Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Disturbance of Tumor Necrosis Factor Alpha-Mediated Beta
Interferon Signaling in Cervical Carcinoma Cells
Anastasia Bachmann, Brigitte Hanke, Rainer Zawatzky, Ubaldo Soto, Jan van Riggelen,
Harald zur Hausen, and Frank Ro¨sl*
Forschungsschwerpunkt Angewandte Tumorvirologie, Abteilung Tumorvirus-Immunologie, Deutsches Krebsforschungszentrum, Heidelberg, Federal Republic of Germany
Received 3 July 2001/Accepted 28 September 2001
In the present study we show that malignant human papillomavirus (HPV)-positive cells lost their ability to
synthesize endogenous beta interferon (IFN-) upon tumor necrosis factor alpha (TNF-␣) treatment. IFN-
transcription, however, was reinducible in nonmalignant HPV-positive cells, which was confirmed in functional protection assays against encephalomyocarditis virus or vesicular stomatitis virus infections. Addition of
neutralizing antibodies against IFN-blocked the antiviral effect, excluding the possibility that other IFN types
were involved. Conversely, both malignant and immortalized cells could be protected against viral cytolysis
when either IFN-, IFN-␣, or IFN-␥was added exogenously. This indicates that only the cross talk between
TNF-␣and the IFN-pathways, and not IFN-␣/and IFN-␥signaling in general, is perturbed in cervical
carcinoma cells. Notably, full virus protection was restricted exclusively to nonmalignant cells, indicating that
the antiviral effect correlates with the growth-inhibitory and virus-suppressive properties of TNF-␣. The
IFN-regulatory factors IRF-1 and p48 (ISGF3␥) emerged as key regulatory molecules in the differential IFN-
response, since their transcription was either absent or only inefficiently enhanced in tumorigenic cells upon
treatment with TNF-␣. Inducibility of both genes, however, became reestablished in cervical carcinoma cells,
which were complemented to nontumorigenicity after somatic cell hybridization. Complementation was
par-alleled by the entire reconstitution of cytokine-mediated IFN-expression and the ability of TNF-␣to exert an
antiviral state. In contrast, under conditions where tumor suppression was not accomplished upon somatic cell
hybridization, neither expression of IRF-1, p48, and IFN-nor antiviral activity could be restored.
Human papillomavirus (HPV)-induced carcinogenesis is a multistep process which is initiated by viral infection (68). At what time and to what extent tumor formation takes place, however, are dependent mainly on the immunological status of the patient. As deduced from epidemiological data, immuno-suppressed individuals or persons with impaired immunocom-petence have a significantly higher risk for developing cervical cancer than corresponding age-matched controls (53). Hence, tumor appearance can be regarded in part as the result of an immunological escape process during which either certain in-ter- and intracellular surveillance mechanisms are functionally abolished or HPV-positive cells are no longer susceptible to immunological control (69). It is therefore reasonable to as-sume that only a physiologically intact communication pathway between inflammatory and HPV-containing cells guarantees a proper antiviral response (52).
Indeed, when fresh biopsies from patients were evaluated by immunohistochemistry, cervical cancer sections were found to be significantly depleted of infiltrating macrophages, T lym-phocytes, and dendritic cells compared with premalignant tis-sue specimens (22, 60, 63, 64). The recruitment of immuno-logical effector cells in turn is mediated by chemokines (chemotactic cytokines) such as monocyte-chemotactic pro-tein-1 (MCP-1) (39, 45, 47). Consistent with the notion that dysregulation in intercellular communication may favor the
outcome of neoplasias, all cervical carcinoma lines tested up to now are devoid of significant inducible MCP-1 expression. Cytokine inducibility, however, can be completely restored in somatic cell hybrids with normal cells (51), which were no longer tumorigenic when heterotransplantated into immuno-compromised animals (61). Tumorigenic segregants derived from the same hybrids again lost MCP-1 expression (28, 51), strongly suggesting that elimination of chemokine expression may provide a selective advantage for tumor formation (52). It should be stressed that this correlation is not restricted to tissue culture conditions, because in situ hybridization studies in combination with immunohistochemistry techniques con-firmed that MCP-1 expression and infiltrating cells of the monocyte/macrophage lineage were detectable only in prema-lignant precursor cells and were absent in high-grade lesions of cancer patients (29, 46).
Recruitment and activation of macrophages can be consid-ered the first line of defense against generalized virus infec-tions and viral spread (for reviews, see references 17 and 19). For example, spontaneous regression of benign warts is accom-panied by a strong infiltration of mononuclear cells (41), where papilloma shrinkage directly correlates with high tumor necro-sis factor alpha (TNF-␣) expression in surrounding macro-phages (20). Hence, TNF-␣ not only may represent a key regulatory cytokine in regression of benign tumors (20) but also could play a pivotal role in the immunological control of dysplastic cervical lesions infected with high-risk HPV types such as HPV type 16 (HPV16) or HPV18. Although not yet directly demonstrated in patients, it is conceivable that mac-rophage-specific TNF-␣synthesis can trigger paracrine MCP-1 * Corresponding author. Mailing address: Angewandte
Tumorvi-rologie, Deutsches Krebsforschungszentrum, Im Neuenheimer Feld 242, 69120 Heidelberg, Federal Republic of Germany. Phone: 49-6221-42-4900. Fax: 49-6221-42-4902. E-mail: F.Roesl@DKFZ.de.
280
on November 8, 2019 by guest
http://jvi.asm.org/
gene expression in immortalized cells (and vice versa) which in turn augments mononuclear cell infiltration as well as secretion of larger amounts of growth-inhibitory cytokines. In support of this assumption was the finding that cocultivation with acti-vated macrophages from normal human volunteers can both suppress viral transcription and induce MCP-1 gene expres-sion, but only in nonmalignant HPV-positive cells (51). Con-versely, cervical carcinoma cells were completely refractory, despite functional TNF-␣ signaling, as monitored by rapid proteolysis of IB␣upon cytokine application (13). The block-age of MCP-1 synthesis and the concomitant resistance to TNF-␣may therefore perturb the intercellular cross talk and favor the accumulation of malignant cells (52).
Another interesting property of TNF-␣is its ability to confer an antiviral state through induction of the beta interferon (IFN-) gene (23, 38). IFN-␣/production is the most rapid host immune response against a variety of viral infections (9). Moreover, it also became evident that the elimination of virus-positive cells is not entirely exerted through major histocom-patibility complex class I-restricted CD8⫹cytotoxic T
lympho-cytes but also relies on appropriate temporal and local expression of cytokines such as TNF-␣and downstream IFN signaling (19, 35).
Considering this scenario in the context of the transforming potential of high-risk HPVs, there are a quite substantial num-ber of studies which show that the IFN signal transduction pathway is a target for viral oncoproteins E6 and E7. For example, HPV18 E6 can interfere with the IFN-␣response in human fibrosarcoma HT1080 cells by impairing Jak-STAT1/2 tyrosine phosphorylation after binding of the ligand to the corresponding receptor (32). Furthermore, E7 expression in spontaneously immortalized human keratinocytes (HaCaT cells) can inhibit p48 (ISGF3␥) protein translocation into the nucleus (3). p48 (ISGF3␥) is part of IFN-stimulated gene fac-tor 3 (ISGF3), a trimerictrans-activating complex formed be-tween p48, STAT1, and STAT2 which induces transcription of IFN-regulated genes via binding to cognate IFN-stimulated response elements (5). However, all of these properties of the viral oncogenes do not provide a reliable explanation of why IFN still has a curative and growth-inhibitory effect on prema-lignant HPV-positive cells (6, 14, 25), despite the fact that E6 and E7 are found to be expressed in those lesions.
To gain insight into IFN signaling and the antiviral activity of TNF-␣, we used cervical carcinoma cells and malignant and nonmalignant somatic cell hybrids derived therefrom as a model system (61). For example, HPV18-positive HeLa cells can be complemented to nontumorigenic phenotype growth after somatic cell hybridization with normal human fibroblasts or keratinocytes. Occasionally, however, rare tumorigenic seg-regants of the same hybrids arise, which is accompanied by the nonrandom loss of chromosomes, in particular of chromosome 11 (for a review, see reference 52). In the case of HPV18-positive cervical carcinoma cells, chromosome 11 indeed seems to play a key role in malignant transformation, since the rein-troduction of the corresponding normal allele via microcell transfer is sufficient to completely suppress tumor formation of either the parental cells or the tumorigenic segregants (52a). Here we demonstrate that, similar to the case for the MCP-1 gene (28, 51), IFN-expression could be induced by TNF-␣
only in nonmalignant HPV-positive cells. The functionality and
specificity of endogenous IFN-production were assessed in antiviral protection assays using encephalomyocarditis virus (EMCV) or vesicular stomatitis virus (VSV) as infectious agents. In contrast, all malignant cells remained protected against viral cytolysis when IFN- was exogenously supple-mented. This indicates that the disturbance of the TNF-␣ -mediated IFN-expression and the loss of an immediate an-tiviral response are additional central events in the multistep progression to cervical cancer, being more the consequence of the in vivo phenotype of the respective host cell rather than a direct effect of E6 and E7 oncoprotein expression.
MATERIALS AND METHODS
Cell lines, hybrid formation, and cytokine treatment.The cervical carcinoma cell lines HeLa, SW756, CaSki, and SiHa; the nontumorigenic somatic cell hybrids made between HeLa cells and normal human fibroblasts (444 cells) and their tumorigenic segregants (CGL3 cells) (61); HeLa⫻CaSki and HeLa⫻
SW756 hybrids (15, 59); SiHa⫻HeLa hybrids (see below); malignant HPK Ia cells (10); and the human lung carcinoma cell line A549 (66) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and 1% penicillin-streptomycin. Modified SiHa cells (so-called universal fuser) (50) harboring dominant (G418 resistance) and recessive (hypoxanthine-phos-phoribosyltransferase-negative) phenotypes were grown in the same medium containing 1 mg of G418 (Gibco BRL) per ml and 10⫺4M 6-thioguanine (Sigma). Somatic cell hybridization between SiHa and HeLa cells was done exactly as described previously (50). For cytokine treatment, the cells were incubated with TNF-␣(Strathman Biotech GmbH, Hannover, Germany), IFN-␣
(Alphaferon3 [natural human IFN-␣; kindly provided by Rentschler, Laupheim, Germany]), IFN-(IFN-1; [natural human IFN-; Rentschler), and IFN-␥
(IF-RC [Thomae, Biberach, Germany]) for different periods of time as described in the figure legends.
