yet been elucidated. We have utilized surface plasmon resonance technology to study the interaction of VCP with C3b and C4b. We measured the kinetics of binding of the viral protein to its target proteins and compared it with human complement regulators factor H and sCR1, assessed the influence of immobilization of ligand on the binding kinetics, examined the effect of ionic contacts on these interactions, and sublocalized the binding site on C3b and C4b. Our results indicate that (i) the orientation of the ligand is important for accurate determination of the binding constants, as well as the mechanism of binding; (ii) in contrast to factor H and sCR1, the binding of VCP to C3b and C4b follows a simple 1:1 binding model and does not involve multiple-site interactions as predicted earlier; (iii) VCP has a 4.6-fold higher affinity for C4b than that for C3b, which is also reflected in its factor I cofactor activity; (iv) ionic interactions are important for VCP-C3b and VCP-C4b complex formation; (v) VCP does not bind simultaneously to C3b and C4b; and (vi) the binding site of VCP on C3b and C4b is located in the C3dg and C4c regions, respectively.
The complement system is an ancient and integral compo-nent of the host’s immunological defense, evolved to combat against all of the invading pathogens, including viruses (9, 27, 38). Recognition of viruses by either of the three complement activation pathways (classical, lectin, or alternative) leads to the formation of C3 convertases, which cleave the central com-plement component C3 into an anaphylatoxic peptide C3a and an opsonic fragment C3b. Once the newly formed C3b mole-cules are deposited on the viral surfaces, they are considered as non-self by the complement system and result in further acti-vation of the pathways and neutralization of viruses through various mechanisms (5, 9). However, during the long coexist-ence of viruses with the host immune system, certain viruses have developed ingenious strategies to evade the complement system. Important examples include the poxviruses, herpesvi-ruses, retroviherpesvi-ruses, paramyxoviherpesvi-ruses, and picornaviruses (2, 13, 31, 33, 37, 46, 52).
Vaccinia virus, a cytoplasmic double-stranded DNA virus, is the most extensively characterized member of the Orthopoxvi-rusgenus (18, 36). It is known to have developed two distinct mechanisms to subvert the host complement system: (i) it encodes a 27-kDa secretory protein, the vaccinia virus comple-ment control protein (VCP), which is homologous to human complement control proteins and is a potent inhibitor of com-plement (23, 32, 42), and (ii) it incorporates the host comple-ment control proteins (CD46, CD55, and CD59) into the outer
envelope of extracellular enveloped virus (EEV), which ren-ders resistance to EEV against complement (53).
VCP is one of the first documented viral immune evasion proteins. It is encoded by the C21L gene and is secreted by the cells infected with vaccinia virus. The primary structure of VCP consists of 263 amino acids, with a 19-amino-acid signal pep-tide sequence. It consists of four tandem copies of short con-sensus repeats (SCRs) or complement control protein (CCP) modules (24), a characteristic structure of host CCPs, and bears 26 to 38% sequence similarity to human CCPs. The nuclear magnetic resonance structure of pairs of VCP modules and the crystal structure of the entire VCP molecule have been determined (15, 39). The structure shows that each SCR folds into a compact 6-strand structure, which is similar to the SCRs of human CCPs. Further, it also showed that the mole-cule has an extended structure from SCR 1 to 3 and a turn between SCR 1 to 3 and SCR 4. The most striking feature revealed in the crystal structure was the charge distribution of the four SCR domains: the SCR domains 1 and 4 carry a positive field around them, whereas the middle two domains are predominantly surrounded by a negative field (39).
Functional studies have shown that VCP is a potent inhibitor of complement activation (23, 32, 42). It is known to bind to complement proteins C3b and C4b and inhibit both the clas-sical and the alternative pathways of complement activation (32, 42). Studies on the mechanism of complement inactivation by VCP have revealed that it acts as a cofactor for factor I-mediated cleavages of C3b and C4b (42), inhibits the forma-tion of the classical/lectin and alternative pathway C3 conver-tases (32), and accelerates the decay of these C3 converconver-tases into their subunits (32). Interestingly, VCP also contains two putative heparin-binding sites; the first site overlaps between
* Corresponding author. Mailing address: National Centre for Cell Science, Pune University Campus, Ganeshkhind, Pune 411007, India. Phone: 91-20-2569-0922. Fax: 91-20-2569-2259. E-mail: arvindsahu @nccs.res.in.
9446
on November 8, 2019 by guest
SCR 1 and SCR 2, and the second site is located in SCR 4 (39). The physiological significance of these heparin-binding sites with respect to complement activation is not clear at present. In addition to in vitro studies on the mechanism of comple-ment inhibition, the role of VCP has also been examined in vivo in experimental animals. It has been shown that recombi-nant vaccinia virus that does not express VCP is attenuated in vivo (17).
Although it is clear that VCP inhibits complement activation by interacting with complement proteins C3b and C4b, the nature of these interactions and the binding mechanisms in-volved are not well understood. Earlier studies have shown that VCP requires all four SCRs for binding to C3b (41, 50), and the crystal structure data suggest the possibility of multiple binding sites for C3b and C4b in VCP (39). Thus, whether VCP binds to C3b at a single or at multiple sites is not clear. Fur-thermore, there is a general belief that the binding of factor I cofactors to C3b and C4b causes conformational changes in these molecules and facilitates the binding of factor I (6, 51). Whether VCP-C3b/C4b complexes undergo structural reorien-tation is not known.
In the present study, we have utilized surface plasmon res-onance (SPR) technology to decipher the mechanism of mo-lecular recognition between VCP and its target proteins C3b and C4b and compared it to human CCPs. Our data show that, unlike host CCPs, interactions of VCP with C3b and C4b follow a simple 1:1 binding mechanism and that these complex formations do not involve conformational changes or multiple-site binding. These interactions, however, are highly dependent on ionic strength, a feature very similar to human CCP-C3b/ C4b interactions. We also present data on the sublocalization of VCP binding sites on C3b and C4b. Our data show that these sites are located in the C3dg and C4c regions of C3b and C4b, respectively. A significant body of knowledge suggests that C3dg acts as a “bridge” between innate and acquired immunity (7, 29, 43). Thus, it is possible that, apart from inhibiting the complement system, VCP may also inhibit the acquired immunity by blocking the C3dg-CD21 interaction. To date, VCP is the only viral protein known to interact with C3dg.
MATERIALS AND METHODS
Expression and purification of recombinant VCP.The vaccinia virus comple-ment control protein cloned inPichia pastoriswas expressed and purified as described earlier with minor modifications (42). In brief, a single colony of recombinantP. pastorisexpressing VCP was inoculated in 10 ml of BMGY medium (100 mM potassium phosphate [pH 6.0], 10 g of yeast extract/liter, 20 g of peptone/liter, 13.4 g of yeast nitrogen base/liter, 0.4 mg of biotin/liter, and 1% glycerol) and incubated overnight at 30°C in a shaking incubator. This inoculum was added to 1 liter of BMGY, followed by incubation for 48 h at 30°C with shaking. The cells were centrifuged, resuspended in 100 ml of BMMY (BMGY containing 0.5% methanol but without 1% glycerol), followed by incubation at 30°C for an additional 24 h with vigorous shaking. After incubation, cells were pelleted and the supernatant containing VCP was collected for purification.
The culture supernatant was sequentially precipitated with 20 and 60% am-monium sulfate at 0°C. The 60% pellet was suspended and dialyzed against phosphate-buffered saline (PBS; 10 mM sodium phosphate [pH 7.4] and 145 mM NaCl) and loaded onto a DEAE-Sephacel column (Sigma, St. Louis, Mo.) preequilibrated with 10 mM sodium phosphate buffer (pH 7.4) containing 250 mM NaCl. The flowthrough was collected, and buffer exchange was performed by using PD-10 desalting columns (Amersham Pharmacia Biotech, Uppsala, Sweden) preequilibrated with 5 mM sodium acetate (pH 4.0). The sample was then loaded onto Mono S 5/5 column (Amersham Pharmacia Biotech). The
bound proteins were eluted with a linear salt gradient of 0 to 1.0 M NaCl. The eluted fractions were analyzed by sodium dodecyl sulfate–11% polyacrylamide gel electrophoresis (SDS–11% PAGE), and the fractions containing VCP were pooled, dialyzed into PBS, and concentrated by ultrafiltration.
