JOURNAL OFVIROLOGY, JUly1994,p.4514-4524 Vol.68,No. 7 0022-538X/94/$04.00+0
Copyright ©D 1994, American SocietyforMicrobiology
Mutations in Accessory DNA
Replicating
Functions Alter the
Relative
Mutation
Frequency
of
Herpes
Simplex
Virus
Type 1
Strains
in
Cultured
Murine Cells
RICHARD B. PYLESANDRICHARD L.THOMPSON*
DepartmentofMolecularGenetics, Biochemistryand
Microbiology, College of
Medicine,University
ofCincinnati, Cincinnati,
Ohio45267-0524 Received16December 1993/Accepted 11April 1994The contribution of the herpes simplex virus type 1 (HSV-1)-encoded uracil DNA glycosylase (UNG), thymidinekinase(TK),and dUTPase to the relative mutantfrequency (RMF)of the virus in culturedmurine cells was examined. Apanelof HSV-1 mutants that lackedsinglyordoublytheUNG, TK,ordUTPaseactivity were generated by disruption of theenzyme coding regionswiththeEscherichia coli
0-galactosidase
(,-gal) geneinstrain17syn+.ToestablishabaselineRMF of strain17syn+,the,8-gal genewasinserted into theUL3focus.
In all of theviruses, the ,-gal insert served as a phenotypic marker of RMF. Amutant plaque wasidentified by the lack of,I-galactivity and,in selectedcases,positiveinsituhybridizationfor,-galsequences. Replication kinetics inNIH 3T3 cellsdemonstrated thatall ofthe mutants replicated efficiently,generating
stocks withequivalent titers.TwoindependentlygeneratedUL3-,I-galviruseswereexamined and established abaselineRMFof -0.5%in both NIH3T3and LM TK- cells. Loss ofdUTPaseactivityresulted inviruseswith
fivefold-increased RMFs,indicatingthat theHSV-1 dUTPasehas anantimutatorfunction.The RMF observed forthetk- viruses was reduced as much as40-fold (RMFof0.02%),suggestingthat the viral TK isamutator
activity. TheRMFoftwoindependent UNG-viruses showednosignificantdifference from the baselineRMF
inlimited passage; however, following successivepassage,the data suggestedthatUNGactivityserves as an
antimutator. These results haveimplicationsfor the naturalhistoryofHSVand thedevelopmentof antiviral
therapies.
Herpes simplex virus (HSV) is one of the most complex animal viruses, encoding over 70 genes within its 152-kb double-strandedDNAgenome(35).Included among theseare a number of enzymatic activities which are involved in the replication of the viral genome (4, 35). As has been shown in other DNAviruses, such asbacteriophage T4 (2, 10, 28, 44), HSV type 1 (HSV-1) has evolved mutation strategies that presumably provide a selective advantage to the virus. In a series of reports, Hall and colleagues have demonstrated that the HSV-1 DNA polymerase (DNA pol; 18-20), like the T4 DNA pol, has a low fidelity ofdeoxynucleoside triphosphate selection that serves as a mutator function (20). Such
poly-merase activities would result in a relative mutant frequency (RMF) dependent upon the nucleotide pool sizes present duringviral DNA replication. Therefore, the virally encoded enzymes that modulate nucleotide pool size and replication fidelity may also play a significant role in viral RMFs. An understandingofthe viralfactorscontributing to the RMF of the virus and thus influencing the rate of development of drug-resistant progeny is crucial to effective antiviral therapy. Regulation of mutation frequencies is of obvious importance
to the success of the virus. For example, naturally occurring drug-resistant progeny are rapidly selected in culture (32, 40, 41) and following antiviral therapy in patients (3, 12, 38, 42). RMFs that are too low or artificially inhibited may limit the viruses'ability to escape a host's immune response or antiviral therapies.Conversely, viral RMFs that are too high would lead
*Corresponding author.
Mailing
address: Dept. of Molecular Ge-netics, Biochemistry andMicrobiology, University of Cincinnati Med-ical Center, P.O. Box 670524, Cincinnati, OH 45267-0524. Phone: (513)558-0062. Fax: (513)558-8474.to anunacceptable level of nonviableprogeny.Antiviral ther-apiesthat include perturbation of viralcontrolsof RMFcould improve theefficiencyof treatment of viraldisease.
HSV providesanexcellent model systemtogainan under-standingofthevirallyencoded functions which contribute to
the RMF, as the virus replicates well in a large variety of cellular environments (17); viralmutants areeasilygenerated, andagreat deal of work has already been completed on the virally encoded enzymes thatpromote efficient DNA
replica-tion.
In this report, a system to establish the viral RMF of a
nonselected transgene,that for 3-galactosidase (,B-gal),inthe
context of viral strains which lackenzymeactivitiesassociated with viral DNAreplication is described. ,B-Galactivityconverts
the chromagen X-Gal (5-bromo-4-chloro-3-indolyl-13-D-galac-topyranoside) intoablueprecipitateandtherefore serves as a
phenotypic markerforindividualplaques. Loss of 3-gal activity duetomutation results in aclear(nonblue) plaque whichhas beengenotypicallyconfirmedbyinsitu hybridization. Specifi-cally, the relative contribution ofthe HSV-1-encoded thymi-dine kinase (TK), dUTPase, and uracil DNA glycosylase (UNG) activities to the viral RMF following propagation in
NIH3T3 and LM TK-celllines was established. The baseline
RMF ofstrain 17syn+ wasdetermined by analysis ofmutants in which the ,B-gal gene was inserted into the UL3 locus. Analysis ofthepassage stocks of thesemutants establishedan
averaged baselineRMFof -0.5% or one mutant in every 200
PFU. In comparison, the RMFs established for the other mutantsdemonstrated that theHSV-1-encoded TK provides a potentmutatoractivity, the viral dUTPase hasanantimutator function, and viralUNGactivity has no appreciable effecton
RMFsinlimitedpassagebut may beanantimutator following
successive rounds ofreplication. Theimplications of these data
4514
on November 9, 2019 by guest
http://jvi.asm.org/
HSV-1 MUTATION FREQUENCY 4515
are discussed with respect to HSV replication and antiviral therapies.
MATERIALS AND METHODS
Cell lines and viruses. Rabbit skin cells (RSC) were cultured as previously described (46). RSC monolayers were used to generate the recombinant viruses, as well as viral stocks and viral genomic DNA, as previously described (46). NIH 3T3 (ATCC CRL 6442) cells are a homogeneous murine fibroblast cell line and were used for passage of the virus panel to examine RMF. NIH 3T3 monolayers were cultured in Dulbec-co's modified Eagle medium (GIBCO/Bethesda Research Laboratories, Gaithersburg, Md.) supplemented with 10% fetal bovine serum at 37°C-5% CO2 in a humidified atmo-sphere. A second murine cell line, LM TK-, was used for a singlepassage of a subset of the virus panel. LM TK- (ATCC CCL 1.3) cells were cultured as described for NIH 3T3 cells.
Virusstrains used in this study were derived from wild-type (wt) HSV-1 strain 17syn+,which was obtained from J. Subak-Sharpe of the Medical Research Council Virology Unit in Glasgow, Scotland. The derivation and history of
17syn+
have been previously described (46). Base pair numbering, restric-tion endonuclease maps, and restriction fragment names are based upon the complete sequence of HSV-1 strain17syn+
compiled by McGeoch and colleagues (29). The methods used forviraltitrations, as well as determination of in vitro replica-tion kinetics, have been described (46).