RNA analysis.RNA was isolated with the RNeasy kit according to the instruc-tions of the manufacturer (Qiagen, Hilden, Germany). Approximately 5g of RNA was separated on 1% agarose gels in the presence of ethidium bromide under nondenaturing conditions (26) and transferred to GeneScreen Plus mem-branes (DuPont, NEN). Specific probes for the hybridization were labeled with [32P]dCTP by random priming (12). The filters were washed in 2⫻SSC (1⫻SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% sodium dodecyl sulfate (SDS) at 68°C and exposed to Kodak films as indicated in the figure legends.
RT-PCR.DNA digestion of total RNA was carried out with 2 U of RQ1-DNase (specific activity, 1 U/l) (Promega, Mannheim, Germany) in 40 mM Tris-HCl (pH 8.0)–10 mM MgSO4–1 mM CaCl2in the presence of 20 U of RNase inhibitor (specific activity, 40,000 U/ml) (Hybaid, Heidelberg, Germany) in a total volume of 100l at 37°C for 10 min. DNase was inactivated by addition of 2l of 0.5 M EDTA (pH 8.0), followed by acidic phenol-chloroform extraction (8). RNA was diluted in RNase-free water. One microgram of DNase-treated RNA was mixed with 0.5g of oligo(dT) primer (Promega) added in a total volume of 12l, heated at 70°C for 10 min, and chilled on ice. The mixture was supplemented with 5⫻reverse transcription (RT) buffer (250 mM Tris-HCl [pH 8.3], 375 mM KCl, 15 mM MgCl2), 10 mM dithiothreitol, 500M deoxynucleo-side triphosphate mix (Roche Diagnostic, Mannheim, Germany), and 20 U of RNase inhibitor and incubated at 42°C for 2 min in a total volume of 19l. After the annealing, 200 U of reverse transcriptase SuperScript II (Gibco BRL) (spe-cific activity, 200 U/l) was added and the reaction mixture was incubated for 1 h at 42°C. The RT products were heated to 72°C for 10 min and chilled on ice. PCRs were performed in a solution containing 10 mM Tris-HCl (pH 8.3), 200
M deoxynucleoside triphosphate mix (Roche Diagnostic), 40 pmol of upstream and downstream primers, 5 U ofTaqpolymerase (Sigma) (specific activity, 5 U/l), and 2l of reverse-transcribed product. The amplification was performed in an MJ Research PTC-200 thermal cycler in a total volume of 50l. For IFN-
detection, the primers 5⬘-GATTCATCTAGCACTGGCTTG-3⬘and 5⬘-CTTCA GGTAAATGCAGAATCC-3⬘(23) were used. The PCR was performed for 40 cycles consisting of 1 min at 94°C, 45 s at 55°C, and 30 s at 72°C. During the last cycle, the extension time was increased to 10 min, and the reaction mixture was rapidly cooled to 4°C. IFN-regulatory factor 3 (IRF-3) expression was monitored using the upstream primer 5⬘-GGTTGCGTTTAGCAGAGGAC-3⬘ and the downstream primer 5⬘-AGGAGATGGTCTGCTGGAAG-3⬘. Amplification (35 cycles) was done at 94°C for 30 s, 57°C for 45 s, and 72°C for 30 s. In the last cycle, the extension time was increased (to 10 min) and then the mixture was rapidly cooled to 4°C. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used
on November 8, 2019 by guest
http://jvi.asm.org/
as an internal control with primers 5⬘-TGGATATTGTTGCCATCAATGAC C-3⬘ and 5⬘-GATGGCATGGACTGTGGTCATG-3⬘ (18). The amplification was performed for 35 cycles of 1 min at 94°C, 45 s at 65°C, and 30 s 72°C, with a prolonged extension time of 10 min in the last cycle before cooling to 4°C. The PCR products were analyzed in 1 to 2% agarose gels.
Antiviral titration assays.Cells were infected with either EMCV or VSV using multiplicities of infections (MOIs) of between 0.1 and 0.2 (37, 66). The cells, seeded in 96-well plates (104cells/well) were pretreated with cytokines as de-scribed in the figure legends. After 24 h, the medium was discarded, fresh medium (37°C) with virus was added, and the plates were incubated at 37°C until the viral lysis in the controls was complete (16 to 20 h). All cells were susceptible to EMCV, except CaSki⫻HeLa hybrids. For those cells, antiviral activity was monitored after infection with VSV. To quantify the cytopathic effect (CPE), the cells were fixed with 4% glutardialdehyde and stained with 1% crystal violet. The dye was solubilized in 33% acetic acid, and the optical density of the eluate was measured at 570 nm in a Labsystem Multiscan MS enzyme-linked immunosor-bent assay reader. To quantify the amount of endogenously synthesized IFN-in 444 cells after treatment with TNF-␣, the supernatants were transferred on A549 indicator cells (66) (3⫻104/well). On the next day, the cells were infected with EMCV. After 26 to 30 h, medium was removed and the cytopathogenic effect was quantified as described above. The amounts of IFN-are expressed as interna-tional reference units per milliliter, using exogenously added Nainterna-tional Institutes of Health human IFN-as a reference.
Neutralization assay.Cells (104) were seeded in 96-well plates. Prior to the addition of TNF-␣, as indicated in the figure legends, the cells were incubated for 1 h at 37°C with antibodies neutralizing 8 U of IFN-(clone 39C; Biotrend, Ko¨ln, Germany) or 10 U of IFN-␣(Hoffmann La Roche Inc., Basel, Switzerland). After 24 h, medium was discarded and the cells were infected with EMCV and incubated at 37°C until viral controls showed complete lysis. Cells were fixed with 4% glutardialdehyde and stained with 1% crystal violet. The eluate was quanti-fied as described above.
EMSAs.For gel retardation, the following oligonucleotides were used: the positive regulatory domain II (PRDII) sequence 5⬘-GGGAAATTCCGGGAA ATTCC-3⬘, which contains two copies of the PRDII core element (1); the PRDIV sequence 5⬘-AATGTAAATGACATAGGAAAACTGA-3⬘ (33); the PRDIII-I sequence 5⬘-GAAAACTGAAAGGGAGAAGTGAAA-3⬘ (33); and an NF-B sequence, 5⬘-AGTTGAGGGGACTTTCCCAGGC-3⬘, derived from the immunoglobulinlight-chain gene (44). The DNAs were synthesized in an Applied Biosystems synthesizer using phosphoramitide chemistry and further purified by high-pressure liquid chromatography. For electrophoretic mobility shift assays (EMSAs), the annealed oligonucleotides were labeled with [␥-32P]ATP (Amersham; 3,000 Ci/mmol) with T4 polynucleotide kinase and gel purified from a 15% polyacrylamide gel. Cellular extracts were prepared as described previously (54), with the only modification being thatN,N-(L-3-trans
-carboxyoxirane-2-carbonyl)-L-leucyl-agmatine (E64),
4-(2-aminoethyl)-benzol-sulfonylfluoride (Pefabloc SC), 1 mM NaF, and 0.2 mM Na3VO4were included as protease inhibitors in concentrations suggested by the manufacturer (Roche). The protein concentration was determined by the Bradford method (Bio-Rad) using defined amounts of bovine serum albumin as standards. The binding of NF-B, activation factor-2 (ATF-2), and c-Jun was performed in a 20-l reaction volume containing 10% glycerol, 12 mM HEPES (pH 7.9), 4 mM Tris-HCl (pH 7.9), 60 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 0.6 mg of bovine serum albumin per ml, 2.0g of poly(dI-dC), and 2 to 4g of nuclear extract. After 5 min, 10,000 cpm of the [␥-32P]ATP 5⬘-end-labeled double-stranded oligonucle-otide probe was added, and the incubation was continued for an additional 30 min at room temperature (58). EMSAs for the IRF family were performed in 20-l reaction volumes containing 10% glycerol, 70 mM Tris (pH 7.5), 30 mM Na acetate, 10 mM EDTA, 1g of poly(dI-dC), and 5g of nuclear extract. After 5 min, 10,000 cpm of the [␥-32P]ATP 5⬘-end-labeled double-stranded oligonu-cleotide probe was added, and the incubation was continued for an additional 15 min at 4°C (7a). The sequence specificity of the binding was routinely controlled in competition experiments by the addition of a 100-fold molar excess of either unlabeled homologous or heterologous oligonucleotides. For monitoring the c-Jun and ATF-2 composition in supershift assays, 2g of polyclonal antibodies directed against c-Jun or ATF-2 was added and the reaction mixture was further incubated for 1 h at 4°C. Selective binding of different IRF members was ana-lyzed by adding 2g of poly- or monoclonal antibodies to the reaction mixture prior to oligonucleotide supplementation. Specifically, the following antibodies (all obtained from Santa Cruz Biotechnology as TransCruz supershift reagents) were used: a c-Jun antibody which recognizes both the nonphosphorylated and phosphorylated forms of c-Jun (epitope corresponding to amino acids 56 to 69 mapping within the amino-terminal domain of the mouse c-Jun protein) (sc-822x), an ATF-2 antibody specific for Thr-71-phosphorylated ATF-2 (sc-8398x),
an IRF-1 antibody specific for the C-terminal region of human IRF-1 (sc-497x), an IRF-2 antibody specific for the carboxy terminus of human IRF-2 (sc-498x), an IRF-3 antibody with an epitope corresponding to full-length IRF-3 of human origin (sc-9082x), and an IRF-7 antibody which recognizes amino acids 1 to 246 of human IRF-7 (sc-9083x). Antibody to p48 (ISGF3␥) was from Transduction Laboratories. The DNA-protein complexes were resolved on 5.5% nondenatur-ing polyacrylamide gels (29:1 cross-linknondenatur-ing ratio), dried, and exposed overnight to Fuji medical X-ray films.