Complement proteins and cleavage products.The human complement pro-teins C3, factor H and factor I, were kindly provided by Michael K. Pangburn (University of Texas Health Center, Tyler, Tex.) and the recombinant human soluble CR1 (sCR1) was a generous gift of Henry Marsh (AVANT Immuno-therapeutics, Inc., Needham, Mass.). C3b was generated by limited trypsin cleav-age and purified on a Mono Q 5/5 column (Amersham Pharmacia Biotech) (42, 44). C4b was purified as follows. Twenty parts of human plasma were mixed with one part inhibitor solution (1 M KH2PO4, 0.2 M Na4EDTA, 0.2 M benzamidine,
and 1 mM phenylmethylsulfonyl fluoride) and precipitated first with 4.5% poly-ethylene glycol and then with 12% polypoly-ethylene glycol at 0°C. The 12% pellet was dissolved in 3.2 mM sodium phosphate (pH 7.4) containing 6.4 mM EDTA, 31.8 mMεamino caproic acid, 6.4 mM benzamidine hydrochloride, and 0.02% sodium azide and then loaded onto a Source Q column (1.2 by 9.5 cm; Amer-sham Pharmacia Biotech). The bound proteins were eluted with a linear gradient of 0 to 0.5 M NaCl. Fractions were spiked with 1 mM PEFA-block (Roche, Mannheim, Germany), and C4b-containing fractions (generated during purifi-cation), identified by SDS-PAGE and immunodiffusion, were pooled and loaded onto a Mono Q 5/5 column (Pharmacia) in 10 mM sodium phosphate (pH 7.9). Bound proteins were eluted with a linear salt gradient of 0 to 0.5 M NaCl and subjected to SDS-PAGE analysis. Homogeneous C4b fractions were pooled and dialyzed into PBS. Experiments were also performed with C4b purchased from Calbiochem (La Jolla, Calif.).
The cleavage products of C3b and C4b were generated as described below. iC3b was generated by incubating 1 mg of trypsin-generated C3b with 166g of factor H and 16g of factor I in 1.2 ml of PBS (pH 7.4) at 37°C for 3 h; C3c and C3dg were generated by incubating 1 mg of trypsin-generated C3b with 166g of sCR1 and 16g of factor I in 1.1 ml of PBS (pH 7.4) at 37°C for 3 h; and C4c and C4d were generated by incubating 1 mg of C4b with 25g of sCR1 and 8g of factor I in 1.0 ml of PBS (pH 7.4) at 37°C for 2 h. The cleavage products of C3b, as well as of C4b, were purified on a Mono Q column as previously described (21, 42). The purity of all of the proteins exceeded 95%, as determined by SDS-PAGE and immunodiffusion analysis.
Site-specific biotinylation of C3b and C4b.C3b and C4b contain a free⫺SH group, which is generated as a result of hydrolysis of the thioester bond present in these proteins (28). The free⫺SH groups in these proteins were labeled by using EZ-Link PEO-maleimide-activated biotin (Pierce, Rockford, Ill.) (45). Briefly, 0.5 mg of C3b or C4b in PBS was dialyzed into 0.1 M sodium phosphate (pH 6.0) containing 5 mM EDTA for 16 h at 4°C. EZ-Link PEO-maleimide-activated biotin was dissolved in 0.1 M sodium phosphate–150 mM NaCl–1 mM EDTA (pH 7.2) at a concentration of 10 mM and then mixed and incubated with C3b or C4b at a 1:70 molar ratio for 30 min at room temperature. Free biotin was removed by passing the reaction mixture through PD-10 column, followed by dialysis for 48 h at 4°C. The C3b (iC3b and C3dg) and C4b (C4d) fragments were obtained by proteolytic cleavages of the same stock of biotinylated C3b and C4b as described above. Monitoring of the biotinylation reactions, cleavages, and purifications was performed by SDS-PAGE, followed by Western blotting with avidin-horseradish peroxidase (HRP) conjugate.
Biotinylation of C3c and C4c.Biotinylation of C3c and C4c was performed by using EZ-Link-Sulfo-NHS-Biotin (Pierce). In brief, 0.2 mg of C3c and C4c were dialyzed against PBS (pH 7.2). EZ-Link-Sulfo-NHS-Biotin was dissolved in PBS (pH 7.2) at a concentration of 2 mM, added to C3c or C4c at a molar ratio of 1:18, and incubated on ice for 2 h. The reaction mixtures were then extensively dialyzed against PBS (pH 7.4) at 4°C, and biotinylation was confirmed by West-ern blot analysis with avidin-HRP conjugate.
Western blot analysis.Site-specific biotinylation of C3b, C4b, and their frag-ments (iC3b, C3dg, and C4d) was confirmed by Western blotting. Labeled pro-teins were separated on SDS-PAGE and electrotransferred onto polyvinylidene difluoride membrane (Bio-Rad, Hercules, Calif.). The membranes were blocked overnight at 4°C with constant rocking with 5% ECL blocking reagent (Amer-sham Biosciences, Buckinghamshire, United Kingdom). The incorporation of biotin was determined by probing the blots with 1:5,000 diluted avidin-HRP conjugate (Bio-Rad) for 2 h at room temperature and a wash with Tris-buffered saline (20 mM Tris, 150 mM NaCl [pH 7.5]) containing 0.05% Tween 20. Labeled proteins were detected by using a SuperSignal West Pico chemilumi-nescent kit (Pierce).
SPR measurements.The kinetics of binding of VCP to C3b, C4b, and their fragments were determined on the SPR-based biosensor BIACORE 2000 (Bia-core AB, Uppsala, Sweden). All of the experiments were performed in PBS-T (10 mM sodium phosphate and 145 mM NaCl [pH 7.4] containing 0.05% Tween
on November 8, 2019 by guest
http://jvi.asm.org/
20) at 25°C unless mentioned otherwise. The addition of 0.05% Tween 20 blocked the nonspecific adsorption of analytes to the sensor chips. In each experiment, ligands were coupled either to Fc-2 or Fc-4, and Fc-1 or Fc-3 served as control flow cells. Experiments were performed either by immobilizing VCP or by immobilizing the target proteins onto the sensor chips; VCP (2000 Rus) was immobilized onto a CM5 chip by using amine-coupling chemistry, while biotinylated-C3b (2300 Rus) and -C4b (2200 Rus) were immobilized onto streptavidin chips (Sensor Chip SA; Biacore AB). Binding was measured at 50
l/min to avoid the mass transport effect. Binding was measured for 120 s, and dissociation was monitored for an additional 180 s. The sensor chips were regenerated with 30-s pulses of 0.2 M sodium carbonate (pH 9.5). Sensograms obtained for the control flow cell were subtracted from the data for the flow cell immobilized with a ligand. The SPR data were analyzed by BIAevaluation software version 3.2 with global fitting. When data did not fit to a simple 1:1 Langmuir binding model it was evaluated by linear transformation analysis as previously described (44, 45, 55) by plotting dRU/dt Vs RU, where RU is the relative response of the biosensor at time t. The apparent equilibrium dissocia-tion constant (KD) was calculated from the equationKD⫽kd/ka, wherekdis the
dissociation rate constant andkais the association rate constant.
Measurement of factor I cofactor activity of VCP for C3b and C4b.A quan-titative analysis of factor I cofactor activity of VCP for C3b and C4b was deter-mined in 10 mM phosphate buffer (pH 7.4) containing 145 mM NaCl. In these assays, 2.7g of C3b or 2.9g of C4b was mixed with 100 ng of factor I and various concentrations of VCP and then incubated at 37°C for 4 h in a total volume of 15l. The reactions were stopped by adding sample buffer containing dithiothreitol and then electrophoresed on an 8% SDS-PAGE gel for determin-ing C3b cleavages and a 9% SDS-PAGE gel for C4b cleavages. The cleavage products were visualized by staining the gel with Coomassie blue. The gels were scanned for densitometric analysis by using ChemiDoc XRS system (Bio-Rad, Segrate, Italy) to calculate the percentage of␣⬘chain. The data obtained was normalized by considering 100%␣⬘chain to be equal to the␣⬘-chain intensity obtained in the absence of factor I (control). The binding data were fit by using nonlinear regression analysis (GraFit; Erithacus Software, London, United King-dom), and a four-parameter logistic analysis was performed to identify the best-fit concentration of VCP causing 50%␣⬘-chain cleavage.