Production of recombinant viruses. The panel of recombi-nantviruses analyzed in this study, with the exceptions of tBd and 1602A, were generated by cotransfection of unit length 17syn+ genomic DNA and modified, cloned
17syn+
DNA fragments. tBd and 1602A are double mutants derived from 17syn+-based,dUTPase-negativestrain dUT-1218, which was provided by V. Preston (14). The viruses were designed to assess thecontributionof each of the specified HSV-1 enzyme activities to the RMF of the virus. Insertional mutations were chosen to minimize genomic perturbations and to provide a readilyassayed marker for RMF.To construct the mutations, fragments of
17syn+
genomic DNA containing the open reading frames (ORF) of interest werecloned into pUC19 by standard methods. Mutation of the selected genes was accomplished by insertion of a 4-kbXbaI
simian virus 40 early promoter-3-gal cassette isolated from plasmid pFJ3 (a kind gift of D. McGeoch [30]). The f-gal cassette was inserted into the genes of interest at sites that disrupted the ORF but left the neighboring ORF unaffected. Upon incorporation into the viral genome, the
,B-gal
insert served as a reporter of recombination and allowed simplified selection ofrecombinants.Cotransfections were performed with LipofectACE (GIBCO/Bethesda Research Laboratories) in subconfluent RSCmonolayers. Briefly, an empirically optimized amount of genomic viral DNA and the specific mutated fragment were incubated for 15 min with 10,ul of LipofectACE in serum-free culture medium (minimal essential medium) at room temper-ature (total volume, 1 ml). This mixture was applied to RSC monolayers which were incubated for 4 h and then fed with normal growth medium (minimal essential medium supple-mented with 5% newborn calf serum). The medium was exchanged with fresh growth medium after approximately 12 h. Plaques developed 36 to 48 h posttransfection and were then overlaid with agarose containing the chromagenic substrate X-Gal (Fisher) at 125 ,ug/ml. Plaques formed by recombinant viruses converted the X-Gal into a blue precipitate. Blue plaques were isolated and plaque purified to homogeneity. The
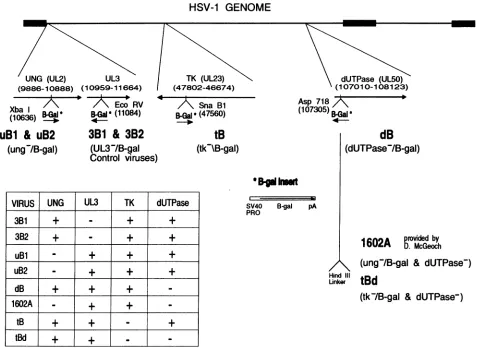
genomic structure of each of the viruses is depicted schemat-ically in Fig. 1. Specifics of the viral mutations are described below.
Two independent control viruses, 3B1 and 3B2
(UL3-
blue isolates 1 and 2), were generated from separate cotransfection cultures. These two viruses carry the Klenow-filledXbaI
,B-gal
cassette inserted at the EcoRV site (bp 11084) within the UL3 ORF (bp 10959 to 11664). The mutation was generated within the cloned17syn+
BamHI
e fragment (bp 2907 to 11820).uBl and uB2 (UNG- blue isolates 1 and 2) are two virus isolates from independent cotransfections carrying the
XbaI
3-gal
cassette attheXbaIsite(bp 10636)
within the UNG ORF (UL2, bp 9886 to 10888). This mutation was also generated in the clonedBamHI
e fragment.Mutation of the
17syn+
TK was accomplished by insertion of the filledXbaI
3-gal cassette at the SnaBI site (bp 47560) within the TK-encoding gene (UL23; bp 47802 to 46674) contained within the cloned17syn+
BamHI
p fragment (bp 45055 to 48634). Recombination of theTK--p-gal
fragment into17syn+
resulted in the isolation of tB(TK- blue).The gene for dUTPase
(UL50,
bp 107010 to 108123) also was mutated by insertion of the filledXbaI
,B-gal
gene at the Asp718 site (bp 107305) which had been blunt ended with Klenow (GIBCO/Bethesda Research Laboratories). Following cotransfection of the dUTPase-1-gal construct and17syn+,
viral strain dB (dUTPase- blue) was isolated. Previous work with engineered HSV-1 mutant strain dUT- 1218, generated by Fisher and Preston, demonstrated that insertion of a 12-bpHindIll
linker at the Asp718 site at bp 107305 in17syn+
eliminated HSV-1-induced dUTPase activity (14).
The two virus isolates that carry double mutations were derived from dUTPase-negative HSV-1 mutant strain dUT-1218. The first double mutant, tBd, was generated by recombination of the
TK--p-gal
construct with unit length dUT- 1218 genomic DNA. This virus lacks both HSV-1-induced TK and dUTPase activities. Virus isolate 1602A was plaque purified from a stock of virus kindly provided by D. McGeoch (30). McGeoch andcolleagues
generated
the origi-nal 1602 stock by cotransfection of a UNG--3-gal construct with dUT-1218 genomic DNA, resulting in a UNG-, dUT-Pase-negative derivative of strain17syn+.
The orientation of the,B-gal
insert in 1602A is opposite to that ofuBl and uB2.Southern blot-restriction fragment length polymorphism analysis was performed as previously described (43) and con-firmed the genomic structures and stock purity of all of the viruses used in this study. Restriction enzymes were purchased from GIBCO/Bethesda Research Laboratories. Digestions were performed under the conditions recommended by the manufacturer.
RMF assay. For RMF analyses, virus passage stocks were generated in either NIH 3T3 or LMTK- monolayers. Passage 1 (P1), P1', and LM TK-
P1
stocks were produced by low-multiplicity-of-infection (MOI) (0.01 PFU per cell) infec-tion of NIH 3T3(P1
andP1')
or LMTK- (LMTK-
P1)
cells. The NIH 3T3P1 stock is also the 96-h time point presented in the replication kinetic experiments. Passage 2 (P2) represents viral stocks generated by low-MOI (0.01 PFU per cell) infec-tion of NIH 3T3 monolayers with theP1
stocks. HP designates viral stocks produced by high-MOI (3 to 5 PFU per cell) infection of NIH 3T3 cells with the original viral stocks.For each of the passages, triplicate cultures were generated, pooled, and subjected to three cycles of freezing and thawing. Aliquots (-2,000 PFU) of each passage were adsorbed onto 100-mm-diameter RSC monolayers for 1 h and then overlaid with culture medium supplemented with 0.03% human im-mune globulin (165-mg/ml stock; Gammar). The plates were
VOL. 68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
4516 PYLES AND THOMPSON
HSV-1 GENOME
_ . x _~~~~~~~~~~~~~
'UNG (UL2) (9886-10888) (
Xba I
B-Gal
(10636) Do
uBl & uB2
(ung-/B-gal)
UL3 10959-11664)
Eco RV B-Gal' (11084)
3B1
&
3B2
(UL31B-gal
Control viruses)
VIRUS
UNG
UL3
TK
dUTPase
3B31
+
-+
+
3B2
+
-+
+
uBl
-+
+
+
uB2
-+
+
+
dB
+
+
+
-1602A
-
+
+
-tB
+
+
-+
tBd
+
+
--I II
! TK (UL23)
(47802-46674)
/A\
Sna B1B-Gal * (47560) -1.
