E.Woodland-Bushman, and E. Kieff. 1986. Definitiveidentificationof amember of the Epstein-Barr
virus nuclear protein 3 family. Proc. Natl. Acad. Sci. USA
79:5916-5920.
12. Heston, L., M. Rabson, N. Brown, and G. Miller. 1982. New
Epstein-Barrvirusvariants from cellularsubclonesofP3J-HR-1
Burkittlymphoma. Nature (London)295:160-163.
13. Jeang, K., and S. D. Hayward. 1983. Organization of the
Epstein-Barrvirusmolecule. III. Location of the P3HR1 dele-tionjunction andcharacterizationof theNotlrepeat units that
form part of the template for an abundant
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.72.296.72.396.2]canoylphorbol-13-acetate-induced mRNA transcript. J. Virol.
48:135-148.
14. Knott,J., D. J. G. Rees, Z. Cheng, and G. G. Brownlee. 1988.
Randomly picked cosmidclones overlap the pyr B and oriC gap
in the physicalmapof E. coli chromosome. Nucleic Acids Res.
16:2601-2612.
15. Maniatis, T., E. F. Fritsch, and J.Sambrook. 1982. Molecular
cloning: alaboratorymanual. Cold Spring Harbor Laboratory,
Cold Spring Harbor,N.Y.
16. Mann, K. P., D. Staunton, and D. A. Thorley-Lawson. 1985.
Epstein-Barr virus-encoded protein foundin plasma membranes
of transformed cells.J. Virol.55:710-720.
17. Mannick, J. B., J.I.Cohen, M.Birkenbach, A. Marchini, and E. Kieff. 1991. TheEpstein-Barr virusnuclear protein encoded by
the leader of the EBNA RNAsis important in B-lymphocyte
transformation. J. Virol. 65:6826-6837.
18. Marchini, A., B. Tomkinson, J. I. Cohen, and E. Kieff. 1991.
BHRF1,the Epstein-Barr virusgenewithhomology toBcl2,is
dispensable for B-lymphocyte transformationandvirus replica-tion. J.Virol. 65:5991-6000.
19. Marsden, H. S., N. D. Stow, V. G. Preston, M. C. Timbury, and N.M. Wilkie. 1978. Physical mapping ofherpes simplex
virus-induced polypeptides. J. Virol.28:624-642.
20. Menezes, J., W. Leibold, G. Klein, and G. Clements. 1975.
Establishment and characterization of an Epstein-Barr virus (EBV)-negative lymphoblastoid B cell line (BJA-B) from an
exceptional, EBV-genome-negative African Burkitt's
lym-phoma. Biomedicine 22:276-284.
21. Miller, G., M.Rabson, and L. Heston. 1984.Epstein-Barr virus with heterogeneousDNAdisrupts latency. J. Virol. 50:174-182.
22. Morse, L. S., T. G. Buchman, B. Roizman, and P. A. Schaffer.
1977. Anatomy of herpes simplex virus DNA. IX. Apparent
exclusionofsomeparental DNAarrangementin thegeneration of intertypic(HSV-1 x HSV-2)recombinants. J. Virol. 24:231-248.
23. Murray, R., M. Kurilla, H. M. Griffin, J. M. Brooks, M.
Mackett, J.A.Arrand,S.R.Burrows, D. J. Moss, E. Kieff, and A. B. Rickinson. 1990. Human cytotoxic T-cell responses
against Epstein-Barr virus nuclear antigens demonstrated by using recombinant vaccinia virus. Proc. Natl. Acad. Sci. USA 87:2906-2910.
24. Nickoloff,J. A., and R. J. Reynolds.1990.Transcription stimu-lateshomologous recombination in mammalian cells. Mol. Cell. Biol. 10:4837-4845.
25. Petti, L., J. Sample, F. Wang, and E. Kieff. 1988. A fifth Epstein-Barr virus nuclear protein is expressed in latently infected growth-transformed lymphocytes. J. Virol. 62:1330-1338.