RESULTS
Kinetic analysis of binding of VCP to C3b and C4b.
Al-though previous studies have identified host proteins C3b and C4b as the target proteins for VCP, the kinetic model of binding involved in the bimolecular interaction of VCP with its target proteins is still not clear.
To gain insight into the interaction of VCP with C3b and
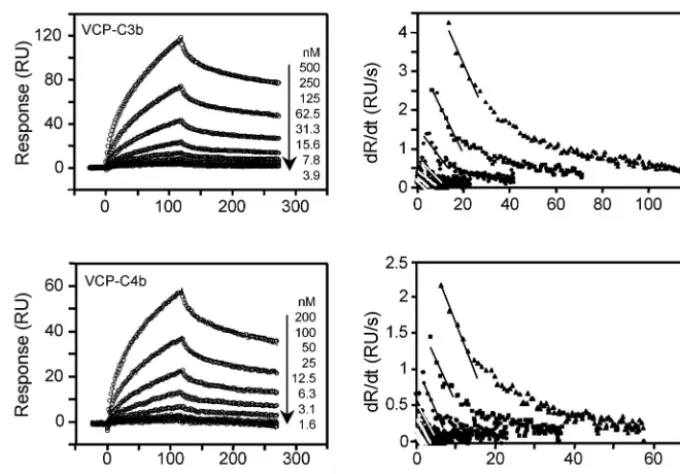
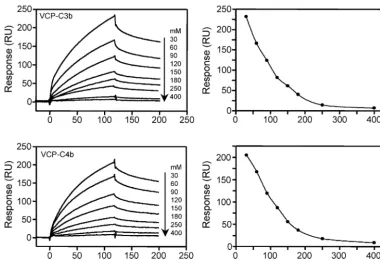
C4b, we utilized SPR technology and compared its interaction to that of human regulators factor H and sCR1 (Fig. 1). VCP was coupled to a sensor chip by using the standard amine-coupling chemistry, and C3b and C4b were flown over the chip to measure binding. VCP bound to both the analytes in a dose-dependent manner (Fig. 2). The binding data (senso-grams) for these interactions could not be fitted to a simple 1:1 binding model. Linear transformation analysis showed nonlin-ear plots, suggesting that these bindings follow complex mod-els. We then attempted to fit these data to complex models to investigate the nature of these interactions. The data showed a close fit to a bivalent analyte model with2values for
VCP-C3b and VCP-C4b interactions of 0.559 and 0.353, respectively (Table 1), suggesting that this is the most likely model of interaction. There was, however, another possibility that amine coupling produced a heterogeneous ligand surface which re-sulted in complex binding. This was further supported by the fact that the data fit well even to the heterogeneous ligand model. The2 value for VCP-C3b interaction was 0.297 and
that for VCP-C4b interaction was 0.264.
To determine whether bivalent binding truly reflected the binding interactions and was not an artifact of the heteroge-neous ligand surface, we designed the following experiment. We oriented C3b and C4b onto a streptavidin chip by labeling their free⫺SH group with biotin (Fig. 3). This approach not only provided a homogeneous ligand surface but also immo-bilized these proteins in their physiological orientation. In complement proteins C3 and C4, Cys988 and Cys991 are
[image:3.603.72.529.67.246.2]in-volved in the formation of the internal thioester bond. During complement activation, proteolytic cleavage of C3 and C4 result in generation of C3b and C4b and exposure of their thioester bond. This bond reacts with hydroxyl or amino groups present on the activating surfaces to form an ester or an amide bond. The covalent attachment of C3b and C4b onto the activating surfaces orients them on these surfaces (Fig. 3A). During this reaction, a free⫺SH group is generated (Fig. 3A).
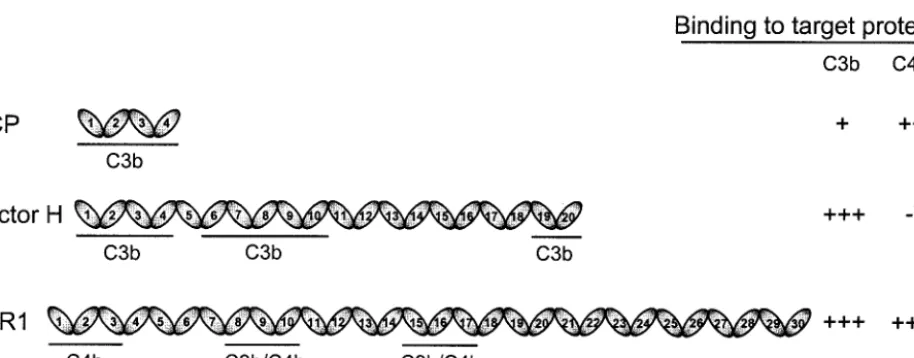
FIG. 1. Schematic representation of structures of VCP, factor H, and sCR1. VCP is entirely composed of four SCR or CCP modules, whereas the human complement regulators factor H and complement receptor 1 are composed of 20 and 30 SCRs, respectively. The SCR domains of each protein are numbered, and the binding domains for C3b and C4b are identified (3, 19, 20, 26, 41, 48, 50). The relative binding activity of VCP to that of factor H and sCR1 is shown on the right. These activities are based on the data presented in Table 1.❋, Factor H binds to C4b only in a buffer with very low ionic strength (47).
on November 8, 2019 by guest
We labeled this⫺SH group with biotin and oriented the la-beled molecules on streptavidin chip (Fig. 3B). Thus, orienta-tion of C3b and C4b in our experimental setup mimicked the physiological orientation of these proteins. The specificity of this labeling was verified by analyzing the reactivity of labeled C3b and C4b with avidin-HRP in a Western blot assay. The results depicted in Fig. 3C and E show that biotin incorpora-tion in C3b and C4b was associated with the␣⬘chains, indi-cating thereby that biotin was incorporated at the thioester site. To further confirm this, we cleaved labeled C3b and C4b molecules with factor I in the presence of either factor H or sCR1. Biotin incorporation was observed only in fragments containing free⫺SH group; in C3b fragments it was associated
with the 68-kDa fragment and C3dg, whereas in C4b fragments it was associated with C4d (Fig. 3C and E).
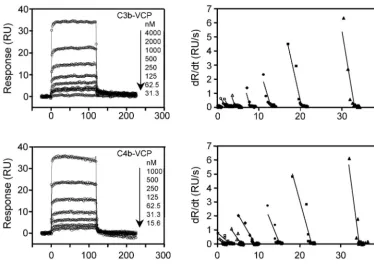
When VCP was flown over streptavidin chips immobilized with oriented C3b or C4b, it bound in a dose-dependent and saturable manner (Fig. 4). The kinetics of binding were dis-tinctly different from those obtained when VCP was immobi-lized by using amine coupling chemistry. Global fitting analysis of the sensograms showed a good fit to a 1:1 binding model (2
values⬍0.9, Table 1) and linear transformation of the binding data showed a single component. Together, these data indi-cated that both VCP-C3b and VCP-C4b interactions follow a simple 1:1 binding model. We then comparedkaandkdof the
[image:4.603.121.461.69.306.2]VCP-C3b interaction with those of the VCP-C4b interactions.
FIG. 2. Analysis of the binding of immobilized VCP to C3b and C4b by SPR. The left panels show sensogram overlays for interactions between immobilized VCP and C3b or C4b. The concentration of analyte injected is indicated at the right of each sensogram overlay. Solid lines correspond to the global fitting of the data simultaneously. Both C3b and C4b data fit to a bivalent analyte model (A⫹B7AB; AB⫹B7AB2; BIAevaluation
3.2). The right panels show linear transformations of the association phase data for the respective sensogram data shown on the left. The straight lines are linear least square fits to the data.