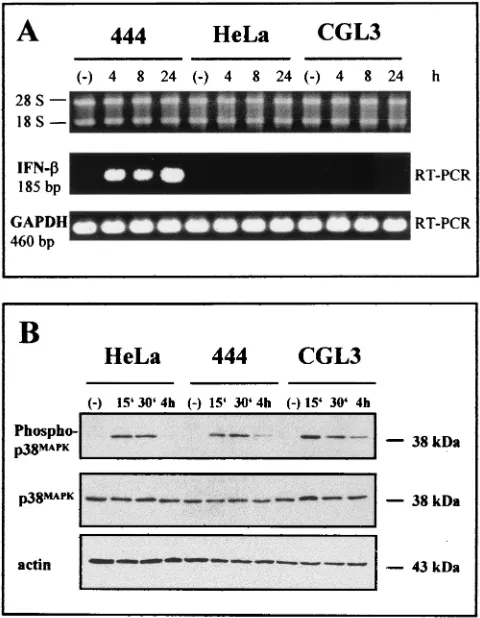
SDS-polyacrylamide gel electrophoresis and Western blotting.The same ex-tracts (25 to 50g) used for the band-shift analyses were separated in 8 to 10% SDS-polyacrylamide gels, electrotransferred to polyvinylidene difluoride mem-branes (Immobilon-P; Millipore Corporation, Bedford, Mass.), and probed with antibodies to IRF-1 (sc-497x; Santa Cruz), IRF-2 (sc-498x; Santa Cruz), and p48 (Transduction Laboratories). The incubation was carried out overnight in Tris-buffered saline supplemented with 5% skim milk powder (Merck), 0.05% Tween 20 (Sigma), and a 1:5,000 (IRF-1 and IRF-2) or 1:500 (p48) dilution of antibod-ies. p38 and phospho-p38 mitogen-activated protein kinase (MAPK) were di-rectly monitored in total cellular extracts obtained after lysis in SDS-polyacryl-amide gel electrophoresis buffer (60 mM Tris, 2% SDS, 10% glycerol, 50 mM FIG. 1. Selective IFN- induction by TNF-␣ in nonmalignant HPV18-positive cells, showing intactness of the MAPK pathway. (A) RT-PCR products generated by IFN-- and GAPDH-specific primer sets (185 and 460 bp, respectively) were separated on 2% agarose gels. The superimposed ethidium bromide-stained gel shows the quality of the RNAs used for the RT-PCR. The positions of the 18S and 28S rRNAs are indicated. Nonmalignant 444 hybrids, their tumorigenic segregants (CGL3 cells), and parental HeLa cells were incubated with 10 ng of TNF-␣ per ml for different periods of time. Lanes (⫺), untreated controls. (B) Western blot analysis of p38 MAPK. Twenty micrograms of total cellular extract per lane was separated in two identical SDS–10% polyacrylamide gels. After transfer, the filters were incubated with either a phosphorylation-specific (Phospho-p38MAPK)
or a phosphorylation-unspecific (p38MAPK) p38 antibody. Equal
load-ing and protein transfer were confirmed by incubatload-ing the upper filter with an actin-specific antibody. The molecular masses are indicated.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.587.302.542.74.383.2]dithiothreitol, 0.1% bromophenol blue, pH 6.8). Antibody dilutions were used as recommended by the supplier (New England Biolabs, Frankfurt, Germany). The bands were visualized with an anti-rabbit immunoglobulin G antibody conju-gated with horseradish peroxidase using the ECL detection system (Amersham). Equal protein transfer and loading were routinely controlled by reincubating the filters with a monoclonal actin-specific antibody (ICN Biomedicals). For reincu-bation with additional antibodies, the filters were stripped with 200 mM NaOH for 5 min at room temperature.
RESULTS
Restoration of the TNF-␣-induced antiviral response in
nonmalignant HPV18-positive somatic cell hybrids:
reexpres-sion of the IFN- gene. To monitor the antiviral effect of
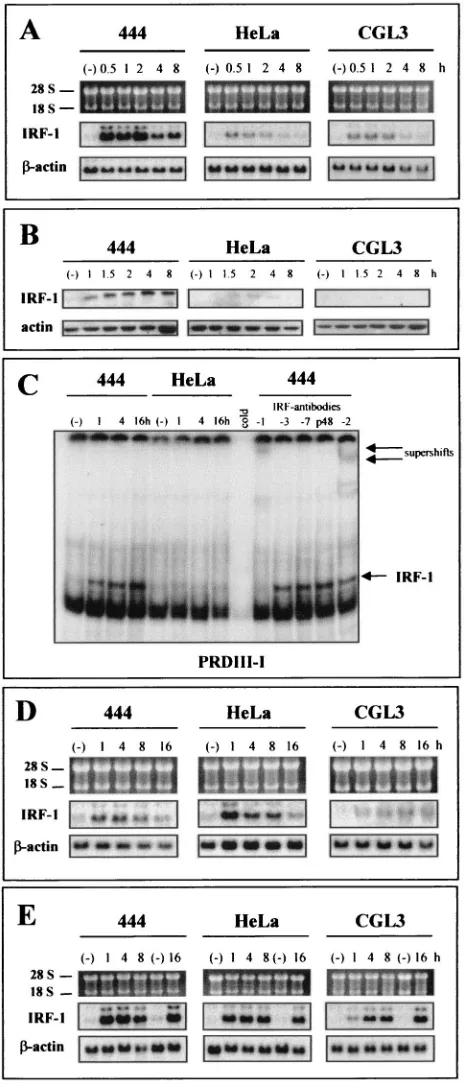
TNF-␣on the IFN-␣/-regulatory pathway, we used HPV18-positive HeLa cells and derived somatic cell hybrids as an experimental model system (61). Tumorigenic HeLa cells can be reverted to nonmalignant growth when fused to primary human fibroblasts (designated 444 cells). Long-term in vitro cultivation, however, leads to rare tumorigenic segregants of the same hybrids (referred to as CGL3 cells), which permits the investigation of cytokine signaling in a cellular environ-ment harboring identical transcription cassettes of HPV18 (49) but where the viral oncoproteins E6 and E7 are expressed in a different genetic background. When such cells were treated with TNF-␣, IFN- mRNA could be detected as a 185-bp RT-PCR fragment in nonmalignant hybrids (444 cells) which became discernible as saturated quantities 4 h after TNF-␣
application and persisted for at least 24 h. When the same experiment was carried out with RNAs obtained from malig-nant segregants (CGL3 cells) or with parental HeLa cells, no IFN-expression occurred (Fig. 1A). The selective inducibility of the IFN-gene could not be attributed to a disturbance of the TNF-␣signal response or receptor engagement in malig-nant cells, because p38MAPKMAPK phosphorylation, a known
hallmark for functional proinflammatory cytokine signaling (65), became visible and disappeared with approximately the same kinetics in all three cell lines without quantitative changes of the net amount of nonphosphorylated p38 (Fig. 1B). Identical loading and protein transfer after Western blot-ting were assessed by reincubablot-ting the same filter with an actin-specific antibody.
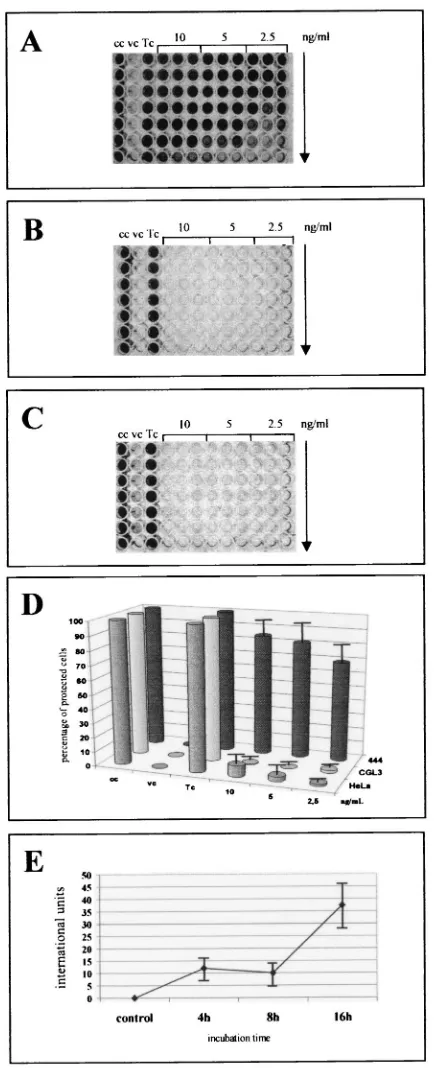
[image:4.587.55.272.78.612.2]To verify that the IFN--specific mRNA was translated into functional protein, a biological assay based on the susceptibil-ity of nonresponsive cells to lysis after infection with EMCV was used (37, 66) (Fig. 2). After being seeded in 96-well plates, the cells were first preincubated with different amounts of TNF-␣ ranging from 10 to 2.5 ng/ml. Only 444 cells were protected (Fig. 2A), while both HeLa cells (Fig. 2B) and the CGL3 tumorigenic segregants (Fig. 2C) were completely sen-sitive to EMCV-mediated cell lysis, even at higher TNF-␣
FIG. 2. TNF-␣confers selective protection against EMCV infec-tion in nontumorigenic HPV18-positive cells. Malignant and nonma-lignant cells grown in microtiter plates were first pretreated for 24 h with serial dilutions (1:2, indicated by the arrow) of TNF-␣(10, 5, and 2.5 ng/ml) and infected with EMCV at an MOI of 0.1 as described in Materials and Methods. To monitor the CPE, the cells were fixed and stained with crystal violet. cc, uninfected control cells; vc, EMCV-infected cells without prior TNF-␣addition; Tc, TNF-␣treated cells not infected with EMCV. (A to C) 444, HeLa, and CGL3 cells, re-spectively. (D) Quantification of the CPE after staining with crystal violet. The dye was eluted, and its absorbance at 570 nm was deter-mined spectroscopically. The bars indicate the percentages of
pro-tected cells relative to the untreated and the TNF-␣-incubated cell controls (cc and Tc, respectively). (E) Determination of IFN- syn-thesis in 444 cells. Supernatants (in twofold serial dilutions) were added to A549 indicator cells, and antiviral activity was determined as described in Materials and Methods. The amounts of IFN-produced at different periods of time (4, 8, and 16 h) after treatment with 10 ng of TNF-␣per ml are expressed as international units per milliliter. All standard deviations are given for three independent experiments per-formed in triplicate.