tB
(tk-\B-gal)
SV40 B-gal
PRO
dUTPase (UL5)
(107010-108123)
Asp 718
/
(107305)B-Gal'
4-dB
(dUTPase-/B-gal)
pA
602A rovKid
1
602A
r
McGeoch
(ung/B-gal
&
dUTPase-)
Hind III
Unker
tBd
(tk-/B-gal
&
dUTPase-)
FIG. 1. Schematic of themutations in the viralpanel. The top line isaschematic representationof the 152-kb HSV-1 genome. Thethick segments are representative of theterminal and internal repeat elements ofthe HSV-1 genome. The arrowsbelow the genome depict the transcripts of interestandtheapproximate locations oftheindicatedORF.Restrictionenzymesites of interestareindicated. Thesiteofdisruption
byinsertionof the,8-galcassetteis indicated.The 3-galcassettecontains the E.coli,3-galgeneunder control of the simianvirus40earlypromoter (SV40PRO)and is terminatedbythe simian virus 40poly(A) signal(pA)andisalsodepicted.The table shows the enzymeactivities encodedby each of theengineeredviruses. Note that control viruses3B1 and3B2are independent isolatesand carry functional UNG, TK,and dUTPase enzymes.
maintained for -48 h, until obvious plaques appeared, and
were then fixedfor 5 min with a solution of2% paraformal-dehyde-0.2% glutaraldehyde-0.01%Nonidet P-40at4°C.The fixed monolayers were rinsed with phosphate-buffered saline (PBS) andthenincubated with X-Gal in buffer [X-Galat 125 ,g/ml in 5 mM
K3Fe(Cn)6-5
mM K4Fe(Cn)6-2mM MgCl2-0.01%NonidetP-40]. Following overnight incubationat37°C, themonolayers were rinsed with PBS and counterstained red with Ponceau S to visualize clear plaques more easily. The plateswere observedat amagnification of X40 with a Nikon TMS microscope. Photomicrographs of selected plates were generated with TMAX-100 film (Kodak) with an image mag-nificationof-X20.The plaques on each plate were counted by using a grid background (4 mm square; -1 microscope field) to map any clearplaques observed.In casesofexcessive cytopathic effect, entire grid squares were eliminated from the counts. Chi-squareanalysiswith the Yatescontinuity factor was performed
on the compiled data. Populations of 2,587 plaques were determinedin advance to besufficient to prevent type I or II
errorswith 95% confidence.
In situ hybridization. The in situ hybridization procedure used to detect the1-gal-specific sequences in viralplaqueswas adapted from the procedure of Smith (39). A ,3-gal-specific probewas generated by randompriming with dUTP labeled with digoxigenin (DIG) byusing the manufacturer's protocol (Genius kit; BoehringerMannheim). Selected X-Gal-stained plates were rinsed with phosphate-buffered saline to remove the Ponceau S counterstain and thenincubated with 125 ,ug of pronase (Boehringer Mannheim) per ml in SSPE (90 mM NaCl, 10mMNaH2PO4, 10mMEDTA, pH7.4) for30minat
37°C. The plates thenwererinsed with SSPE and treated with
0.2 NHCl for 10 minatroomtemperature.Following 5 min of postfixation in 4% paraformaldehyde, the plates were rinsed extensively with SSPE and then prehybridized foratleast2 hat 42°C in a solution of 50% formamide (Fisher)-SSPE-0.1% sodium dodecyl sulfate-5X Denhardt's solution (lx Den-hardt's solution is 0.2% Ficoll[type400;Sigma], 0.2% polyvi-nylpyrrolidone, and0.2% bovine serum albumin [fraction V; Sigma]-20 ,gof denatured herring sperm DNA (Sigma) per ml. Hybridization of the DIG-labeled 1-gal probe (added at
-500ng/ml) proceeded overnight (16 h) at 42°C. The plates
I
J.VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.69.546.81.431.2]HSV-1 MUTATION FREQUENCY 4517
werewashed in 2x SSC(Ix SSC is 0.15 MNaCland 0.015 M
sodiumcitrate)at42°Cwith severalchanges of buffer, followed
byseveral room temperature washes in 0.1x SSC.
Immunohistochemicaldetection of the hybridized probe was
performed by using anti-DIG Fab fragment conjugated to
alkaline phosphatase (Boehringer Mannheim). The plates were preincubated in a 1% blocking solution (1% [wt/vol]
Genius kitblockingreagentin100 mMTris-150mM NaCl, pH 7.5 [Genius buffer 1]) for 30 min at room temperature. The
anti-DIG Fab fragmentwasaddedat a1:250 dilution in the 1%
blocking solution and incubated for1 to2h.Following several
washes with Genius buffer 1, the plates were treated with a
prepared solution of Histomark Red (Kirkegaard & Perry).
Following incubation for 1 h to overnight, a red precipitate
formedinthepresence of the alkalinephosphatase conjugate.
Byusing the previouslygeneratedgridmap to locate the clear
(nonblue) plaques, the plateswere reexamined and scored for
the presence of the ,B-galgene.
RESULTS
Recombinant virus panel. The genomic structures of the
recombinant viruses are shown schematically in Fig. 1.
Inser-tion of the -4-kb 3-galcassette served threefunctions in the
study. First, the ,B-gal gene was used to disrupt the enzyme
ORF without affecting the neighboring ORFs. Second, upon
incorporation into the 17syn+ genome, (3-galactivity servedas aphenotypic marker ofrecombination, expediting virus puri-fication. Third, the ,B-galgene served as a "phenotypic sink" for mutations acquired during replication of the virus.
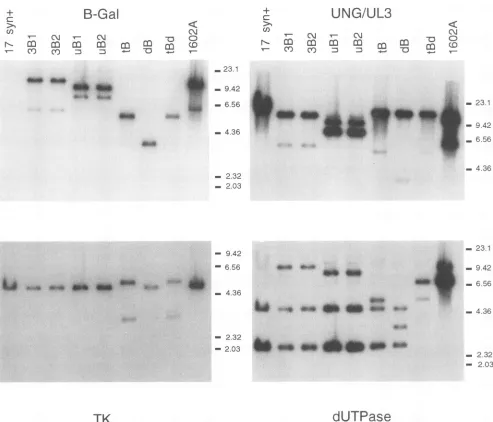
Following plaque purification of the recombinant viruses, viral DNA was prepared and analyzed by Southern blot-restriction fragment length polymorphism analysis as previ-ously described (43). A representative Southern blot of Asp718-digested viral DNAs washybridized successivelytothe indicated 32P-labeled probes and is shown in Fig. 2. By this
assay, all of the viral stocks were found to be pure, with no contaminating wild-typebandsevidentfollowing hybridization with the mutation locus-specific probe.