TABLE 1. Kinetic and affinity data for the interactions of VCP and human complement control proteins factor H (fH) and sCR1 with C3b, C4b, and their fragments
Ligandb Analyte k
d1(1/s)/ka1(1/Ms) SE (kd1/ka1) KD1c kd2(1/s)/ka2(1/Ms)a SE (kd2/ka2) KD2 2
VCP C3b 9.61⫻10⫺2/9.58⫻103 2.52⫻10⫺3/179 10M 1.03⫻10⫺3/1.5⫻102 1.08⫻10⫺5/2.75 0.69⫻10⫺5 0.559
VCP C4b 0.103/3.17⫻104 2.93⫻10⫺3/482 3.2M 1.34⫻10⫺3/3.83⫻102 1.71⫻10⫺5/5.88 0.35⫻10⫺5 0.353
C3b VCP 0.554/7.66⫻105 2.5⫻10⫺2/6.38⫻104 0.724M NA NA NA 0.887
C4b VCP 0.38/2.42⫻106 1.1⫻10⫺2/9.69⫻104 0.157M NA NA NA 0.526
C3b CR1 5.74⫻10⫺2/4.4⫻106 13 nM*
C3b fH 5.98⫻10⫺2/1.1⫻106 54.4 nM*
C4b CR1 4.17⫻10⫺2/3.8⫻106 11 nM*
C3dg VCP NA NA 0.179M† NA NA NA 0.111
C4c VCP 3.93⫻10⫺4/2.66⫻103 4.16⫻10⫺5/61 0.148M NA NA NA 0.122
ak
a2(1/Ms)⫽ka2(1/RUs)⫻100⫻molecular weight of the ligand. NA, not applicable.
bVCP was immobilized on CM5 chip by using standard amine coupling chemistry. C3b, C4b, and C4dg were site specifically biotinylated by labeling their free⫺SH groups and immobilized on SA chips. C4c was biotinylated by labeling its amino groups and immobilized on an SA chip.
cData were calculated by global fitting analysis (BIA evaluation 3.2); *, data were calculated by linear transformation analysis (35, 44); †, data were calculated by steady-state analysis (BIAevaluation 3.2).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.603.46.541.579.684.2]on November 8, 2019 by guest
There was a 3.2-fold increase inkaand a 1.5-fold decrease inkd
for the VCP-C4b interaction compared to the VCP-C3b inter-action. Together, this resulted in a 4.6-fold increase in the affinity of VCP for C4b compared to C3b (Table 1).
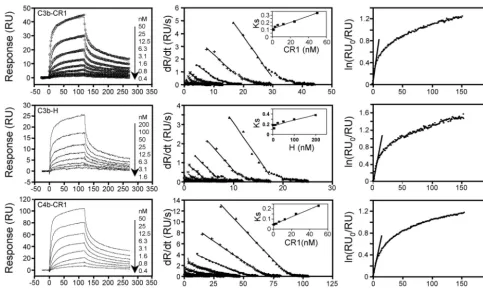
Next, we analyzed binding of oriented C3b and C4b with multivalent proteins sCR1 and factor H (Fig. 1). As expected, binding of sCR1 to C3b and C4b and factor H to C3b did not follow a 1:1 binding model (Fig. 5). Because the data did not fit to a 1:1 model, the association and dissociation rate constants were calculated by linear transformation analysis as previously described (44, 45, 55). The affinities of sCR1 and factor H for C3b were 56- and 13-fold greater, respectively, than VCP, whereas that of sCR1 for C4b was 14-fold greater than that of VCP.
Comparison of the factor I cofactor activity of VCP for C3b
and C4b.The data presented above indicated that VCP has a
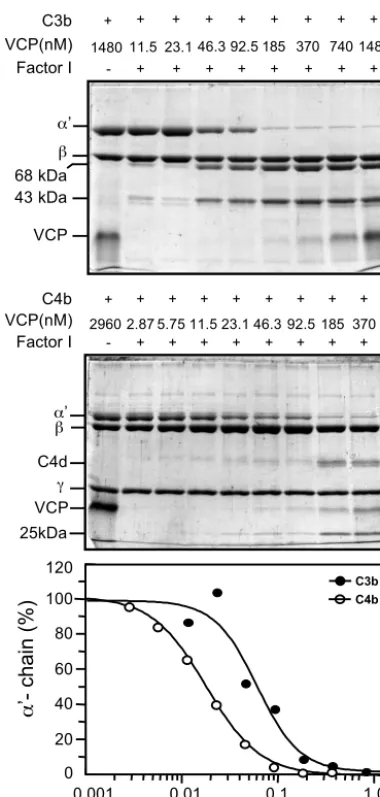
[image:6.603.108.482.69.332.2]4.6-fold-higher affinity for C4b than C3b. In order to test whether this difference in affinity is also reflected in its func-tion, we measured relative factor I cofactor activity of VCP for C3b and C4b. Figure 6 shows a comparison of cofactor activity of VCP in factor I-mediated cleavage of C3b and C4b. In this assay, equimolar concentrations of C3b or C4b were incubated with factor I, and various concentrations of VCP in a physio-logical ionic strength buffer. The reaction mixtures were run on SDS-PAGE gels and cleavage of the␣⬘chain of C3b and C4b were quantitated by densitometric analysis. The concentration of VCP required to cleave 50% of␣⬘chain of C3b and C4b was
FIG. 4. Analysis of binding of VCP to immobilized C3b and C4b. The left panels show overlay plots of the binding of immobilized C3b and C4b to VCP. Various concentrations of VCP were injected over a streptavidin chip immobilized with C3b or C4b. Solid lines represent the global fitting of the data to a 1:1 Langmuir binding model (A⫹B7AB; BIAevaluation 3.2). The right panels show linear transformations of the association data for the respective sensogram data shown on the left. The straight lines are linear least-square fits to the data.
FIG. 3. Site-specific biotinylation of C3b and C4b. The free⫺SH groups of C3b and C4b were labeled with PEO-maleimide biotin, and labeled proteins were cleaved into iC3b or C3c and C3dg and C4c and C4d and analyzed by SDS-PAGE and Western blotting as described in Materials and Methods. C3b cleavage in the presence of factors H and I results in cleavage of the␣⬘chain into N-terminal 68-kDa and C-terminal 43-kDa fragments; the appearance of these fragments indicates the generation of iC3b, whereas C3b cleavage in the presence of sCR1 and factor I results in cleavage of the␣⬘chain into N-terminal 25-kDa fragment, C3dg, and C-terminal 43-kDa fragments, which indicates the generation of C3c and C3dg. C4b cleavage in the presence of sCR1 and factor I results in cleavage of the␣⬘chain into N-terminal 25-kDa, C-terminal 16-kDa, and central C4d fragments; the appearance of these fragments indicates the generation of C4c and C4d. (A) Diagram showing covalent attachment of C3b and C4b to the activating surface. (B) Diagram depicting the experimental design. Site-specific biotinylated C3b and C4b were immobilized on a streptavidin chip (SA chip). (C) Coomassie blue staining (left) and Western blot (right) of biotinylated C3b and its cleavage products. Lane 1, biotinylated C3b; lane 2, biotinylated C3b cleaved with factors I and H; lane 3, biotinylated C3b cleaved with factor I and sCR1. (D) Schematic representation of C3b, C3c, and C3dg structures. Arrows indicate factor I-mediated cleavages generated in the presence of cofactors and closed balloon indicates the location of⫺SH group labeled with biotin. (E) Coomassie blue staining (left) and Western blot (right) of biotinylated C4b and its cleavage products. Lane 1, biotinylated C4b; lane 2, biotinylated C4b cleaved with factor I and sCR1. (F) Schematic representation of C4b, C4c, and C4d structures. Arrows indicate factor I-mediated cleavages generated in the presence of sCR1, and the closed balloon indicates the location of⫺SH group labeled with biotin.
on November 8, 2019 by guest
http://jvi.asm.org/
0.059 and 0.018M, respectively, indicating that better binding of VCP to C4b leads to better cofactor activity of VCP for C4b.