on November 8, 2019 by guest
http://jvi.asm.org/
concentrations. These data illustrate that the antiviral activity of TNF-␣was reconstituted in nonmalignant hybrids even in the presence of the viral oncoproteins but was lost in tumori-genic segregants or parental HeLa cells. To estimate how much IFN-was definitively synthesized, the supernatants of nonmalignant hybrids were removed and tested for IFN- ac-tivity against EMCV on A549 indicator cells, where TNF-␣
itself had no antiviral activity (66). Concordant with the RT-PCR data (Fig. 1A), biologically active IFN-was first detect-able after 4 h, leading to an average accumulation of IFN-
ranging between 18 and 33 IU at 16 h after TNF-␣ adminis-tration (Fig. 2E). To ensure that endogenous IFN-synthesis was responsible for the protective effect against EMCV infec-tion, the same assay was performed in the presence of neutral-izing IFN-antibodies added 1 h prior to TNF-␣ supplemen-tation. Figure 3 shows that only the addition of antibodies against IFN-, and not addition of an IFN-␣-specific anti-serum, significantly inhibited the TNF-␣-induced antiviral ac-tivity. This supports the notion that IFN-was in fact the key effector protein which selectively protected nontumorigenic cells against EMCV infection.
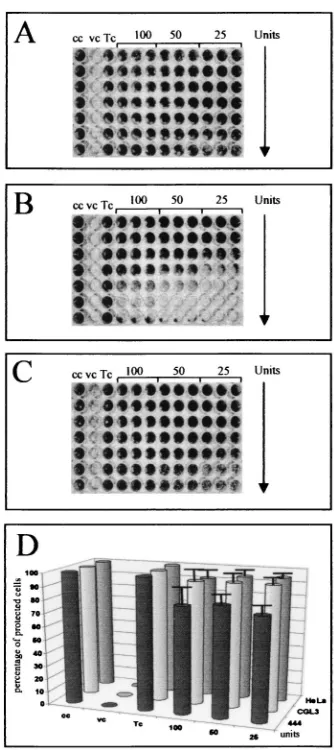
Exogenous IFN- supplementation protects HPV-positive
cells from EMCV infection independently from the in vivo
phenotype.Since it has been reported that the expression of
[image:5.587.336.503.70.445.2]viral oncogenes can interfere with IFN signaling (for a review, see reference 30), it was mandatory to examine whether ma-lignant and nonmama-lignant cells still reacted selectively when IFN-was added exogenously. In this case, all cell lines could be protected against EMVC-mediated cytolysis independently of whether IFN-(Fig. 4) or IFN-␣or IFN-␥(A. Bachmann et al., unpublished observation) was added to the tissue culture medium. Note that protection was successful even after appli-cation of less than 25 U of IFN-per ml, which was in agree-ment with our preceding data measuring the bioavailability of 444 cell-secreted IFN-at 16 h after TNF-␣addition (compare Fig. 2E and 4A). Contrary to previous observations (3, 32, 42, 48), the present data unambiguously demonstrate that, as far as the antiviral activity is concerned, both IFN-␣/and IFN-␥
can still protect HPV18-positive cells against EMCV infection (Fig. 4D), even after ongoing oncogene expression (see Dis-cussion).
IRF-1 and p48 (ISGF3␥) are selectively induced in
nonma-lignant cells upon TNF-␣ treatment.To gain further insight
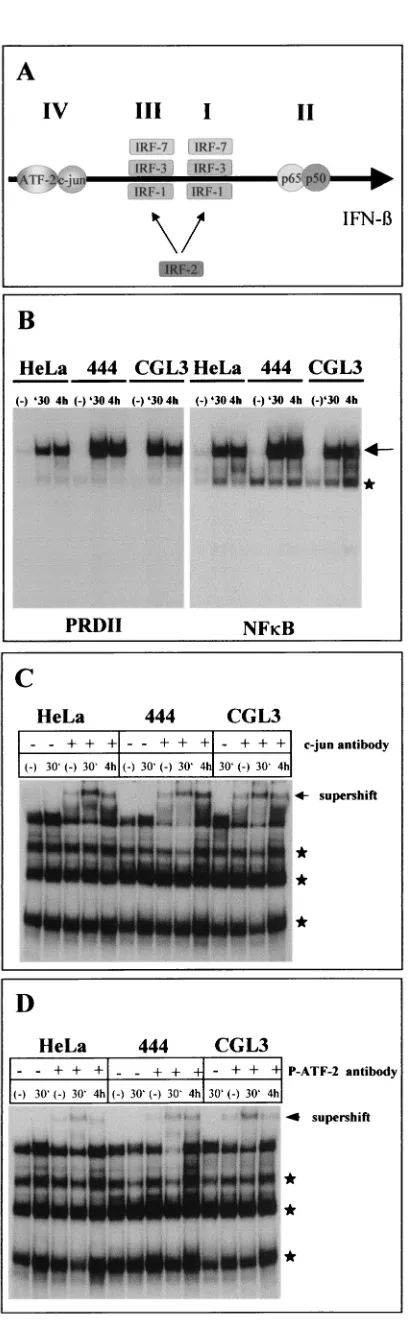
[image:5.587.47.285.76.194.2]into the differential regulation of the IFN- gene in nonma-lignant hybrids, we next examined the transcription factors involved in IFN-induction. One major key regulatory factor which is activated after addition of proinflammatory cytokines is NF-B (31). NF-B binds to the IFN-promoter as a part of a coordinately assembled multiprotein complex called the en-hanceosome (for a review, see reference 36). At the DNA level, the IFN-upstream region is composed of four PRDs (PRDI to -IV) which act synergistically in stimulating tran-scription (17, 24, 36) (Fig. 5A shows a schematic overview). Using parts of PRDII in comparison with an NF-B site ob-tained from the immunoglobulinlight-chain gene (44) for EMSAs, both oligonucleotides showed approximately the FIG. 3. EMCV protection is mediated by IFN-. 444 cells were
pretreated with different concentrations of TNF-␣ prior to EMCV infection in the presence of antibodies sufficient to neutralize IFN-␣ (␣), IFN-(), or both (␣ ⫹ ) as described in Materials and Methods. cc, uninfected control cells; vc, EMCV-infected cells without prior TNF-␣addition; Tc, TNF-␣-treated cells not infected with EMCV.
FIG. 4. Overall protection of HPV18-positive cells after exogenous IFN-supplementation. Malignant and nonmalignant cells grown in microtiter plates were first pretreated for 24 h with serial dilutions (1:2, indicated by the arrow) of IFN-(100, 50, and 25 U) and infected with EMCV at an MOI of 0.1 as described for Fig. 2. (A to C) 444, HeLa, and CGL3 cells, respectively. (D) Quantification of the degree of protection. The bars indicate the percentages of IFN--protected cells relative to untreated and IFN--incubated control cells used as a reference (indicated as cc and Tc, respectively). Standard deviations are given for three independent experiments performed in triplicate.
on November 8, 2019 by guest
http://jvi.asm.org/
same affinity in all three cell lines investigated. NF-B binding already became discernible 30 min after TNF-␣addition (Fig. 5B), which makes it unlikely that the absence of IFN- induc-ibility was due to a failure in cytokine signaling towards the NF-B branch. When the same set of experiments were carried out with PRDIV containing thecis-regulatory sequences for c-Jun and ATF-2, a more complex binding pattern appeared. When the origins and specificities of the various bands were examined by addition of c-Jun (Fig. 5C) and phosphorylation-specific ATF-2 (Fig. 5D) antibodies in supershift EMSAs, both transcription factors again revealed roughly the same binding kinetics.
A completely different picture emerged when we monitored the expression of IRF-1, which was originally identified as a critical mediator of the IFN response (for a review, see refer-ence 62). As depicted in Fig. 6A, TNF-␣ treatment led to a strong induction of IRF-1 transcription exclusively in nontu-morigenic hybrids (444 cells), whereas the gene was only mar-ginally elevated in the malignant cells (CGL3 and HeLa cells). The effect became even more pronounced when IRF-1 expres-sion was examined by Western blot analysis (Fig. 6B). When EMSAs were performed with oligonucleotides encompassing the PRDIII-I region (Fig. 5A), significant IRF-1 binding was obtained only with nuclear extracts derived from TNF-␣ -treated 444 cells (Fig. 6C, left). The authenticity of IRF-1 was verified after addition of a specific antibody, which leads to a disappearance of the induced band. IRF-3, IRF-7, and p48 do not bind at this region after short-term TNF-␣application (4 h), since the binding pattern was not affected after addition of the respective antibodies (Fig. 6C, right). Conversely, consis-tent with the EMCV infection assays described above (Fig. 4), IRF-1 could be activated independent of the cell phenotype when either IFN-or IFN-␥was exogenously supplemented (Fig. 6D and E, respectively). No selective induction or over-expression in tumorigenic cells was seen in analysis of the expression of IRF-2 (Fig. 7A), a transcription factor which antagonizes the function of IRF-1 by competing for the same binding site (PRDI and -II) (for a schematic overview, see Fig. 5A) (62). The same was true for IRF-3 (2), encoded by a transcriptional activator gene, which was constitutively tran-scribed and not further stimulated after TNF-␣ application (Fig. 7B).