Following hybridization with the radiolabeled 4-kb (-gal probe, all of the mutant viruses demonstrated the expected fragment(s).Nohybridizationwasdetected in the 17syn+ lane. Addition of theAsp718 siteand the 4-kb (3-galsequence atthe UL3 locus (Fig. 1) resulted intwo novel bandsof5,732 and 10,994bpevidentfollowing hybridizationwith either the (-gal or UNG-UL3 probe (lanes 3B1 and 3B2). The wild-type Asp718 fragment, which contains the UL3 and UNG (UL2)
genes, is 12,610bp longandis evident onthe UNG-UL3 blot (for example, the 17syn+ lane).The smallerbands evident in
the tB, dB, and tBd viral samples (which have unaltered
UNG-UL3regions) are dueto
hybridization
of the viral (-galsequences to the pUC19 vector included in the UNG-UL3 probe.
Hybridization of
Asp718-digested
uBI and uB2 DNAswiththe
(-gal
or UNG-UL3 probe resulted in twofragments
of7,524 and 9,202 bp (lanes uBI and uB2 on the
3-gal
and UNG-UL3blots). These novelfragments again resulted frominsertion of theAsp718siteand the 4-kb
3-gal
sequence. Mostof the
3-gal
cassette iscontained inthe9,202-bp
fragment,
as shown following hybridization to the,B-gal probe.
Following
hybridization to the UNG-UL3 probe, which should bind
primarily to the 7,524-bpfragment, the intensities of
hybrid-ization are reversed.
Mullaney
et al. demonstrated that a 17syn+-derived virus, designated in1601, which carried the same UL2 locus mutation, was UNG- butfully
replication
competent andconcludedthat the HSV-1 UNG isdispensable
forreplication in cultured cells (30). The 3-gal insertion at the UNG locus in double mutant 1602A is in the opposite orien-tation to the insertion in uBI and uB2. Following Asp718 digestion of 1602A viral DNA, two novel fragments of 6,200 and 10,526 bp were evident ((3-gal and UNG-UL3 blots).
Insertion of the (3-galcassette into the TK ORF resulted in two novel fragments of3,599 and 5,493 bp followingAsp718 digestion oftB and tBd viral DNAs (TK blot). The expected Asp718 wild-type 4,959-bp fragment was evident in all of the
otherviruses following hybridization to the TK-specific probe
(bp 47053 to 47893). Three independent measures of TK
activity, including arabinosylthymine resistance, phosphoryla-tion of[3H]thymidine, and ['4C]thymidine plaque
autoradiog-raphy, demonstratedthat the mutation in tB eliminated
HSV-1-induced TKactivity (31a).
Owing to the cloning procedure used to disrupt the dUT-Pase gene, the Asp718 site of insertion (bp 107305) was regenerated at each end of the 3-gal insert, resulting in the presenceof the3,549-bp, promoterless(-galgene (Fig. 2,(3-gal blot, lane dB). Mutationatthis sitehas previously been shown toeliminate HSV-1 dUTPase activity (14,50).Hybridizationof
the dUTPase-specific probe (bp 105105 to 110095) demon-strated the two expectedwild-type bands(Asp718 fragments u and o, 2,772 and 4,680 bp, respectively) in all of the viruses
except thetwodoublemutants.These bandswereexpected in
dB, as the insertion is released by digestion withAsp718. In
additiontothesetwofragments, the fragments containing the
3-gal
sequence arevisible inthe recombinant virus lanesalso,
because ofthe presenceofvector sequencesin the dUTPase probe.
Double mutantstBd and 1602Awere derived from HSV-1 strain 17syn+ dUTPase-negative mutant dUT-1218 (14), in
whichtheAsp718 siteatbp 107305wasconverted to aHindlll site(Fig. 1). This mutation resulted infusionof the Asp718 u and ofragments, producinganovelfragment of7,464bp (lanes tBd and1602A).Probing of this blot and others with additional regions of the HSV-1 genome demonstrated no unexpected genomic perturbations (data notshown).
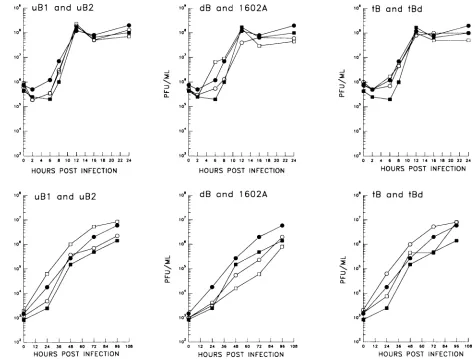
Replication kinetics of the viruses in NIH 3T3 cells. To assess the RMF of the HSV-1 strains, it was necessary to
establish that the mutant viruses replicated equivalently in
cultured cells. Single-step replication kinetic analyses with
NIH 3T3 monolayers confirmed that no general replication defectwas present in any of the viruses (Fig. 3, top panels). Sincetheeffect ofanyreplication defect would be expectedto become more pronounced with successive rounds of replica-tion, multistep kinetics were compiled following low-MOI (0.01 PFU per cell) infection of NIH 3T3 monolayers. The results of these assays demonstrated no difference in the replication abilities of the viruses. In all cases, the mutant viruses attainedpeak titersroughly equivalenttothoseof3B1 and3B2(Fig. 3, lowerpanels),and in otherexperiments they attained peak titers equivalenttothoseof strain 17syn+ (data not shown).
Together, these data confirmed that each ofthe enzymes andthe UL3 geneproductaredispensableforviralreplication
in NIH 3T3cells,in agreementwith resultsreported previously
for manyothercell lines(1,14,23, 30).Inaddition,noselective
advantageordisadvantagewasdetectedinthe viruses
contain-ing the
1-gal
insert, allowingaccurate estimation of the viral RMFon the basis ofphenotypiclossof(-gal
activity.RMFassay.Previous examinations ofthefrequencyofviral mutants in stocks ofwild-type HSV-1 strains have
demon-strated a range ofRMFsvarying from 0.001 to >1% (18-20, 31, 40,41).Inthese studies,mutantplaqueswereidentifiedby resistance toanti-HSVcompoundsin the culture media used. VOL. 68? 1994
on November 9, 2019 by guest
http://jvi.asm.org/
4518 PYLES AND THOMPSON
B-Gal
N
m
m
m
mm
mm
(DCO n A S -D
Cl) N-)
UNG/UL3
CM r CMN
m
COCOm
CO Cr) :D
C\m
-4- ~ Tr _.23.1
_9.42
6.56
b
_ j 23.1-4.36 -* w* -94
6.56
_ 4.36 - 2.32
- 2.03
9.42 - 23.1
- 6.56
- 4.36
jp~~~ml
ILI.~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~.,
Wl 'mww IEa d
** N..
A._X
._t
- 9.42 - 6.56
- 4.36
- 2.32
- 2.03 01
TK
- 2.32
2.03
dUTPase
FIG. 2. Southern blot-restriction fragment length polymorphism analysis of the viruses employed in this study. Following digestion with restrictionenzymeAsp718, the viralDNAfragmentswereseparatedin0.8%agaroseand transferredtonitrocelluloseaspreviouslydescribed(43). The indicated 32P-labeled probes describedinthetextwerehybridized successivelytothe blottodemonstrate thegenomicstructuresof the viruses. Thesamplesourceisindicatedabove the blot. Migrationof the DNA size markers(in kilobasepairs)is denotedontheside of each blot. In other studies, drug resistance was mapped totheviral TK
and DNApolgenes (5, 36), suggesting two possible types of mutations quantitated by this method. Hall and colleagues
werethe first tomap amutatorfunctionto aspecific
HSV-1-encoded protein, demonstrating that antimutator strains of HSV-1 encodea modifiedDNApol with increased fidelity of
deoxynucleoside triphosphate selection (20). Inthese studies,
stocks of HSV-1weregenerated following infection ofcultured
cells at an MOI of0.01 PFUpercell and then plated in the presence of antiviral compounds directed to the viral TK
activity. Mutants were quantitated by the number of plaques
that formed in the presenceof the antiviral compound. This systemisnotsuitableforassessmentof the contribution of the
viral TK toRMFs. In addition, TKmutations may also be a
disadvantagetothevirus (13,22,45) and thereforebe selected
against. Accurate estimation of theRMFis complicated by the
factthat thenonmutantplaques didnotform, allowing onlyan
estimation of the totalpopulation assayed.