Effect of NaCl on binding of VCP to C3b and C4b.VCP is
structurally and functionally similar to host CCPs. Previous mutation analysis of charged residues of CCPs, as well as C3b, have highlighted the importance of ionic interactions in CCP-C3b/C4b complex formations (4, 6, 11, 25, 30, 40). In order to understand whether electrostatic interactions also play a crit-ical role in binding of VCP to C3b and C4b, we measured binding at different salt concentrations. VCP immobilized on a CM5 sensor chip was allowed to bind to C3b or C4b in the presence of different NaCl concentrations. The buffer used for binding was 10 mM phosphate (pH 7.4) containing 0.05% Tween 20 and various concentrations of NaCl ranging from 30 to 400 mM. It is clear from the data that binding declined with increases in the NaCl concentration and was entirely abolished at 400 mM NaCl (Fig. 7). We have verified these data by performing experiments in the reverse orientation, wherein C3b or C4b were oriented on the sensor chip and VCP was allowed to bind at different salt concentrations (data not shown). The data confirmed that both VCP-C3b and VCP-C4b complex formations are strongly dependent on salt concentra-tion. These data indicate that, like host CCPs, ionic
interac-tions are also critical in the formation of C3b and VCP-C4b complexes.
Does VCP bind simultaneously to C3b and C4b? Because
VCP interacts with C3b, as well as C4b, we sought to deter-mine whether VCP could interact simultaneously with C3b and C4b. To address this issue, we evaluated the binding of VCP or VCP preincubated with C4b to C3b immobilized on a strepta-vidin chip. A positive control was formed by analyzing binding of VCP preincubated with anti-VCP monoclonal antibody (MAb) NCCS 67.2 to immobilized C3b. This MAb binds to VCP (KD⫽ 3.8 nM [determined by SPR analysis]) but does
[image:7.603.50.534.71.362.2]not inhibit the factor I cofactor activity of VCP for C3b and C4b (data not shown). In this assay, the expected response was if both C3b and C4b bound simultaneously to VCP then there would be a buildup of response in VCP-C4b complex binding compared to binding of VCP alone. Binding of VCP alone yielded a maximum response of 22 RU, whereas binding of VCP-C4b complex (VCP preincubated with C4b) yielded a response of 19 RU, demonstrating that there was no buildup in response (Fig. 8A). As expected, the binding of VCP-MAb complex (positive control) showed a buildup in response with a maximum signal of 61 RU. The MAb and C4b by themselves yielded no response. Similar results were obtained when the
FIG. 5. Binding of sCR1 and factor H to immobilized C3b and C4b. The left panels show overlay plots for binding of sCR1, factor H to immobilized C3b, and sCR1 to immobilized C4b. Various concentrations of analytes were injected over streptavidin chips containing biotinylated C3b or C4b. Solid lines shown in the C3b-CR1 panel represent the global fitting of the data to a bivalent analyte model (A⫹B7AB; AB⫹B
7AB2; BIAevaluation 3.2). The middle panels show the linear transformation of the association phase data of the respective sensogram data
shown on the left. The straight lines are linear least-square fits to the data. The inset shows values ofks(determined from the slope of the fits of
the association data) replotted against analyte concentration. The slope of this plot providedka. The right panels show the linear transformation
of the highest concentration of the dissociation phase data of the respective analyte shown on the left. The slope of the fits provided the off rates (kd).
on November 8, 2019 by guest
experiment was performed by using the converse arrangement (Fig. 8B). Thus, binding of VCP and VCP-C3b complex to immobilized C4b showed maximum responses of 39 and 40 RU, respectively, whereas VCP-MAb binding (positive con-trol) resulted in a maximum response of 270 RU. The MAb and C3b alone did not show any binding to C4b. These data indicate that VCP does not complex simultaneously with C3b and C4b.
Localization of VCP binding site on C3b and C4b.In the
present study, we also attempted to sublocalize the binding site of VCP on C3b and C4b. For this purpose, we characterized binding of VCP to physiologic fragments of C3b (C3c and C3dg) and C4b (C4c and C4d) with SPR. Biotinylated C3dg
and C4d were generated by cleaving the site-specific biotinyl-ated C3b and C4b with sCR1 and factor I (Fig. 4), whereas biotinylated C3c and C4c were generated by cleaving the un-labeled C3b and C4b with factor I and sCR1 and labeling them with EZ-Link-Sulfo-NHS-Biotin. The labeled C3dg/C4d (1100 RU/880 RU) and C3c/C4c (1300 RU/850 RU) were immobi-lized on Fc-2 and Fc-3 of streptavidin chips, and Fc-1 served as a control. Binding was studied by flowing VCP in PBS (pH 7.4). VCP bound to C3dg and C4c but not to C3c and C4d (Fig. 9). Binding to both C3dg and C4c was dose dependent. TheKD
values for C3dg and C4c were 0.18 and 0.15M, respectively. Binding of VCP to C4c instead of C4d, as one may expect, is not unusual since a recent study reported that CR1 binds to C4c but not C4d (8). From the data presented here it is evident that the nature of interaction of VCP with C3b is different than that of C4b.
DISCUSSION
Previous studies have established VCP as a virulence deter-minant of vaccinia virus. Using a skin lesion model it has been shown that vaccinia virus mutants that do not express VCP are attenuated in rabbits and guinea pigs (17). It is believed that this in vivo effect is due to enhanced complement-mediated neutralization of the mutant virus as well as the generation of specific inflammatory response at the site of infection due to the lack of VCP-mediated inactivation of the complement sys-tem. Although it is clear that VCP inactivates complement by interacting with complement proteins C3b and C4b, the nature of mechanisms involved is not clear. SPR analyses make it possible to probe the mechanism of protein-protein interac-tion; therefore, in the present study we have utilized this tech-nology to unravel the molecular mechanisms underlying the interaction between VCP and its target proteins C3b and C4b. Our real-time kinetic data obtained from SPR measure-ments of binding of C3b and C4b to VCP immobilized by using amine-coupling chemistry indicate that these interactions do not follow a 1:1 binding model. Linear transformation of the data showed that the reactions are multistep (Fig. 2). Such reactions could reflect the presence of multiple binding sites with different affinities, a conformational change, or a more complex model. The multistep binding model gains some sup-port from the previous studies. (i) Based on the earlier mu-tagenesis data on membrane cofactor protein and the crystal structure of VCP, it has been proposed that a number of sites on VCP might interact with C3b and C4b (39). (ii) A study on factor H-C3b interaction suggested that binding of factor H to C3b causes a conformational change in C3b (51). Thus, there is a possibility that VCP and/or the target molecules (C3b and C4b) undergo structural reorientation upon binding.
The multistep binding, however, could simply be due to heterogeneity in the analyte and/or the surface under test. Both C3b and C4b are known to form dimers in solution. Such dimers are formed either due to thioester linkage formation between the ␣⬘ chains and/or the disulfide bond formation between the free⫺SH groups of C3b and C4b (54). We have ruled out the first possibility by purifying C3b and C4b over a gel filtration column. Prior to SPR experiments, C3b and C4b were passed over Superose 12 column (Amersham Pharmacia Biotech; two columns linked in a series), and the absence of
120 100 80 60 40 20 0
0.001 0.01 0.1 1.0
VCP conc.(
µ
M)
’- chain (%)
C3b C4b 68 kDa ’ VCP 43 kDa C3b 11.5 VCP(nM) + + + +
Factor I - + + + + +
23.1 46.3 92.5 185 370 740 1480
+ + + + + + + + 1480 VCP C4d 25kDa ’ C4b 2.87 VCP(nM) + + + +
Factor I - + + + + +
5.75 11.5 23.1 46.3 92.5 185 370
+ + +
+ + + +
[image:8.603.68.258.69.467.2]+ 2960
FIG. 6. Comparison of factor I cofactor activity of VCP for C3b and C4b. Equimolar concentrations of C3b or C4b and factor I were incubated in 10 mM sodium phosphate (pH 7.4) containing 145 mM NaCl with increasing concentrations of VCP at 37°C for 4 h (upper and middle panels). Cleavage products were visualized by running the samples on 8% and 9% SDS-PAGE gels for C3b and C4b, respectively, and staining with Coomassie blue. The intensities of the␣⬘chains of C3b and C4b were determined by densitometric analysis and are rep-resented graphically (lower panel).