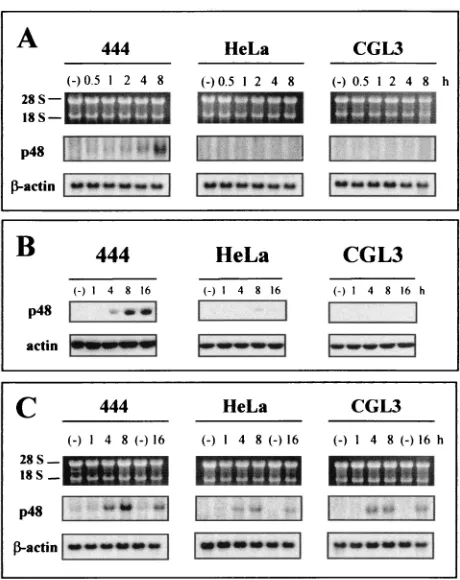
p48 (ISGF3␥) is another IRF family transcription factor and is the major binding component of ISFG3 (a multimeric com-plex between STAT1 and -2) (5). Although p48 (ISGF3␥) seems to be more involved in the autocrine amplification of the so-called delayed IFN-␣/ response (24), knockout
experi-FIG. 5. EMSAs of NF-B, c-Jun, and ATF-2 at the IFN- up-stream regulatory region. (A) Schematic overview of the IFN- -regu-latory region (enhanceosome). I to IV, PRDs and the relative binding positions of c-Jun; ATF-2; IRF-1, -2, -3, and -7; and NF-B (p50/p65). (B to D) HeLa, 444, and CGL3 cells were treated with TNF-␣for 30 min and 4 h. Lanes (⫺), untreated controls. Nuclear extracts were prepared for EMSAs with a PRDII and NF-B probe (B) or an oligonucleotide probe harboring PRDIV (C and D). PRDIV EMSAs were performed either in the absence (⫺) or in the presence (⫹) of supershift antibodies directed against c-Jun (C) or phosphorylated ATF-2 (D). Specific bands are marked by an arrow. Asterisks indicate nonspecific binding.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.587.62.266.59.719.2]ments have revealed that p48⫺/⫺mice were impaired in their
antiviral activity against EMCV and VSV infections (27). To assess the role of p48 (ISGF3␥) in the perturbation of
TNF-␣-mediated IFN signaling in our experimental system, p48 expression was examined. As depicted in Fig. 8A, only nonma-lignant cells revealed significant p48 (ISGF3␥) mRNA levels at 4 and 8 h after TNF-␣ treatment, while the gene remained transcriptionally silent in the malignant counterparts. Since it has been reported that E7 of HPV16 can potentially interfere with p48 (ISGF3␥) translocation into the nucleus (3), cells were fractionated and the nuclear extracts were monitored by Western blot analysis. To exclude cytoplasmic contamination, the quality of nucleus-cytoplasm separation was controlled by incubation of the filter with a cytoplasmic (pyruvate kinase M2) marker protein (Bachmann et al., unpublished data) (34). Similar to the case for IRF-1 (Fig. 6B), nuclear p48 (ISGF3␥) accumulation occurred only in nonmalignant cells (Fig. 8B), while gene expression was inducible with similar kinetics in all three cell line when IFN- or IFN-␥was administered (Fig. 8C).
[image:7.587.47.279.75.619.2]Complementation of a nontumorigenic phenotype between different cervical carcinoma cell line restores antiviral activity.
FIG. 6. Selective IRF-1 induction by TNF-␣ in nonmalignant HPV18-positive cells. (A) Transcriptional analysis of IRF-1 after TNF-␣treatment for 0.5, 1, 2, 4, and 8 h. Total RNA (5g/lane) was separated on 1% agarose gels. Filters were consecutively hybridized with probes specific for IRF-1 and -actin. IRF-1 was exposed to Kodak Biomax film and-actin was exposed to Kodak X-Omat film for 1 day. The positions of the 18S and 28S rRNAs are indicated. (B) Western blot analysis of nuclear extracts (50g/lane) after TNF-␣ application. After electrotransfer, the filters were incubated with an-tibodies directed against IRF-1. Equal loading was assessed with an actin-specific antibody. (C) Left, EMSAs using an oligonucleotide probe harboring PRDIII-I. HeLa and 444 cells were treated with
FIG. 7. IRF-2 and IRF-3 expression in HPV18-positive cells. (A) IRF-2 Western blot analysis of nuclear extracts (50g of protein/lane) of each cell line treated with TNF-␣as indicated. After electrotransfer, the same filters were consecutively incubated with antibodies raised against IRF-2 and actin. (B) RT-PCR products of IRF-3 and GAPDH (399 and 460 bp, respectively) after separation on 2% agarose gels. The quality of the RNAs used for the RT-PCR is shown at the top. The positions of the 18S and 28S rRNAs are indicated. Cells were incu-bated for 16 h in the presence of 10 ng of TNF-␣per ml. Lanes (⫺), untreated controls.
TNF-␣for 1, 4, and 16 h. Lanes (⫺), untreated controls. Right, PRD III-I EMSAs with TNF-␣-treated (4 h) 444 extracts in the presence of supershift antibodies directed against IRF-1, -2, -3, and -7 and p48. The specific IRF-1 and supershift bands are indicated by arrows. (D and E) Same as panel A but after treatment with IFN-(10 U/ml) or with IFN-␥(10 U/ml). The IFN application was extended to 16 h.
on November 8, 2019 by guest
http://jvi.asm.org/
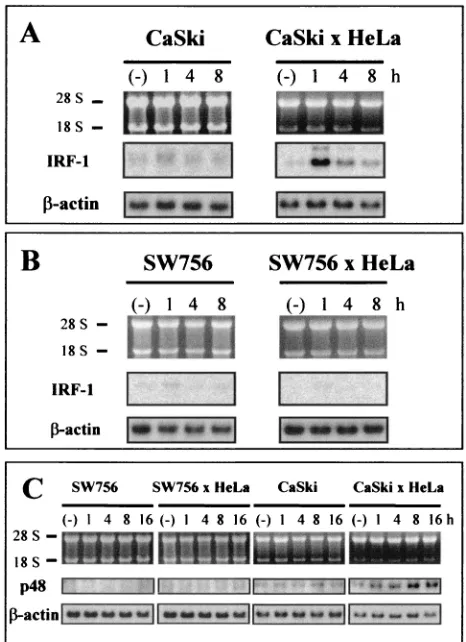
Based on the preceding experiments, we reasoned that cervical carcinoma cells in general may lack TNF-␣-induced antiviral activity, which should correlate with IRF dysregulation and tumorigenicity. To test this prediction, HPV16-positive CaSki cells and HPV18-positive SW756 cells were seeded in 96-well plates and then tested in EMCV and VSV infection assays. Both the CaSki and SW756 malignant cell lines were unable to resist viral infection after pretreatment with TNF-␣(Fig. 9A, upper panels). As shown by RT-PCR analysis, deficiency of viral resistance was again paralleled by an absence of IFN-
expression (Fig. 9B). Conversely, as already shown for HeLa cells (Fig. 3), protection could be achieved when either
IFN-␣/or IFN-␥was exogenously supplemented, which was again paralleled by IRF-1 and p48 expression (Bachmann et al., unpublished observations). When other malignant cells, such as tumorigenic variants of in vitro-immortalized HPV16-posi-tive human keratinocytes (HPK Ia cells) (10) were tested, these cells were also found to be highly sensitive to EMCV and VSV infection (Table 1). Only HPV16-positive SiHa cervical carcinoma cells, which had low tumorigenic potential in animal
experiments, were partially protected against EMCV infection after TNF-␣treatment (ranging between 30 to 40%), but these cells were again completely sensitive when infected with VSV (Table 1).
In a recent study we have demonstrated that tumorigenicity of HeLa and CaSki cells can be entirely suppressed after so-matic cell hybridization. In contrast, hybrid formation between HeLa and SW756 resulted in cell clones which were still ma-lignant after heterotransplanation into immunocompromised animals (59). Utilizing this complementation system in the context of TNF-␣-mediated IFN-signaling, antiviral activity and protection against both EMCV and VSV infection could be completely restored in nonmalignant CaSki ⫻ HeLa hy-FIG. 8. Selective p48 (ISGF3␥) induction by TNF-␣in
nonmalig-nant HPV18-positive cells. (A) Transcriptional analysis of p48 after TNF-␣treatment for 0.5, 1, 2, 4, and 8 h. Total RNA (5g/lane) was separated on 1% agarose gels. Filters were consecutively hybridized with probes specific for p48 (ISGF3␥) and-actin. Exposure was done on Kodak Biomax films (p48) or Kodak X-Omat films (-actin) for 3 days and 1 day, respectively. The positions of the 18S and 28S rRNAs are indicated. (B) Western blot analysis of nuclear extracts (25g/ lane) at different times after TNF-␣application. After electrotransfer, the filters were incubated with a polyclonal p48 antibody. Equal load-ing was confirmed with an actin-specific antibody. (C) Same as panel A but after treatment with IFN-␣ (10 U/ml). IFN-␣ application was extended to 16 h.
FIG. 9. Virus protection in nontumorigenic hybrids between cervi-cal carcinoma cells. (A) CaSki, SW756, SW756⫻HeLa, and CaSki⫻ HeLa hybrids were first pretreated for 24 h with serial dilutions (1:2, indicated by the arrow) of TNF-␣(10, 5, and 2.5 ng/ml) and infected with EMCV at an MOI of 0.1 as described for Fig. 2. CaSki⫻HeLa hybrids were infected with VSV at an MOI of 0.2. cc, uninfected control cells; vc, infected cells without prior TNF-␣ addition; Tc, TNF-␣treated cells not infected with virus. (B) RT-PCRs of RNAs obtained from SW756, CaSki, and somatic cell hybrids using HeLa cells as a fusion partner after treatment with TNF-␣ for 16 h. For details, see the legend to Fig. 1A. Lanes (⫺), untreated controls.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.587.48.278.70.360.2]brids, while SW756 ⫻ HeLa hybrids remained sensitive to EMCV infection after TNF-␣treatment (Fig. 9A, lower pan-els). When IRF-1 and p48 (ISGF3␥) transcription in these cell lines was monitored by Northern blotting, both genes were found to become significantly reinduced by TNF-␣in nonma-lignant CaSki⫻HeLa cells but not in SW756⫻HeLa hybrids, where only marginal transcription was detectable (Fig. 10). IRF-1 and p48 (ISGF3␥) reexpression resulted in complete restoration of IFN- transcription, as confirmed by RT-PCR analyses (Fig. 9B).