To establish the RMF of HSV-1 strain17syn+derivatives in cultured murinecells,asystemwasdevelopedthatemploys a
nonselected phenotypicmarker(3-gal). Stocksof virusstrains carrying this markerwere prepared as described in Materials
and Methods following infection (0.01 PFU percell) of NIH 3T3monolayers. Aliquotsof-2,000PFU ofthepassagestocks
were assayedon RSCmonolayersfor the ,B-gal phenotype of individual PFU byhistochemical stainingwith X-Gal. A
mu-tantwasidentifiedasaclearplaque (Fig. 4, toppanels).
Toconfirm that clearplaquesobservedinthepassagestocks weretrue mutants(,3-gal activitynegative) and didnot
repre-sent contamination with the parental wild-type virus (P-gal
genenegative),insituhybridizationwasperformedonselected
plates. Control 17syn+ plaques did not cross-react with the probe, indicating that the in situ hybridizations were specific
for ,B-gal sequences (data not shown). Ascan be seen in the bottompanelsofFig. 4, mutantplaquesareidentifiedbylack
of the blue X-Galprecipitate (darklystained cellsattheplaque +
(I)
N.-J.VlIROL.
*we 4*0 :.
4i"M
0"'..
I
ik oiw.oo
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.62.555.74.496.2]HSV-1 MUTATION FREQUENCY 4519
and uB2
2 4 6 8 10 12 14 16 18 2022 HOURS POST INFECTION
-j
UA-
a-102
0 1 2 24 36 48 60 72 84 96 108 HOURS POST INFECTION
dB and 1602A
2 4 6 8 10 12 14 16 18 20 22 24 HOURS POST INFECTION
10s
10
103
108
104
10
a-10 t CE
104
1 2 24 36 48 60 72 84 96 108 HOURS POST INFECTION
HOURS POST INFECTION
tB and tBd
12 24 36 48 60 72 84 96 108
[image:6.612.73.548.77.436.2]HOURS POST INFECTION
FIG. 3. Replication kinetic analyses withNIH3T3 cells. Triplicate cultures of NIH 3T3 cellswere infectedatanMOIof 3to5 (top panels)
or0.01 (lower panels)PFUpercell. At theindicatedtimespostinfection,the stockswereharvested and subjectedtothree cycles of freezing and thawing,andthenthe viraltitersweredeterminedonRSCmonolayers. The resultsareplotted against the 3B1 and 3B2 dataoneach graph.For
eachvirus,the titer of the inoculum isindicatedas the zero-time point.Symbols: 0,3B1; *,3B2;0,uBI, tB, and dB;Fa,uB2, tBd, and 1602A.
rim) due to the loss of,B-gal activity but immunohistochemi-cally stained red (gray cells within theplaque) following in situ hybridization with the DIG-labeled ,B-gal probe. For each virus, at least 30% of the previously mapped mutant plaques
were tested for the presence of (-gal sequences. All of the
plaques tested contained 3-gal sequences by this assay and therefore could not be attributed to contaminating wild-type virus. These data confirmed that the RMFs established for all ofthe viralmutantswereaccurateand notinflated because of contaminating wild-type virus.
RMFof HSV-1 followingpassageinmurine cells. Thedata compiled from RMF analysis of two independent passage
stocks generated from low-MOI infections ofNIH 3T3 cells (designated P1 and P1') are summarized in Fig. 5. Statistical significance of the differences in the data was examined by
using chi-square analysis with the Yates continuity factor to
compare the RMF of each virus with thewild-type blue RMF establishedby analysisof 3B1 and 3B2.Differencesresultingin P values of <0.05 were considered significant and are indi-cated.
To establish the baseline RMF of a 17syn+ derivative in which theUNG, TK, and dUTPase geneswereunaltered, the
passage stocks of control viruses 3B1 and 3B2wereexamined.
These strains carried the (-gal insert at the UL3 locus (bp 10959to11664), whichwaschosen because it is dispensable for viral replication in cultured cells (1) and does not encode a
known enzyme activity involved in DNA replication. Two
independent viruses were isolated to limitthe possibility that unselected, second-site mutations would alter the findings.
Analysis of the P1 and P1' stocks of 3B1 and 3B2 demon-strated the HSV-1 baseline RMF in NIH 3T3 cells to be between 0.4 and 0.6%or one mutant in 178to243 PFU (Fig. 5). This finding is consistent with the range of frequencies reported for HSV-1 strains (18-20, 31, 40, 41). Chi-square analysis demonstrated no statistically significant difference between 3B1 and 3B2 for either the P1 or P1' passage.
Comparison of P1 and P1' stock RMFs demonstrated the reproducibility of theassay,with estimatedPvaluesof 0.54 and 0.37 for 3B1 P1 versusP1'and 3B2 P1 versusP1', respectively.
Although no selective advantage or disadvantage of (-gal
activitywasobserved in thereplicationkineticanalysis (Fig. 3), itwaspossiblethatmutant plaque frequencycould beaffected by successive replication. To control for such a possibility, a
high-MOI passage (HP) stockwas generated from NIH 3T3 cellsfollowinginfection with 3to5PFUpercell. Nosignificant difference wasfound between the RMFs of 3B1 and 3B2 P1,
-j Z:2
0--j :2
N0l
V()L.68, 1'994
on November 9, 2019 by guest
http://jvi.asm.org/
4520 PYLES AND THOMPSON
Xf-,
_fA~~~
-Z4~~
.44~ ~ ~
AC
44
o *,~ ~ re^.Bu " ++*bFD '' e . 5~i
uS.# tS Sv.
eS' ,. Z ''': *5,eV.*<~
FIG. 4. Photomicrographsof theRMFplaqueassay.Analysisofthepassagestockswascompleted byplatingof-2,000PFU/100-mm-diameter
RSCmonolayer. Plaquesweremaintainedin mediumcontaininganti-HSVserumandthen,at 48hpostinfection,fixed andstained with X-Gal.
13-Gal activityresultedinablueprecipitate,asseenin thetop two panels (cells with ,B-gal activityappear dark and are located at theplaquerim). Plaqueswhich didnotproduce 1-galactivity (mutantsindicatedby arrowheads)also became visible in thetoptwopanelsaftercounterstainingwith
Ponceau S. To confirm that the clear plaqueswere true mutants and not wild-type contaminants, in situ hybridizationwith a DIG-labeled,
1-gal-specific probewasperformed.Followingimmunolocalization of the DIG, positiveinsitu hybridizationresultedinstainingof theplaque center(labeledcellsappeargrayatthecenterof theplaque).The bottom twopanels representtrue mutantsthat contained the 13-gal genebut
lacked 13-gal activity.