on November 8, 2019 by guest
http://jvi.asm.org/
dimers was confirmed by SDS-PAGE. Amine coupling method, however, is known to produce heterogeneity in the ligand surface due to differential attachment of ligands. Thus, to rule out this possibility, we oriented C3b and C4b onto streptavidin chips by labeling Cys988 and Cys991, respectively
(Fig. 3), which are involved in the thioester bond formation. This strategy not only allowed us to generate a homogeneous ligand surface but also simulated the in vivo orientation of these proteins (Fig. 3A and B); C3b and C4b bound to the streptavidin chips simulated C3b and C4b deposition on the activating surface, and VCP in solution mimicked binding of soluble VCP to deposited C3b and C4b. The data shown in Fig. 4 clearly demonstrate that the binding reactions between VCP and C3b, as well as C4b, follow a 1:1 binding model. Our data, therefore, indicate that these reactions do not involve multiple site interactions or conformational changes and are less com-plex than previously thought. The simple 1:1 interaction seems to be a general phenomenon between viral homologs of com-plement control proteins and C3b/C4b since a similar binding mechanism was observed between kaposica (the Kaposi’s sar-coma-associated herpesvirus homolog of CCP) and C3b/C4b (J. Mullick and A. Sahu, unpublished observation). How com-plement regulators (e.g., CR1, factor H, MCP, and C4BP) function as factor I cofactors in the degradation of C3b and C4b is not clear at present. It has been suggested, at least in the case of factor H, that binding of this molecule induces a con-formational change in C3b that facilitates the binding of factor I (51). It is clear from our data that VCP does not induce conformational change either in C3b or in C4b; thus, it seems
that structural reorientation in C3b and C4b is not necessary for factor I cofactor activities of viral homologs of CCPs.
VCP has previously been shown to inhibit both the classical and the alternative pathways of the complement system (32, 42). In a comparative analysis it was shown that VCP was 34-fold more active in inhibiting the classical pathway than the alternative pathway (42). It was suggested that the greater effect of VCP on the classical pathway was due to its dual effect on C3b and C4b. It is, however, possible that the greater effect of VCP on the classical pathway could be in part due to higher affinity of VCP for C4b. The comparative affinity data of VCP for C3b and C4b presented in Table 1 indicate that its affinity for C4b is 4.6-fold better than that of C3b. Furthermore, its higher affinity for C4b is also reflected in its factor I cofactor activity; VCP showed 3.3-fold greater factor I cofactor activity for C4b than C3b (Fig. 6). Based on these data we suggest that the greater activity of VCP against the classical pathway is due to its dual effect on C3b and C4b, as well as to its higher affinity for C4b.
There is now a consensus that ionic interactions form an essential component of the binding interface between CCPs (factor H, CR1, C4BP, and MCP) and C3b/C4b. This premise is based on the following two observations: (i) these interac-tions show ionic strength dependence and (ii) extensive mu-tagenesis data show that the negatively charged residues on C3b and C4b and positively charged residues on CCPs are important for CCP-C3b/C4b interactions (4, 6, 11, 25, 30, 40). Because VCP is structurally and functionally similar to CCPs, we sought to determine whether VCP utilizes a similar
mech-FIG. 7. Effect of NaCl concentration on the binding of VCP to C3b and C4b. Sensogram overlays for binding of immobilized VCP to C3b (125 nM) and C4b (100 nM) in the presence of various concentrations of NaCl are shown. On the left, the binding response (RU) is plotted against time. On the right, the maximum binding response obtained for each buffer condition is plotted against the NaCl concentration.
on November 8, 2019 by guest
[image:9.603.102.482.71.335.2]anism to stabilize the binding with C3b and C4b. The data obtained here clearly show that binding of VCP to C3b and C4b is highly dependent on ionic interactions (Fig. 7). Thus, our data suggest that long-range electrostatic forces and ion pairing are critical in the formation of stable VCP-C3b/C4b complexes. The crystal structure of the entire VCP molecule revealed that the SCR domains 1 and 4 are highly positively charged, whereas the middle two SCR domains are surrounded by a net negative field (39). Based on our data and the premise that positively charged residues of CCPs play a predominant role in ionic interactions, it could be speculated that SCRs 1 and 4 of VCP play a significant role in ionic interactions. However, a previous study on the monkey poxvirus homolog of VCP, which is devoid of a large portion of SCR 4, showed that this protein contains complement inhibitory activity (49). Therefore, it is likely that ionic interactions in VCP occur mainly through SCR 1.
SPR analyses allow measure of bi- and multimolecular com-plex formation and thus serve as a powerful tool for examining
surface topography. Therefore, in the present study, we also sought to determine whether VCP could bind simultaneously to C3b and C4b. We measured binding of VCP or VCP preincu-bated with C4b to C3b-biotin immobilized on a streptavidin chip. We observed that preincubation of VCP with C3b did not result in an increase in mass on the chip (Fig. 8A). These results were also validated by using a converse arrangement wherein VCP or VCP preincubated with C3b was allowed to bind to immobilized C4b (Fig. 8B). These data indicate that C3b and C4b binding to VCP are not independent events; thus, VCP may not be able to execute the dual functions of inactivating C3b and C4b together. Whether the lack of simultaneous binding of C3b and C4b to VCP is due to competition for the same binding site or sterically hindered access to these sites due to close proximity is not clear and requires further investigation.
Our data on the localization of VCP binding site on C3b and C4b has revealed that these sites are located in the C3dg and C4c regions, respectively (Fig. 9). It should be pointed out here that, to date, VCP is the only viral protein known to interact with C3dg (38). The other C3-interacting viral proteins, which have been studied for their interaction with C3 fragments, are glycoprotein C of herpes simplex virus types 1 and 2, and these are known to bind to C3c (21, 31). There is now a large body of evidence implicating the importance of interaction between C3dg (a proteolytically cleaved fragment of C3) with its recep-tor CR2 (CD21), which is present on B cells and follicular dendritic cells, in the modulation of antibody-mediated hu-moral response (7, 29, 43). For example, mice deficient either in C3 or in C4 protein showed impaired antibody response to suboptimal doses of T-dependent antigen, and their responses were characterized by reduced number and size of germinal centers (10, 12). Similarly, mice containing disrupted CR2 lo-cus showed impaired antibody response to T-dependent gens (1, 34). In addition, the administration of CR2 anti-body or the soluble form of CR2 suppressed the in vivo immune response (14, 16). Whether binding of VCP to C3dg inhibits its interaction with CR2 is not clear at present. How-ever, such interaction would be of great benefit to the virus because then VCP would not only inhibit the innate (comple-ment activation) but would also inhibit the acquired immune response (generation of specific antibody production).