DISCUSSION
Extending our previous studies to unravel immunological escape mechanisms during HPV-induced carcinogenesis (for a review, see reference 52), we found that the inducibility of the IFN-gene by TNF-␣was eliminated in tumorigenic HPV18-positive HeLa cells but was reconstituted in nonmalignant HeLa⫻fibroblast hybrids (Fig. 1). Restoration of IFN- ex-pression resulted in successful protection of cells against infec-tion with EMCV or VSV (37) (Fig. 2). Furthermore, the fail-ure of TNF-␣to induce an effective antiviral response in 444 cells in the presence of neutralizing antibodies against IFN-
(but not against IFN-␣) confirmed a direct involvement of autocrine secreted IFN-as an antiviral mediator elicited by TNF-␣ (Fig. 3). Of particular interest also was the fact that malignant cells remained protected against virus when either IFN-␣/ or IFN-␥was directly supplemented into the tissue culture medium (Fig. 4). These data provide compelling evi-dence that IFN signaling and the induction of an antiviral function operate equally well in all cell lines and independently from the proliferative phenotype in immunocompromised an-imals.
The observation that TNF-␣-mediated induction of IFN-
was restricted to nontumorigenic hybrids strongly suggests that the cross talk between the TNF-␣pathway towards transcrip-tional activation of the IFN- gene is disturbed in cervical
carcinoma cells. This may have considerable implications for immune evasion processes during progression to cervical can-cer (52, 67). TNF-␣represents an important regulatory cyto-kine with immunomodulatory and growth-inhibitory functions in nonmalignant HPV-positive keratinocytes (35, 51). TNF-␣
[image:9.587.304.537.73.394.2]both suppresses transcription of the viral E6 and E7 oncogenes and induces the expression of MCP-1, exclusively in nontu-morigenic cells (51). MCP-1 belongs to a superfamily of small secretory proteins called chemokines (39, 47), which recruit and activate mononuclear cells, the first line of defense against viral infection (45). Activated macrophages in turn not only secrete additional TNF-␣, thereby amplifying the cytokine re-sponse, but also are capable of inducing IFN-␣/, which have strong antiviral functions, in their target cells (23, 38). It should be emphasized that IFN- induction represents the earliest antiviral response which occurs by an protein synthesis-inde-pendent pathway (for reviews, see references 17 and 19). Since IFN-␣is not able to be induced in cells lacking both copies of the IFN- gene (IFN-⫺/⫺ cells), it is thought that IFN-
FIG. 10. Restoration of IRF-1 and p48 (ISGF3␥) expression in nonmalignant CaSki⫻HeLa hybrids. (A and B) Transcriptional anal-ysis of IRF-1 in CaSki (A) and SW756 (B) cells and the corresponding HeLa hybrids after TNF-␣treatment for 1, 4, and 8 h. Total RNA (5 g/lane) was separated on 1% agarose gels. Filters were consecutively hybridized with probes specific for IRF-1 and-actin. (C) Same as panels A and B but after hybridization with a p48 (ISGF3␥)-specific probe. Exposure was done on Kodak Biomax films (p48) or Kodak X-Omat films (-actin) for 3 days and 1 day, respectively. Lanes (⫺), untreated controls.
TABLE 1. Summary of results of EMCV and VSV protection studies in relation to the proliferative phenotype
of the cells in nude mice
Cell linea Tumorigenicityb Protection by TNF-␣
HPV-18
444 ⫺ ⫹
-444 ⫺ ⫹
CGL3 ⫹ ⫺
HeLa ⫹ ⫺
SW756 ⫹ ⫺
HPV-16
SiHa ⫹ ⫹/⫺
CaSki ⫹ ⫺
HPK Ia late passage ⫹ ⫺
Hybrids
CaSki⫻HeLa ⫺ ⫹c
SW756⫻HeLa ⫹ ⫺
SiHa⫻HeLa ⫹ ⫺
aHPV 16- or 18-positive cervical carcinoma cells and derived cell hybrids. bFormation of tumors after subcutanous inoculation of 107cells in both flanks of 6-week-old female nude mice (59).
cCaSki⫻HeLa hybrids were susceptible only to VSV infections.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:9.587.42.281.101.268.2]binding to its cognate receptor is a prerequisite for activation of further IFN-␣production (11). Consequently, loss of both TNF-␣-mediated MCP-1 and IFN- inducibility in tumori-genic cells concomitant with the absence of a negative regula-tory effect on viral E6 and E7 expression could provide an explanation for the observed depletion of immunological ef-fector cells in dysplastic lesions (22, 29, 46, 60, 63, 64), which not only diminishes the immediate-early antiviral response but also may increase the incidence of cervical cancer.
Although both E6 and E7 can counteract the function of regulatory proteins involved in the ultimate IFN response (3, 40, 42, 48), it was amazing that tumorigenic cells still respond to exogenous IFN treatment (Fig. 4). As reported recently, HPV18 E6 affects IFN-␣ signaling by reducing Jak-STAT1/2 tyrosine phosphorylation in human fibrosarcoma cells (32). Additionally, HPV16 E6 can bind to IRF-3, thereby impairing Sendai virus-induced activation of IFN-and 2⬘,5⬘ -oligoadeny-late synthetase transcription (48). However, it must be stressed that both processes were not completely perturbed by E6, strongly indicating that, at least in the latter case, other factors (such as IRF-1 [see below]) can functionally substitute for IRF-3. Nonetheless, because TNF-␣ can selectively suppress HPV transcription in nontumorigenic cells (51), it was still conceivable that the reduction of oncogene expression to threshold levels may partially allow an IFN- response. To clarify this point, we used modified nontumorigenic HeLa⫻
fibroblast hybrids which were additionally transfected with an HPV18 E6-E7 transcription cassette under the control of the
-actin promoter. In those transfectants, only endogenous transcription, and not the-actin-driven E6-E7 transcription, became suppressed upon cytokine treatment. Nevertheless, TNF-␣-treated -actin 444 cells were still protected against EMCV infection, arguing against a direct involvement of viral oncogene expression in the outcome of the antiviral response (Bachmann et al., unpublished results).
By compiling the results of experiments which monitor tran-scription factors engaged in the differential regulation of IFN- gene expression (36), the following picture emerged. Using duplicated parts of PRDII in comparison to an NF-B binding site derived from the immunoglobulin light chain (44), TNF-␣addition resulted in similar binding patterns when nuclear extracts were analyzed in EMSAs (Fig. 5B). Therefore, the absence of IFN- inducibility in tumorigenic cells cannot be attributed to inefficient cytokine signaling towards NF-B activation, since no obvious differences in affinity and binding kinetics could be discerned. An equivalent situation was found with PRDIV-derived oligonucleotides harboring the recogni-tion sequences for c-Jun and ATF-2. Binding of the latter to PRDIV was paralleled by a threonine-specific phosphorylation at position 71 (Fig. 5B), which occurred in the same temporal relationship as detected for the phosphorylation of the up-stream MAPK p38MAPK(Fig. 1B). p38MAPKrepresents a
ma-jor effector MAPK of ATF-2, which becomes transiently acti-vated after addition of proinflammatory cytokines such as TNF-␣(65).
Regarding IRF-1, however, strong inducibility and DNA binding to PRDIII-I after TNF-␣treatment was achieved only in nonmalignant cells, while the corresponding protein was not detectable or was barely detectable in the tumorigenic coun-terparts (Fig. 6A, B, and C). The reason for this discrepancy in
gene regulation is presently not understood. Suppression or inefficient expression of IRF-1, however, can be epigenetically modified by different degrees of chromatin condensation (55). This notion was reinforced by a recent study demonstrating that HPV16 E7 can recruit to the IFN- promoter region a histone deacetylase which blocks IRF-1 trans activation on corresponding reporter constructs (42). Whether an altered nucleosomal organization may account for inefficient IRF-1 transcription upon TNF-␣treatment remains to be elucidated. In any case, IRF-1 dysregulation in tumorigenic HPV-positive cells might be of potential biological interest, especially in light of the fact that IRF-1 can act as a tumor suppressor under specific conditions (for a review, see reference 62). Since over-expression of IRF-1 induces apoptosis (62), it will be worth-while in further studies to test whether the ectopic expression of a dominant-negative mutant of IRF-1 alters the growth properties of 444 cells towards malignancy in nude mice.
IRF-1 is functionally counteracted by IRF-2 by competition for the same binding site within the IFN- promoter (21). Moreover, IRF-2 has a considerably longer half-life than IRF-1 (approximately 8 h versus 30 min) (62) and is present in uninduced cells [Fig. 7A, lanes (⫺)], probably to prevent un-controlled IFN- synthesis. As further outlined in Fig. 7A, IRF-2 remained constitutively expressed throughout the cell cycle, and no obvious quantitative difference in the protein amount could be seen when extracts from malignant and non-malignant cells were compared. An analogous situation was found for the transcription of the IRF-3 gene, whose expres-sion was also not further augmented after TNF-␣ addition (Fig. 7B). Conversely, like the case for IRF-1, only nonmalig-nant cells retained their ability to selectively synthesize p48 (ISGF3␥) (Fig. 8A). p48 (ISGF3␥) represents the DNA bind-ing component of a trimeric complex (ISGF3) which induces, jointly with STAT1 and STAT2, the transcription of antiviral genes such as those for the 2⬘,5⬘-oligoadenylate synthetase or double-stranded RNA-dependent protein kinase R (5). Al-though p48 (ISGF3␥) is structurally related to IRF-1 and binds to the IFN- promoter (24), the protein does not execute a redundant function in the cell but rather complements IRF-1 in inducing both IFN-␣/and IFN-␥responses (21). The pref-erential accumulation of p48 (ISGF3␥) in the nuclear fraction of 444 cells (Fig. 8B) is in contrast to recent results showing that HPV16 E7 can block p48 (ISGF3␥) translocation into the nucleus in spontaneously immortalized human keratinocytes (HaCaT cells) after IFN-␣treatment (3). Since this study in-vestigated the role of E7 in IFN-␣signaling in cells not asso-ciated with a natural HPV infection, it is likely that the out-come of the response may reflect the biological properties of the respective model system, the dosage of the transduced exogenous viral oncogene, the nature of the exogenous stim-ulus, and/or the phenotype of the host cell.