26. Rabson, M., L. Gradovilie, L. Heston, and G. Miller. 1982. Nonimmortalizing P3J-HR-1 Epstein-Barr virus:adeletion
mu-tantof itstransformingparent,Jijoye. J.Virol. 44:834-844. 26a.Rabson, M.,L.Heston,and G. Miller. 1983. Identification ofa
rare Epstein-Barr virus variant that enhances early antigen
expression in Raji cells.Proc. Natl. Acad. Sci. USA 80:2762-2766.
27. Rickinson, A., L. S. Young, and M. Rowe.1987.Influence of the Epstein-Barr virus nuclear antigen EBNA 2 on the growth
phenotypeof virus-transformedBcells. J.Virol. 61:1310-1317.
27a.Ricksten, A., B. Kallin, H. Alexander, J. Dillner,R. Fahraeus, G.Klein, R. Lerner, and L. Ramos.1988. BamHI Eregion of the Epstein-Barr virusgenomeencodes threetransformation
asso-ciated nuclearproteins. Proc. Natl. Acad. Sci. USA 85:995-999.
28. Roizman, B., and F. J. Jenkins. 1985. Genetic engineering of novelgenomesof largeDNAviruses. Science 229:1208-1214.
29. Sample, J., L. Young, B. Martin, T. Chatman, E. Kieff, A. Rickinson, and E.Kieff. 1990. Epstein-Barr virustypes 1and2
differ in their EBNA3A, EBNA 3B,and EBNA 3C genes.J.
Virol. 64:4084-4092.
30. Shulman, M.J., L. Nissen, and C. Collins. 1990. Homologous recombination in hybridoma cells: dependence on time and fragment length. Mol. Cell. Biol. 10:4466 4472.
31. Skare,J., J. Farley,J. L. Strominger, K.0.Fresen, M.S. Cho,
and H. Zur Hausen.1985.Transformationby Epstein-Barr virus requiressequencesin theregion of BamHI fragmentsYandH. J. Virol. 55:286-297.
32. Swaminathan, S.,B. Tomkinson, and E. Kieff. 1991.
Recombi-nantEpstein-Barr virus with small RNA (EBER)genesdeleted transforms lymphocytes and replicates in vitro. Proc. Natl. Acad. Sci. USA88:1546-1550.
33. Thomas, B. J., and R. Rothstein.1989. Elevatedrecombination
ratesintranscriptionally activeDNA. Cell 56:619-630.
34. Thomas, K.R.,and M. R.Capecchi. 1987. Site-directed muta-genesis bygenetargetinginmouseembryo-derivedstemcells. Cell 51:503-512.
35. van Santen, V., A. Cheung, and E. Kieff. 1981. Epstein-Barr virus RNA: size and direction oftranscriptionof virusspecific cytoplasmic RNAs inatransformed cell line.Proc. Natl. Acad. Sci. USA 78:1930-1934.
36. vanZiji, M.,W.Quint, J. Briaire,T. deRover,A.Gielkens,and A.Berns.1988.Regenerationofherpesviruses frommolecularly clonedsubgenomicfragments. J. Virol. 62:2191-2195. 36a.Wang, F., C.Gregory, C.Sample, M. Rowe,D. Liebowitz,R.
Murray, A. Rickinson, and E. Kieff. 1990. Epstein-Barr virus latentmembraneprotein(LMP1) and nuclearproteins2 and3C
areeffectors ofphenotypic changesinBlymphocytes:EBNA-2 and LMP1 cooperativelyinduceCD23. J. Virol. 64:2309-2318.
37. Wang, F., A. Marchini,and E. Kieff. 1991.Epstein-Barrvirus (EBV) recombinants: use ofpositive selection markers to
res-cue mutants in EBV-negative B-lymphoma cells. J. Virol.
65:1701-1709.
38. Wigler, M., A. Pellicer, S. Silverstein, and R. Axel. 1978. Biochemicaltransfer ofsingle-copyeucaryoticgenesusingtotal cellularDNAasdonor. Cell 14:725-731.




![FIG. 8.TlE3-100LMP1derivedderivedLCLshownseparated1]), Analysis of progeny LCLs for expression of lytic (A) and (B) proteins](https://thumb-us.123doks.com/thumbv2/123dok_us/1310460.84352/9.612.72.296.72.396/fig-derivedderivedlclshownseparated-analysis-progeny-lcls-expression-lytic-proteins.webp)