P1',orHPstocks.However,amarginally significantdifference (P = 0.047) was demonstrated when the 3B1 and 3B2 HP stocks were compared. The results of the HP stocks are
presented graphically in Fig.6. A second passage (P2)ofthe P1 stockalsowascompleted byinfectingNIH 3T3 cellsat0.01
PFU per cell. The data from the P2 stocks (Fig. 6) were
consistent with the RMFs established for the P1, P1',and HP stocks.
The contribution of the HSV-1 UNG, dUTPase, and TK activities tothe viral RMF. UNGactivityremovespotentially
mutagenic uracil residues fromDNAthatarepresentbecause
of misincorporation by DNA pol or spontaneous cytosine
deaminations(27).Ifcytosinedeaminationsarenotrepaired,a
C-to-Ttransition is expectedto occur, suggestingadirect role for UNG in determining theviral RMF. The data compiled
from examination of theP1and P1'passagestocks ofuBland uB2 demonstrated that the HSV-1 UNG activity does not
appreciablyaffect the RMFfollowinglimitedpassagein NIH 3T3 cells. The RMFsestablished for both uBl anduB2were
equivalenttothe baseline RMF(0.4to0.5%or onemutant in 188to267PFU; Fig. 5). Consistent with the observation that the MOI hadnosignificant effectontheRMFof 3B1or3B2,
.A.
J. VIROL.
94
17;I
:.. ':" .:'
i, ':
*:
U
V.,.
I4." (P-;-v
j -i. #,:.
..t.
14
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.94.527.78.526.2]HSV-1 MUTATION FREQUENCY 4521
3B1 3B2 uB1
m uB2
5 dB 1602A tB tBd
0 0.5 1 1.5
% MUTANT
2.5
--37/5197 ~159598
26240
3B2 2220I4528 3B2
uBl . -.57/5829|
uB2 64/ 053
dB 1602A
tB /5665*
t1/73094l
tBd a
0 0.5 1 1.5 2 % MUTANT
Co 3B1 3B2 uBl uB2 dB 1602A tB
tBd - 1_
, --, ---+
-25 -20 -15 -10 -5 0 5 10
FOLD DIFFERENCE FROMBASELINE
FIG. 5. Estimated RMF ofP1 and P1' viral stocks generated in NIH3T3 cells. The results of themutantplaqueassayappliedto two
independently generated low-MOI (0.01 PFUpercell)passagestocks
(P1 and P1') arepresented. The statistical significance of the differ-encesinthe datawasanalyzed by chi-square analysis with the Yates
continuityfactor. Each of the RMFswascompared with the RMFs of
the control 3B1 and 3B2 virus stocks. The RMFs thatwerefoundtobe significantly different from those of the controls (P < 0.05) are
indicated with asterisks. The results arepresentedasthe percentages
of thepopulationsthatwerefoundtobemutants.The fractionnextto
each barrepresentsthe totalplaquecountwith the number ofmutants
observed divided bythe total number ofplaques. The lower graph demonstrates the fold difference from the baseline RMF (0.5%) for each of the virus stocks.
nodifferencewasobserved between the RMFs ofthe HP stock
and the low-MOI stocks of uBl or uB2 (Fig. 5 and 6). Successive passagesofuBl and uB2 (P2 inFig. 6) resulted in stocks with RMFs that were significantly increased (twofold; Fig. 6, lower graph) comparedwith the uBl and uB2 RMFs observed in theP1 and P1' passages (P1 stocksversusthe P2
stock,P = 0.001).
Itwasof interesttodetermine iftheviraldUTPaseactivity, which should alsoreduce dUTPmisincorporation,would
con-tributetothe RMF. Asignificantincrease in thefrequencyof clearplaquesfrom the baseline RMFwasobserved intheP1 and P1' stocksofdUTPase-negativeviruses dB and 1602A(2.5 and 2%, respectively; Fig. 5).This finding clearlyestablished that the HSV-1-encoded dUTPase provides an antimutator function. In these stocks, a mutant plaque occurred as fre-quentlyas 1 in40, significantly moreoften thanin3B1 or3B2 (P < 0.00001). These results are not attributed easily to unselected, secondary mutations since dB and 1602A were
derivedfrom differentparentalviruses(17syn+ and dUTPase-negative 17syn+ mutantdUT-1218, respectively)andcarrythe
3-galgene atdifferent loci (Fig. 1).
The P2 stock of dB demonstrated a significant increase in RMF from the RMF established in the P1 stocks (P = 0.03;
381 31321 uBl
uB2
dB~
1602A tB
tBd _-
--30 -25 -20 1 5 -10 -5 FOLD DIFFERENCE FROM BASELINE
O 5 10
FIG. 6. Estimated RMF of P2 and HP viral stocks generated in NIH3T3 cells. As described in the text,aP2 stockwasgenerated by
low-MOI (0.01 PFUpercell) infection of NIH 3T3 cells with the PI
stocks of each virus. The original virus stocks were also used to generate an HP stock following high-MOI (3 to 5 PFU per cell) infection of NIH 3T3 cells. The results of theRMFassaysof these viral
stocksarepresentedasdescribed in thelegendtoFig.5.
Fig. 6). This RMFwasthe highest observed inanyof the NIH
3T3 viruspassagestocks. The 1602A P2 stock also hadaslight
butnotsignificant increase in the RMF compared with the P1 stocks (P = 0.17). The RMFs in the HP stocks of dB and
1602A were not significantly different from those of the low-MOI passage stocks (Fig. 5 and 6).
HSV TKactivity primarilyconverts thymidine toTMP but alsophosphorylates deoxycytidine and nucleoside analogs that
are the basis forsome successful antiherpes therapies.
Exam-ination of the TK- virus passagestocks clearly demonstrated a mutator function associated with this enzyme activity. The
P1 and P1' stocks of the two mutantswhich lack TK, tB and tBd, were found to contain a minimal number of mutant plaques (Fig. 5). The P1 RMFswere foundtobe 5-to20-fold less than the baseline (Fig. 5, lower graph). Having estab-lished an appreciable RMF increase associated with loss of dUTPase activity in both dB and 1602A, we found that the mutatoractivity ofTKwas even moredramatic,sincea50-fold
differencein RMFwasobservedbetween tBd (TKand dUT-Pase negative) and 1602A (UNG and dUTPase negative) in theP1 stocks.The RMFs established for the tB and tBd P2 and HPstockswereconsistentwith the RMFs of the P1 stocks(Fig.
5 and 6).
ViralRMFs following passagein LM TK- cells. To deter-mine if the observed RMFswereaffected significantly bythe cellular environment provided by NIH 3T3 cells, a low-MOI
passageof selected viruseswasperformedwith LM TK- cells. LM TK- cellswerechosentodetermineiftheloss ofacellular
enzyme activity involved in nucleotide pool size regulation would measurably affect the viral RMF. The results of the RMFanalysis ofthe passagestocks arepresented inFig. 7.