Another important aspect of our analysis is the correlation between nontumorigenicity and the antiviral activity induced by TNF-␣. Considering our preceding experiments, we realized that HeLa⫻fibroblast hybrids, which were converted to ma-lignancy via ectopic c-fos expression (58), have almost com-pletely lost their ability to block EMCV cytolysis after TNF-␣
addition (Bachmann et al., unpublished observations). We therefore went on to use an additional cell system which is based on the fact that fusion of two malignant cells results in
on November 8, 2019 by guest
http://jvi.asm.org/
nonmalignant hybrids when different tumor suppressor genes are affected. Complementation to nontumorigenicity cannot be accomplished when the same gene or pathway is defective (7, 43). Accordingly, when HPV18-positive HeLa cells or HPV16-positive CaSki cells, both of which are highly suscep-tible to viral infection after pretreatment with TNF-␣(Fig. 9A, upper panels), were fused, the resulting hybrids were nontu-morigenic after inoculation into immunocompromised ani-mals. In contrast, hybrid formation between HeLa cells and TNF-␣-unresponsive HPV18-positive SW756 cervical carci-noma cells (Fig. 9A, lower panels) failed to suppress tumor formation (59). When those hybrid clones were challenged with viral infection after TNF-␣ treatment, antiviral activity could be completely restored in nonmalignant CaSki⫻HeLa hybrids, whereas SW756⫻HeLa hybrids (and SiHa⫻HeLa hybrids) remained sensitive (Fig. 9A; Table 1). Reconstitution of a functional antiviral response was therefore not a peculiar-ity of the initially utilized HeLa⫻fibroblast hybrid system but rather correlated with nonmalignancy and the capability to reexpress IRF-1 and p48 (ISGF3␥) (Fig. 10) and, in turn, IFN- (Fig. 9B). This supports the notion that the antiviral response of TNF-␣is determined by the in vivo phenotype of the respective HPV-positive host cell line rather than by on-cogene expression per se.
Besides immunostimulatory and antiviral activities, the re-striction of endogenous IFN-production to nontumorigenic cells also involves an additional interesting feature which may explain the long latency period between viral infection and the final progression to cervical cancer (67, 68). IFN- synthesis inversely correlates with angiogenesis as well as with cell pro-liferation and, under some circumstances, with terminal differ-entiation (4). Notably, IFN- can down-regulate angiogenic factors such as basic fibroblast growth factor (56), interleukin-8 (57), and matrix metalloproteases (16), all of which are neces-sary to promote tumor growth and metastasis. Furthermore, upon analysis of human keratinocytes in medium either favor-ing or preventfavor-ing terminal differentiation, IFN- expression was detectable only in cells without proliferating cell nuclear antigen (PCNA). Similar results were obtained from the im-munohistological evaluation of normal tissue sections, where IFN-production was restricted to nondividing suprabasal cell layers (4). It will therefore be the aim of further studies to analyze these additional properties of IFN-in the context of HPV-induced carcinogenesis in greater detail.
ACKNOWLEDGMENTS
We thank Eric Stanbridge (University of California, Irvine), Claudia Denk (DKFZ, Heidelberg, Germany), and Matthias Du¨rst (University of Jena, Jena, Germany) for providing cell lines. The technical help of Anita Weyland is appreciated.
REFERENCES
1.Algarte, M., H. Kwon, P. Genin, and J. Hiscott.1999. Identification by in vivo genomic footprinting of a transcriptional switch containing NF-B and Sp1 that regulates the IB␣promoter. Mol. Cell. Biol.19:6140–6153. 2.Au, W.-C., P. A. Moore, W. Lowther, Y.-T. Juang, and P. M. Pitha.1995.
Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA92:11657–11661. 3.Barnard, P., and N. A. J. McMillan.1999. The human papillomavirus E7
oncoprotein abrogates signaling mediated by interferon-␣. Virology259:
305–313.
4.Bielenberg, D. R., M. F. McCarty, C. D. Bucana, S. H. Yuspa, D. Morgan,
J. M. Arbeit, L. M. Ellis, K. R. Cleary, and I. J. Fidler.1999. Expression of interferon-is associated with growth arrest of murine and human epider-mal cells. J. Investig. Dermatol.112:802–809.
5.Bluyssen, H. A. R., J. E. Durbin, and D. E. Levy.1996. ISGF3␥p48, a specificity switch for interferon activated transcription factors. Cytokine Growth Factor Rev.7:11–17.
6.Bornstein, J., Y. Ben-David, J. Atad, B. Pascal, M. Revel, and H. Abramovici.
1993. Treatment of cervical intraepithelial neoplasia and invasive squamous cell carcinoma by interferon. Obstet. Gynecol. Surv.4504:252–260. 7.Chen, T. M., G. Pecoraro, and V. Defendi.1993. Genetic analysis of in vitro
progression of human papillomavirus-transfected human cervical cells. Can-cer Res.53:1167–1171.
7a.Chen, C.-J., T.-T. Lin, and J. E. Shively.1996. Role of interferon regulatory factor-1 in the induction of biliary glycoprotein (cell CAM-1) by interferon-gamma. J. Biol. Chem.271:2818–28188.
8.Chomczynski, P., and N. Sacchi.1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Bio-chem.162:156–159.
9.Doly, J., A. Civas, S. Navarro, and G. Uze.1998. Type I interferons: expres-sion and signalization. Cell. Mol. Life Sci.54:1109–1121.
10.Du¨rst, M., S. Seagon, S. Wanschura, H. zur Hausen, and J. Bullerdiek.1995. Malignant progression of an HPV16-immortalized human keratinocyte cell line (HPK Ia) in vitro. Cancer Genet. Cytogenet.85:105–112.
11.Erlandsson, L., R. Blumenthal, M. L. Eloranta, H. Engel, G. Alm, S. Weiss, and T. Leanderson.1998. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr. Biol.8:223–226.
12.Feinberg, A. P., and B. Vogelstein.1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem.
132:6–13.
13.Finzer, P., U. Soto, H. Delius, A. Patzelt, J. F. Coy, A. Poustka, H. zur Hausen, and F. Ro¨sl.2000. Differential transcriptional regulation of the monocyte-chemoattractant-protein-1 (MCP-1) gene in tumorigenic and non-tumorigenic HPV 18 positive cells: the role of the chromatin structure and AP-1 composition. Oncogene19:3235–3244.
14.Gangemi, J. D., L. Pirisi, M. Angell, and J. W. Kreider.1994. HPV replica-tion in experimental models: effects of interferon. Antiviral Res.24:175–190. 15.Geisen, C., C. Denk, J. H. Ku¨pper, and E. Schwarz.2000. Growth inhibition of cervical cancer cells by the human retinoic acid receptor beta gene. Int. J. Cancer85:289–295.
16.Gohji, K., I. J. Fidler, R. Tsan, R. Radinsky, A. C. Eschenbach, T. Tsuruo, and M. Nakjima.1994. Human recombinant interferon-beta and -gamma decrease gelatinase production and invasion by human KG-2 renal carci-noma cells. Int. J. Cancer58:380–384.
17.Goodbourn, S., L. Didcock, and R. E. Randell.2000. Interferons: cell sig-nalling, immune modulation, antiviral responses and virus countermeasures. J. Gen. Virol.81:2341–2364.
18.Griffiths, D. J., P. J. Venables, R. A. Weiss, and M. T. Boyd.1997. A novel exogenous retrovirus sequence identified in humans. J. Virol.71:2866–2872. 19.Guidotti, L. G., and F. V. Chisari.2000. Cytokine-mediated control of viral
infections. Virology273:221–227.
20.Hagari, Y., L. R. Budgeon, M. D. Pickel, and J. W. Kreider.1995. Association of tumor necrosis factor-␣gene expression and apoptotic cell death with regression of Shope papillomas. J. Investig. Dermatol.104:526–529. 21.Harada, H., T. Fujita, M. Miyamoto, Y. Kimura, M. Maruyama, A. Furia, T.
Miyata, and T. Taniguchi.1989. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell58:729–739.
22.Hughes, R. G., M. Norval, and S. E. M. Howie.1988. Expression of major histocompatibility class II antigens by Langerhans’ cells in cervical intraepi-thelial neoplasia. J. Clin. Pathol.41:253–259.
23.Jacobsen, H., J. Mestan, S. Mittnacht, and C. W. Dieffenbach.1989. Beta interferon subtype 1 induction by tumor necrosis factor. Mol. Cell. Biol.
9:3037–3042.
24.Kawakami, T., M. Matsumoto, M. Sato, H. Harada, T. Taniguchi, and M. Kitagawa.1995. Possible involvement of the transcription factor ISGF3␥in virus-induced expression of the IFN-gene. FEBS Lett.358:225–229. 25.Khan, M. A., W. H. Tolleson, J. D. Gangemi, and L. Pirisi.1993. Inhibition
of growth, transformation, and expression of human papillomavirus type 16 E7 in human keratinocytes by alpha interferons. J. Virol.67:3396–3403. 26.Khandjian, E. W., and C. Meric. 1986. A procedure for Northern blot
analysis of native RNA. Anal. Biochem.159:227–232.
27.Kimura, T., Y. Kadokawa, H. Harada, M. Matsumoto, M. Sato, Y. Kashi-wazaki, M. Tarutani, R. S. P. Tan, T. Takasugi, T. Matsuyama, T. W. Mak, S. Noguchi, and T. Taniguchi.1996. Essential and non-redundant role of p48 (ISGF3␥) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells1:115–124.
28.Kleine, K., G. Ko¨nig, J. Kreuzer, D. Komitowski, H. zur Hausen, and F. Ro¨sl.1995. The effect of the JE (MCP-1) gene, which encodes the monocyte chemoattractant protein-1, on the growth of HeLa cells and derived somatic cell hybrids in nude mice. Mol. Carcinogenesis14:179–189.