A central question in the biology of vaccinia virus is, what role does complement play in controlling its infection and how does VCP help to subvert it? Previous in vitro studies have clearly shown that infectivity of both the infectious forms of vaccinia virus, i.e., intracellular immature virus (17, 53), as well as EEV (53), was destroyed by complement when anti-vaccinia virus antibodies were present during the assay. It is important to point out here that although the incorporation of host com-plement regulators confers resistance to EEV against the al-ternative pathway, this form is still susceptible to complement-mediated neutralization by the classical pathway (53). Thus, for successful propagation of the virus, it would be advanta-geous for the virus to encode a complement regulatory protein that would effectively inhibit the classical pathway. The data obtained thus far make it clear that VCP effectively inhibits the classical pathway-mediated inactivation of both intracellular immature virus and EEV (17, 53). It is noteworthy that this viral protein is a more effective regulator of the classical path-way than even the host complement regulators factor H and
FIG. 8. Simultaneous binding of C3b and C4b to VCP. (A) VCP (0.6M), VCP (0.6M) preincubated with C4b (0.56M), or VCP (0.6M) preincubated with anti-VCP MAb (0.4M [this antibody does not inhibit the functional activity of VCP]) at 25°C for 30 min was injected over a streptavidin chip containing biotinylated C3b, and the association and dissociation phases were monitored. (B) VCP (0.6 M), VCP (0.6M) preincubated with C3b (0.6M), or VCP (0.6 M) preincubated with anti-VCP MAb (0.2M) at 25°C for 30 min was injected over a streptavidin chip containing biotinylated C4b, and binding and dissociation were monitored.
on November 8, 2019 by guest
http://jvi.asm.org/
C4-binding protein (22, 42). The data presented here indicate that the greater effect of VCP against the classical pathway is due to its higher affinity for C4b than C3b (Fig. 4 and Table 1). How VCP regulates complement-mediated inactivation of vaccinia virus in vivo is not clear at present. A prior study had shown that VCP deletion mutant produced smaller skin lesions than the wild-type virus in rabbits (17). Interestingly, lesions produced by mutant and wild-type viruses were similar for the first few days but reduced in size after day 5 in the mutant virus, and this timing correlated well with the appearance of anti-vaccinia virus antibody. It is therefore likely that VCP protects vaccinia virus from the classical pathway of comple-ment during the late phase of infection. In the present study we have shown that VCP interacts with C3dg fragment of C3 (Fig. 9). Thus, as discussed above, it is also possible that VCP-mediated inhibition of specific antibody generation leads to protection of vaccinia virus from the classical pathway of com-plement activation. Together, these observations suggest that structural determinants of VCP important in interacting with C3b and C4b may act as potential targets for future therapies and drug development. The initial belief that multiple-site interactions exist between VCP and C3b and C4b (39) sug-gested that such an endeavor would need blocking of multiple interactions and thus would not be a realistic approach. How-ever, our data suggest that these interactions follow a simple one-site model and therefore targeting VCP would be feasible.
ACKNOWLEDGMENTS
We thank John D. Lambris (Department of Pathology and Labora-tory Medicine, University of Pennsylvania, Philadelphia, Pa.) for con-tinuous support; Michael K. Pangburn (Department of Biochemistry, University of Texas Health Science Center, Tyler, Tex.) for support and the generous gift of complement proteins C3, factor H, and factor I; Henry Marsh (AVANT Immunotherapeutics, Inc., Needham, Mass.) for providing sCR1; and Gabriela Canziani (Protein Interaction Facility, University of Utah, Salt Lake City) for discussion and advice on the maintenance of Biacore. We also thank Sharanabasava Halli-hosur for excellent technical assistance.
This study was supported by the Wellcome Trust Overseas Senior Research Fellowship in Biomedical Science in India to A.S.
REFERENCES
1. Ahearn, J. M., M. B. Fischer, D. Croix, S. Goerg, M. Ma, J. Xia, X. Zhou, R. G. Howard, T. L. Rothstein, and M. C. Carroll.1996. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B-cell response to T-dependent antigen. Immunity4:251–262.
2. Alcami, A., and U. H. Koszinowski.2000. Viral mechanisms of immune evasion. Trends Microbiol.8:410–418.
3. Alsenz, J., J. D. Lambris, T. F. Schulz, and M. P. Dierich.1984. Localization of the complement-component-C3b-binding site and the cofactor activity for factor I in the 38-kDa tryptic fragment of factor H. Biochem. J.224:389–398. 4. Arnaout, M. A., N. Dana, J. Melamed, R. Medicus, and H. R. Colten.1983. Low ionic strength or chemical cross-linking of monomeric C3b increases its binding affinity to the human complement C3b receptor. Immunology48:
229–237.
5. Bernet, J., J. Mullick, A. K. Singh, and A. Sahu.2003. Viral mimicry of the complement system. J. Biosci.28:249–264.
6. Blom, A. M., J. Webb, B. O. Villoutreix, and B. Dahlback.1999. A cluster of positively charged amino acids in the C4BP alpha-chain is crucial for C4b binding and factor I cofactor function. J. Biol. Chem.274:19237–19245. FIG. 9. Binding of VCP to immobilized C3b and C4b fragments. The left panels show sensograms for interactions between VCP and C3b or C4b fragments. VCP (1.3M) was injected over a streptavidin chip immobilized either with C3b fragments (C3c-biotin and C3dg-biotin) or with C4b fragments (C4c-biotin and C4d-biotin). The right panels show overlay plots for interactions between immobilized C3dg or C4c and VCP. The small arrow in the C3dg-VCP panel indicates the time point used for evaluating steady-state affinity data. Solid lines shown in the C4c-VCP panel represent the global fitting of the data to a 1:1 Langmuir binding model (A⫹B7AB; BIAevaluation 3.2).
on November 8, 2019 by guest
7. Carroll, M. C., and D. T. Fearon. 2004. Regulation by complement of acquired immunity, p. 327–333.InJ. E. Volanakis and M. M. Frank (ed.), The human complement system in health and disease. Marcel Dekker, Inc., New York, N.Y.
8. Clemenza, L., and D. E. Isenman.2004. The C4A and C4B isotypic forms of human complement fragment C4b have the same intrinsic affinity for com-plement receptor 1 (CR1/CD35). J. Immunol.172:1670–1680.
9. Cooper, N. R.1998. Complement and viruses, p. 393–407.InJ. E. Volanakis and M. M. Frank (ed.), The human complement system in health and disease. Marcel Dekker, Inc., New York, N.Y.
10. Da Costa, X. J., M. A. Brockman, E. Alicot, M. Ma, M. B. Fischer, X. Zhou, D. M. Knipe, and M. C. Carroll.1999. Humoral response to herpes simplex virus is complement-dependent. Proc. Natl. Acad. Sci. USA96:12708–12712. 11. DiScipio, R. G.1981. The binding of human complement proteins C5, factor B,1H and properdin to complement fragment C3b on zymosan. Biochem. J.199:485–496.
12. Fischer, M. B., M. Ma, S. Goerg, X. Zhou, J. Xia, O. Finco, S. Han, G. Kelsoe, R. G. Howard, T. L. Rothstein, E. Kremmer, F. S. Rosen, and M. C. Carroll.1996. Regulation of the B-cell response to T-dependent antigens by classical pathway complement. J. Immunol.157:549–556.
13. Gewurz, B. E., R. Gaudet, D. Tortorella, E. W. Wang, and H. L. Ploegh.2001. Virus subversion of immunity: a structural perspective. Curr. Opin. Immu-nol.13:442–450.
14. Hebell, T., J. M. Ahearn, and D. T. Fearon.1991. Suppression of the immune response by a soluble complement receptor of B lymphocytes. Science254:
102–105.
15. Henderson, C. E., K. Bromek, N. P. Mullin, B. O. Smith, D. Uhrin, and P. N. Barlow.2001. Solution structure and dynamics of the central CCP module pair of a poxvirus complement control protein. J. Mol. Biol.307:323–339. 16. Heyman, B., E. J. Wiersma, and T. Kinoshita.1990. In vivo inhibition of the
antibody response by a complement receptor-specific monoclonal antibody. J. Exp. Med.172:665–668.
17. Isaacs, S. N., G. J. Kotwal, and B. Moss.1992. Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neu-tralization of infectivity and contributes to virulence. Proc. Natl. Acad. Sci. USA89:628–632.
18. Johnston, J. B., and G. McFadden.2003. Poxvirus immunomodulatory strat-egies: current perspectives. J. Virol.77:6093–6100.
19. Jokiranta, T. S., J. Hellwage, V. Koistinen, P. F. Zipfel, and S. Meri.2000. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J. Biol. Chem.275:27657–27662.
20. Klickstein, L. B., T. J. Bartow, V. Miletic, L. D. Rabson, J. A. Smith, and D. T. Fearon.1988. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J. Exp. Med.168:1699–1717.
21. Kostavasili, I., A. Sahu, H. M. Friedman, R. J. Eisenberg, G. H. Cohen, and J. D. Lambris.1997. Mechanism of complement inactivation by glycoprotein C of herpes simplex virus. J. Immunol.158:1763–1771.