*P2
HP
L_-_
1_1SQ/5783
2.5 3 3.5
5/631.4
/5427* 1 7/.dQ%tiR*
m
_ -.L---
-VOL. 68, 1994
c-
----I
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.320.556.79.334.2] [image:8.612.57.300.79.348.2]4522 PYLES AND THOMPSON
3B3 uBl
o dB > 1602A
tB
tBd
02/5161r
I "
05-
0-1-
---2-
2. 0.5 1 1.5 2 2.5 3% MUTANT
3B1 uBl cn dB
CC
> 1602A tB tBd
-45 -35 -25 -15 -5
FOLD DIFFERENCEFROM BASELINE
FIG. 7. Estimated RMFs of viral stocks generate cells. A low-MOI (0.01 PFUpercell) infection ofLM
performed with the indicated viruses, as described ir RMFs of the resulting stocks were assayed, and t presented graphically asindicated in the legendto Fi1
differences from the LM TK-- 3B1 stock are denot asterisks. Significantdifferences between the RMFest
P1 stock generatedin NIH 3T3 cells and the RMF ir
cell-generatedviral stockare indicatedwith double asi
The RMF ofthe 3B1 stock generated from] infection was the lowest of the RMFs observed
(Fig. 5, 6, and 7); however, the difference frc established in the NIH 3T3 P1 stockswas not s
stocks versus LM TK- P1 stock, P = 0.12).
confirm an averaged baseline RMF for 17syn+ -0.5%. Analysis of the uBI passage stock gene TK- cells demonstrated asignificant decrease ii ofmutant plaques (Fig. 5 versus Fig. 7; P1 stoc] TK- P1 stock, P = 0.005). This reductionwas c( aslight antimutator effect for UNG activityand the NIH3T3 cellular environmentwasbetter abl sate the UNGmutation.
Both the dB and 1602A LM TK- cell passag(
that demonstrated that the antimutator functi(
with dUTPase activitywas not specificto NIH 3 LM TK- dBstock RMFwassignificantly lowert
observedinthe dB NIH 3T3 P1 stock(P= 0.033 fivefold greaterthanthat of the LM TK- 3B1 s This findingwasconsistent with the generaltrer RMF inthe LM TK- cell environment. Incontra LM TK- stock had a significantly increased RN
with the NIH 3T3 P1 stocks (P1 stocks versus
stock,P= 0.0002; Fig.5versusFig. 7),
suggestinj
TK- cellular environment was less able to coi
mutations present in 1602A. In the 1602A LM mutant was observed approximately every 27 pl
Fig. 7). Despite the lack of cellular TK, both th
stocks showed RMFs equivalent to those established in NIH
3T3 cells
(Fig.
7).
DISCUSSION
*189/515* The evidence presented in this report demonstrates that the
I HSV-1-encoded TK and dUTPase
significantly
contribute to viral RMFs following viralpropagation
in two murine celllines. In
addition,
arole forthe HSV-1 UNG indetermining
3.5 4 the viral RMF was
suggested by
the serial passage of twoindependent
UNG- viral stocks. The baseline viral RMF of0.5% was established in stocks of two
independent
17syn+
UL3-p3-gal
strains that encode unaltered UNG, TK, and_ X dUTPase activities
(Fig. 5, 6,
and7).
It should be noted thatthe function of the HSV-1 UL3 gene product has not been
established;
therefore,
it ispossible
that thisgeneproduct
plays
a role in controlling the viral RMF. However, the baseline RMF established withthese twovirusesis consistent with the
RMFs
reported
for otherwild-type
HSV-1 strains(18-20, 31,
40,
41).
Thereproducibility
of thisassayis evidentby
compar-ison oftwo
independent
passagesfollowing
low-MOI infection(P1
andP1' inFig.
5). Passage
of 3B1 intheLM TK- celllinealso demonstrated that the established baseline RMFwasnot
specific
tothe NIH 3T3 cellular environment(Fig.
7).
5 15 The HSV-1 strains examined in this study that lacked a
functional TK
activity
had thehighest-fidelity replication
ofLM TK any of the viruses examined. The tB passage stocks
demon-TK-cellswas strated the fewestmutantsof any of the passage stocks andhad
i thetext. The RMFs that were up to 40-fold reduced from the baseline.
the results are Analysis of double mutant tBd, which lacked both TK and
g. 5. Significant dUTPase activities, demonstrated the
significance
of the mu-ted with single tatorfunction associated with TKactivity,
since viruseslacking
tablished in the dUTPase activity had RMFs up to fivefold greater than the
n the LM TK- baseline. It is possible that the RMFs observed for tB and tBd terisks. were due to a compensatory mutation. It is unlikely that this is the case,
however,
becausetBwasderiveddirectly
from17syn+
while tBd was
generated
from17syn+-derived,
dUTPase-negative
strain dUT- 1218. It ismorelikely, therefore,
that theLM TK- cell reduced RMFs are dueto the loss ofTK
activity.
for this virus A
possible
explanation
for the increased RMF associated )m the RMF witha functional HSV-1TKmaybe found in the broad range ,ignificant (P1 ofsubstratesrecognized by
this enzyme(reviewed
inreference These results 16). In addition tophosphorylating
nucleotides which arederivatives of useful for DNA
replication,
the TKactivity
phosphorylates
crated in LM nucleoside
analogs
which can lead to termination of DNA n the number synthesis(6-8).
Thenucleotide-binding
site in theTKprotein
ksversus LM has been indicated(16), suggesting
that TK activities with onsistentwith limited substratespecificity
could beengineered
and then suggests thatassayed
in this system. Infact, drug-resistant
HSV-1 strainsle tocompen- encoding TK
proteins
with reduced substratespecificity
al-ready
have beenreported (26, 47). Analysis
of these alteredes had RMFs TKgenes ina strainwith a knownbaselineRMFshouldhelp
on associated elucidate themechanismbywhichTKcontributestotheRMF. T3 cells. The The
explanation
for the antimutator function associatedhanthe RMF with the viral dUTPase is less obvious. As noted above, the
§)
butwasstill stocks ofdUTPase-negative
virus dB hadRMFsfivefoldhigher
,tock
(Fig. 7).
thanthebackground.Consistent withthesefindings,dUTPaseid of reduced activity has been shown to serve an antimutator function in 1st, the1602A other systems (34, 37).Theactivity of dUTPase resultsinthe
vF compared reduction of dUTP pools and indirectly increases available
LM TK- P1 dTTP pools (9, 24, 25). On the basis of the reduced RMF
g that theLM which results from loss ofTKactivity, reductions in the dTTP mpensate the pool donoteasily explainthe alteredRMF. Conversion ofthe TK- stock, a dUTPpool todUMP mayreduceRMFs,butmisincorporation laques
(3.8%;
ofdUTP would be expected tobe repaired by the cellularor te tB and tBd viralUNGactivityin the cellduringinfection(11, 27).
IfuracilI
-1-J. VIROL.
787
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.61.294.80.339.2]HSV-1 MUTATION FREQUENCY 4523
residues are not removed, a mutation does not necessarily occur; however, the presence of uracil residues in DNA may affect the proper functioning of regulatory sequences (15, 48). It is therefore unlikely that either of these possibilities fully explains the contribution of dUTPase. Identification of the regions of the HSV-1 dUTPase which are important for enzymeactivity and viral characteristics is in progress and may help to elucidate the contribution of this enzyme to the viral RMF.