29.Kleine-Lowinski, K., R. Gillitzer, H. Ku¨hne-Heid, and F. Ro¨sl.1999. Mono-cyte-chemoattractant protein-1 (MCP-1) gene expression in cervical
on November 8, 2019 by guest
http://jvi.asm.org/
epithelial hyperplasia and cervical carcinomas. Int. J. Cancer82:6–11. 30.Koromilas, A. E., S. Li, and G. Matlashewski.2001. Control of interferon
signalling in human papillomavirus infection. Cytokine Growth Factor Rev.
12:157–170.
31.Lenardo, M. J., C.-M. Fan, T. Maniatis, and D. Baltimore.1989. The in-volvement of NF-B in-interferon gene regulation reveals its role as a widely inducible mediator of signal transduction. Cell57:287–294. 32.Li, S., S. Labrecque, M. C. Gauzzi, A. R. Cuddihy, A. H. T. Wong, S.
Pellegrini, G. J. Matlashewski, and A. E. Koromilas. 1999. The human papillomavirus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activations by interferon-␣. Oncogene18:5727–5737. 33.Lopez, S., and S. Navarro.1998. Transcriptional repression of type I IFN
genes. Biochimie80:689–701.
34.Maehama, T., A. Patzelt, M. Lengert, K.-J. Hutter, K. Kanazawa, H. zur Hausen, and F. Ro¨sl.1998. Selective down-regulation of human papilloma-virus transcription by 2-deoxyglucose. Int. J. Cancer76:639–646. 35.Majewski, S., J. Malejczyk, and S. Jablonska.1996. The role of cytokines
and other factors in HPV infection and HPV-associated tumors. Papilloma-virus Rep.7:143–155.
36.Maniatis, T., J. V. Falvo, T. H. Kim, T. K. Kim, C. H. Lin, B. S. Parekh, and M. G. Wathelet.1998. Structure and function of the interferon-beta enhan-ceosome. Cold Spring Harbor Symp. Quant. Biol.63:609–620.
37.McNeill, T. A.1981. Interferon assay. J. Immunol. Methods46:121–127. 38.Mestan, J., W. Digel., S. Mittnacht, H. Hillen, D. Blohm, A. Moeller, H.
Jacobsen, and H. Kirchner.1986. Antiviral effects of recombinant tumour necrosis factor in vitro. Nature (London)323:816–819.
39.Miller, M. D., and M. S. Krangel.1992. Biology and biochemistry of the chemokines: a family of chemotactic and inflammatory cytokines. Crit. Rev. Immunol.12:17–46.
40.Nees, M., J. M. Geoghegan, T. Hyman, S. Frank, L. Miller, and C. D. Woodworth.2001. Papillomavirus type 16 oncogenes downregulate expres-sion of interferon-responsive genes and upregulate proliferation-associated and NF-B-responsive genes in cervical keratinocytes. J. Virol.75:4283– 4296.
41.Oguchi, M., J. Komura, H. Tagami, and S. Ofuji. 1981. Ultrastructural studies of spontaneously regressing plane warts. Macrophages attack verru-ca-epidermal cells. Arch. Dermatol. Res.270:403–411.
42.Park, J.-S., E.-J. Kim, H.-J. Kwon, E.-S. Hwang, S.-E. Namkoong, and S.-J. Um.2000. Inactivation of interferon-regulatory factor-1 tumor suppressor protein by HPV E7 oncogene. J. Biol. Chem.275:6764–6769.
43.Pereira-Smith, O. M., and J. R. Smith.1988. Genetic analysis of indefinite division in human cells: identification of four complementation groups. Proc. Natl. Acad. Sci. USA85:6042–6045.
44.Pierce, J. W., M. Lenardo, and D. Baltimore.1988. Oligonucleotide that binds nuclear factor NF-kappa B acts as a lymphoid-specific and inducible enhancer element. Proc. Natl. Acad. Sci. USA85:1482–1486.
45.Rappolee, D. A., and Z. Werb.1992. Macrophage-derived growth factors. Curr. Top. Microbiol. Immunol.181:87–140.
46.Riethdorf, L., S. Riethdorf, K. Gu¨tzlaff, F. Prall, and T. Lo¨ning.1996. Differential expression of the monocyte chemoattractant protein-1 in human papillomavirus-16 infected squamous intraepithelial lesions and squamous cell carcinomas of the cervix uteri. Am. J. Pathol.149:1469–1476. 47.Rollins, B. J.1991. JE/MCP-1: an early-response gene encodes a
monocyte-specific cytokine. Cancer Cells3:517–524.
48.Ronco, L. V., A. Y. Karpova, M. Vidal, and P. M. Howley.1998. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev.12:2061–2072.
49.Ro¨sl, F., E.-M. Westphal, and H. zur Hausen.1989. Chromatin structure and transcriptional regulation of human papillomavirus type 18 DNA in HeLa cells. Mol. Carcinogenesis2:72–80.
50.Ro¨sl, F., T. Achtsta¨tter, T. Bauknecht, G. Futterman, K. J. Hutter, and H. zur Hausen.1991. Extinction of the HPV18 upstream regulatory region in cervical carcinoma cells after fusion with non-tumorigenic human keratino-cytes under non-selective conditions. EMBO J.10:1337–1345.
51.Ro¨sl, F., M. Lengert, J. Albrecht, K. Kleine, R. Zawatzky, B. Schraven, and H. zur Hausen.1994. Differential regulation of the JE gene encoding the monocyte chemoattractant protein (MCP-1) in cervical carcinoma cells and derived hybrids. J. Virol.68:2142–2150.
52.Ro¨sl, F., K. Kleine-Lowinski, and H. zur Hausen.1999. The possible role of chemokines in HPV-linked carcinogenesis, p. 207–225.InB. J. Rollins (ed.), Chemokines and cancer. Humana Press Inc., Totowa, N.J.
52a.Saxon, P. J., E. S. Srivatsan, and E. J. Stanbridge.1986. Introduction of human chromosome 11 via microcell transfer controls tumorigenic expres-sion of HeLa cells. EMBO J.5:3461–3466.
53.Schneider, A., and L. A. Koutsky.1992. Natural history and epidemiological features of genital HPV infection, p. 25–52.InN. Munoz et al. (ed.), The epidemiology of human papillomavirus and cervical cancer. International Agency for Research on Cancer, Lyon, France.
54.Schreiber, E., P. Matthias, M. M. Mu¨ller, and W. Schaffner.1989. Rapid detection of octamer binding proteins with ‘mini-extracts’ prepared from a small number of cells. Nucleic Acids Res.15:6419–6436.
55.Shestakova, E., M.-T. Bandu, J. Doly, and E. Bonnefoy.2001. Inhibition of histone deacetylase induces constitutive derepression of the beta interferon promoter and confers antiviral activity. J. Virol.75:3444–3452.
56.Singh, R. K., M. Gutman, C. D. Bucana, R. Sanches, N. Llansa, and I. J. Fidler.1995. Interferons alpha and beta downregulate the expression of basic fibroblast growth factor in human carcinomas. Proc. Natl. Acad. Sci. USA92:4562–4566.
57.Singh, R. K., M. Gutman, N. Llansa, and I. J. Fidler.1996. Interferon-
prevents the up-regulation of interleukin-8 expression in human melanoma cells. J. Interferon Cytokine Res.16:577–584.
58.Soto, U., B. C. Das, M. Lengert, P. Finzer, H. zur Hausen, and F. Ro¨sl.1999. Conversion of HPV 18 positive non-tumorigenic HeLa-fibroblast to invasive growth involves loss of TNF-␣mediated repression of viral transcription and modification of the AP-1 transcription complex. Oncogene18:3187–3198. 59.Soto, U., C. Denk, P. Finzer, K.-J. Hutter, H. zur Hausen, and F. Ro¨sl.2000.
Genetic complementation to non-tumorigenicity in cervical carcinoma cells correlates with alterations in AP-1 composition. Int. J. Cancer86:811–817. 60.Spinillo, A., P. Tenti, R. Zappatore, F. De Seta, E. Silini, and S. Guaschino.
1993. Langerhans’ cell counts and cervical intraepithelial neoplasia in women with human immunodeficiency virus infection. Gynecol. Oncol.48:
210–213.
61.Stanbridge, E.1984. Genetic analysis of tumorigenicity in human cell hy-brids. Cancer Surv.3:335–350.
62.Taniguchi, T., N. Tanaka, K. Ogasawara, S. Taki, M. Sato, and A. Takaoka.
1999. Transcription factor IRF-1 and its family members in the regulation of the host response. Cold Spring Harbor Symp. Quant. Biol.64:465–472. 63.Tay, S. K., D. Jenkins, P. Maddox, N. Hogg, and A. Singer.1987. Tissue
macrophage response in human papillomavirus infection and cervical intra-epithelial neoplasia. Br. J. Obstet. Gynaecol.94:1094–1097.
64.Viac, J., I. Guerin-Reverchon, Y. Chardonnet, and A. Bremond.1990. Lang-erhans cells and epithelial cell modifications in cervical intraepithelial neo-plasia: correlation with human papillomavirus infection. Immunobiology
180:328–338.
65.Wysk, M., D. D. Yang, H.-T. Lu, R. A. Flavell, and R. J. Davis.1999. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tu-mor necrosis factor-induced cytokine expression. Proc. Natl. Acad. Sci. USA
96:3763–3768.
66.Yousefi, S., M. R. Escobar, and C. W. Gouldin.1985. A practical cytopathic effect/dye-uptake interferon assay for routine use in the clinical laboratory. Am. J. Clin. Pathol.83:735–740.
67.zur Hausen, H.1994. Disrupted dichotomous intracellular control of human papillomavirus infection in cancer of the cervix. Lancet343:955–957. 68.zur Hausen, H., and F. Ro¨sl.1994. Pathogenesis of cancer of the cervix. Cold
Spring Harbor Symp. Quant. Biol.59:623–628.
69.zur Hausen, H.2000. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst.92:690– 698.