22. Kotwal, G. J.1994. Purification of virokines using ultrafiltration. Am. Bio-technol. Lab.12:76–77.
23. Kotwal, G. J., S. N. Isaacs, R. Mckenzie, M. M. Frank, and B. Moss.1990. Inhibition of the complement cascade by the major secretory protein of vaccinia virus. Science250:827–830.
24. Kotwal, G. J., and B. Moss. 1988. Vaccinia virus encodes a secretory polypeptide structurally related to complement control proteins. Nature
335:176–178.
25. Krych, M., R. Hauhart, and J. P. Atkinson.1998. Structure-function analysis of the active sites of complement receptor type 1. J. Biol. Chem.273:8623– 8629.
26. Krych, M., D. Hourcade, and J. P. Atkinson.1991. Sites within the comple-ment C3b C4b receptor important for the specificity of ligand binding. Proc. Natl. Acad. Sci. USA88:4353–4357.
27. Lachmann, P. J.2002. Microbial subversion of the immune response. Proc. Natl. Acad. Sci. USA99:8461–8462.
28. Lambris, J. D., A. Sahu, and R. Wetsel.1998. The chemistry and biology of C3, C4, and C5, p. 83–118.InJ. E. Volanakis and M. Frank (ed.), The human complement system in health and disease. Marcel Dekker, Inc., New York, N.Y. 29. Lee, S. H., J. U. Jung, and R. E. Means.2003. “Complementing” viral infection: mechanisms for evading innate immunity. Trends Microbiol.11:
449–452.
30. Liszewski, M. K., M. Leung, W. Cui, V. B. Subramanian, J. Parkinson, P. N. Barlow, M. Manchester, and J. P. Atkinson.2000. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J. Biol. Chem.275:37692–37701.
31. Lubinski, J., T. Nagashunmugam, and H. M. Friedman.1998. Viral inter-ference with antibody and complement. Semin. Cell Dev. Biol.9:329–337. 32. Mckenzie, R., G. J. Kotwal, B. Moss, C. H. Hammer, and M. M. Frank.1992.
Regulation of complement activity by vaccinia virus complement-control protein. J. Infect. Dis.166:1245–1250.
33. Means, R. E., S. M. Lang, Y. H. Chung, and J. U. Jung.2002. Kaposi’s sarcoma associated herpesvirus immune evasion strategies. Front Biosci.
7:e185–e203.
34. Molina, H., V. M. Holers, B. Li, Y. Fung, S. Mariathasan, J. Goellner, J. Strauss-Schoenberger, R. W. Karr, and D. D. Chaplin.1996. Markedly impaired humoral immune response in mice deficient in complement recep-tors 1 and 2. Proc. Natl. Acad. Sci. USA93:3357–3361.
35. Morton, T. A., D. B. Bennett, E. R. Appelbaum, D. M. Cusimano, K. O. Johanson, R. E. Matico, P. R. Young, M. Doyle, and I. M. Chaiken.1994. Analysis of the interaction between human interleukin-5 and the soluble domain of its receptor using a surface plasmon resonance biosensor. J. Mol. Recognit.7:47–55.
36. Moss, B.2001.Poxviridae: the viruses and their replication, p. 2849–2883.In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott/The Williams & Wilkins Co., Philadelphia, Pa.
37. Mullick, J., J. Bernet, A. K. Singh, J. D. Lambris, and A. Sahu.2003. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) open read-ing frame 4 protein (kaposica) is a functional homolog of complement control proteins. J. Virol.77:3878–3881.
38. Mullick, J., A. Kadam, and A. Sahu.2003. Herpes and pox viral complement control proteins: “the mask of self.” Trends Immunol.24:500–507. 39. Murthy, K. H., S. A. Smith, V. K. Ganesh, K. W. Judge, N. Mullin, P. N.
Barlow, C. M. Ogata, and G. J. Kotwal.2001. Crystal structure of a com-plement control protein that regulates both pathways of comcom-plement acti-vation and binds heparan sulfate proteoglycans. Cell104:301–311. 40. Oran, A. E., and D. E. Isenman.1999. Identification of residues within the
727–767 segment of human complement component C3 important for its interaction with factor H and with complement receptor 1 (CR1, CD35). J. Biol. Chem.274:5120–5130.
41. Rosengard, A. M., L. C. Alonso, L. C. Korb, W. M. Baldwin, III, F. Sanfil-ippo, L. A. Turka, and J. M. Ahearn.1999. Functional characterization of soluble and membrane-bound forms of vaccinia virus complement control protein (VCP). Mol. Immunol.36:685–697.
42. Sahu, A., S. N. Isaacs, A. M. Soulika, and J. D. Lambris.1998. Interaction of vaccinia virus complement control protein with human complement pro-teins: factor I-mediated degradation of C3b to iC3b1inactivates the
alter-native complement pathway. J. Immunol.160:5596–5604.
43. Sahu, A., and J. D. Lambris.2001. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Im-munol. Rev.180:35–48.
44. Sahu, A., A. M. Soulika, D. Morikis, L. Spruce, W. T. Moore, and J. D. Lambris.
2000. Binding kinetics, structure-activity relationship, and biotransformation of the complement inhibitor compstatin. J. Immunol.165:2491–2499.
45. Sarrias, M. R., S. Franchini, G. Canziani, E. Argyropoulos, W. T. Moore, A. Sahu, and J. D. Lambris.2001. Kinetic analysis of the interactions of com-plement receptor 2 (CR2, CD21) with its ligands C3d, iC3b, and the EBV glycoprotein gp350/220. J. Immunol.167:1490–1499.
46. Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden.2003. Poxviruses and immune evasion. Annu. Rev. Immunol.21:377–423. 47. Seya, T., K. Nakamura, T. Masaki, C. Ichihara-Itoh, M. Matsumoto, and S.
Nagasawa.1995. Human factor H and C4b-binding protein serve as factor I-cofactors both encompassing inactivation of C3b and C4b. Mol. Immunol.
32:355–360.
48. Sharma, A. K., and M. K. Pangburn.1996. Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc. Natl. Acad. Sci. USA93:10996–11001. 49. Smith, S. A., N. P. Mullin, J. Parkinson, S. N. Shchelkunov, A. V. Totmenin,
V. N. Loparev, R. Srisatjaluk, D. N. Reynolds, K. L. Keeling, D. E. Justus, P. N. Barlow, and G. J. Kotwal.2000. Conserved surface-exposed K/R-X-K/R motifs and net positive charge on poxvirus complement control proteins serve as putative heparin binding sites and contribute to inhibition of mo-lecular interactions with human endothelial cells: a novel mechanism for evasion of host defense. J. Virol.74:5659–5666.
50. Smith, S. A., R. Sreenivasan, G. Krishnasamy, K. W. Judge, K. H. Murthy, S. J. Arjunwadkar, D. R. Pugh, and G. J. Kotwal.2003. Mapping of regions within the vaccinia virus complement control protein involved in dose-de-pendent binding to key complement components and heparin using surface plasmon resonance. Biochim. Biophys. Acta1650:30–39.
51. Soames, C. J., and R. B. Sim.1997. Interactions between human comple-ment components factor H, factor I and C3b. Biochem. J.326:553–561. 52. Stoiber, H., C. Speth, and M. P. Dierich.2003. Role of complement in the
control of HIV dynamics and pathogenesis. Vaccine21(Suppl. 2):S77–S82. 53. Vanderplasschen, A., E. Mathew, M. Hollinshead, R. B. Sim, and G. L. Smith.1998. Extracellular enveloped vaccinia virus is resistant to comple-ment because of incorporation of host complecomple-ment control proteins into its envelope. Proc. Natl. Acad. Sci. USA95:7544–7549.
54. Volanakis, J. E., and M. M. Frank.1998. The human complement system in health and disease. Marcel Dekker, Inc., New York, N.Y.
55. Willis, S. H., A. H. Rux, C. Peng, J. C. Whitbeck, A. V. Nicola, H. Lou, W. F. Hou, L. Salvador, R. J. Eisenberg, and G. H. Cohen.1998. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol.72:5937–5947.