UNG activitywasexpectedtoincrease thefidelity of DNA replication by removal of uracil because of misincorporation and from spontaneous deamination of cytosine residues in DNA (27). Such deaminations are potentially mutagenic, as theyresult inaG:C-to-A:Ttransitionif not repaired, and may be of great concern following extended periods of HSV-1 latencyinahost(30).Inother systems, UNG activity has been demonstrated to be more effective on the G:U mismatch resulting from deaminations (49) and important for reduction of mutation frequencies (21, 34, 37). In support of a similar antimutator function for the HSV-1 UNG, successive passages ofuBl and uB2resulted inasignificant increase in the RMF (Fig. 6, P2)comparedwith both the P1 and P2 stocks of 3B1 and 3B2. It is attractive to hypothesize that in a UNG-background with time or successive passage, the number of cytosinedeaminations would build up, leading to an increase in the RMF. Analysis of successive passages (P3, P4, etc.) may lend support to this theory. It also would be interesting to establish the RMFof virus recovered from reactivations fol-lowing increasing periods of latency; however, preliminary studies have been impeded by the minimal amounts of virus recovered.
dUTPase-negative, UNG- double mutant 1602A was
ex-pected to have an increased RMF because of an increased probability of dUTP misincorporation and a reduced ability to
remove such bases. Consistent with these predictions, the 1602A passagestocks had some of thehighest RMFs of any of the stocks examined. It is possiblethat the NIH 3T3 cellular environment is abletocompensatepartiallyforthe viralUNG mutation, since passage in LM TK- cells resulted ina 1602A stock with thehighestRMFobserved(onemutantin 27PFU; Fig. 7).
The RMF assay reported here provides a more versatile system for assessment of the contribution ofvirally encoded factors, for several reasons. The results obtained with this systemarebased uponloss of,B-gal activity,which shouldnot
have affected viral replication, providing no selective advan-tageordisadvantage.Previous studiesassayedfor mutations of the TK gene (18-20, 31, 40, 41), which, although dispensable for viral replication in cultured cells, has been shown in this workto contribute dramatically to the viral RMF. The RMF assayinprior studies selected fora
drug
resistancephenotype
to quantitate altered progeny, which may have limited the scoring of mutations. The RMF differences could bedue,
in part, to the use of differentwild-type
HSV-1 strains in the different studiesas wellas the cell lines used togenerate the passage stocks. Inthisstudy,
allofthevirusesexaminedwere derived originally from the HSV-1 17syn+ strain and were passaged in two murine cell lines to demonstrateclearly
the contribution of thevirallyencodedUNG,
TK,and dUTPasetothe viralRMF.
Apossiblelimitation of this assay is the effect ofthecontext
of the
p-gal
gene in the viral genome.Specific regions
of the genome may bemore orless pronetomutation,
leading
in this assay to greater or fewer clearplaques. However,
dUTPase-negativeviruses dB and 1602A,which carryn-gal
insertionsat oppositeendsof theunique long
region (Fig.
1),
demonstratednodifference between their RMFs, suggesting that the context has alimited effect or these two regions of the genome have similar mutation potentials.
The reported RMFs in this study were not artificially inflated because of contamination of the passage stocks with wild-type, 3-gal gene-negative virus, since in situ hybridization demon-strated that100% of the mutant plaques tested contained
p-gal
sequences. The MOI also can be eliminated as a possible concern,since RMFs of stocks generated by infection with 0.01 PFU per cell(Fig. 5) were consistent with the RMFs observed followinghigh-MOI passage (3 to 5 PFU per cell; Fig. 6). The RMFs ofthe 3B1 and 3B2 HP stocks were not significantly different from the P1 RMFs (Fig. 5 and 6), also suggesting that there was no selection for or against viruses which lacked a functional ,B-gal gene.
HSV-encoded TK, dUTPase, and UNG activities are dis-pensable for efficient viral replication (14, 23, 30); however, all three enzymes have been shown to play important roles in viral replication in vivo (13, 32, 33). Although no replication defi-ciency of dUTPase-negative or UNG- viral strains has been observed in vitro, it is likely that these enzymes promote increased fidelity of DNA synthesis. Having established that HSV-1 encodes several genes which affect the viral RMF in addition to the viral DNApol (20), antiviral therapies which inhibit the function of mutator activities or enhance the activity of antimutators may increase the effectiveness of present therapies.
ACKNOWLEDGMENTS
This research was supported by Public Health Service grants NS 25879 from theNationalInstitute ofNeurologicalandCommunicative Disordersand Stroke andAl 22667 and Al 32121 from the National Institute ofAllergyandInfectious Diseasesto L.Stanberryand R.L.T. R.B.P.wastherecipient ofanAlbert J. RyanFoundationpredoctoral fellowship.
The advice and viruses provided by D. McGeoch were greatly appreciated.
REFERENCES
1. Baines, J. D., and B. Roizman. 1991. The open readingframes UL3, UL4, UL10, and UL16 aredispensablefor thereplication of herpes simplex virus 1in cell culture. J. Virol. 65:938-944. 2. Bernstein, C., H. Bernstein, S. Mufti, and B. Strom. 1972.
Stimulation of mutationinphageT4by lesionsin gene 32 andby thymidine imbalance. Mutat. Res. 16:113-119.
3. Burns, W. H., et al. 1982. Isolation and characterisation of resistant herpes simplex virus after acyclovir therapy. Lancet i:421-423.
4. Challberg, M. D., and T. J. Kelly. 1989. Animal virus DNA replication.Annu. Rev.Biochem.58:671-717.
5. Coen, C.M., and P. A. Schaffer. 1980. Two distinct loci confer resistancetoacycloguanosineonherpes simplexvirus type 1. Proc. Natl. Acad.Sci. USA77:2265-2269.
6. Coent,D. M. 1990.Antiviraldrug resistance.Ann.N. Y.Acad.Sci. 616:224-237.
7. Coen,D.M.1991.Theimplicationsof resistancetoantiviral agents forherpesvirusdrug targets andtherapy. Antiviral Res. 15:287-300.
8. Crumpacker, C. S. 1989. Molecular targets of antiviraltherapy.N. Engl. J. Med. 321:163-172.
9. Daikoku, T., N. Yamamoto,K.Maeno,andY. Nishiyama. 1991. Role of viral ribonucleotide reductase in the increase of dTTP poolsize inherpes simplex virus-infectedVero cells. J. Gen. Virol. 72:1441-1444.
10. Drake, J. W. 1969. Genetic control of mutationratesin bacterio-phageT4. Nature(London)221:1128-1132.
11. Duker, N., and C. L. Grant. 1980. Alterations in the levels of deoxyuridinetriphosphatase, uracil-DNAglycosylaseand AP
en-donucleaseduringthe cellcycle.Exp. Cell Res. 125:493-497.
VOL. 68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
4524 PYLES AND THOMPSON
12. Erlich,K. S., J. Mills,P. Chatis,G.J.Mertz, D. F. Busch, S. E. Follansbee,R.M.Grant,andC. S.Crumpacker. 1989. Acyclovir-resistant herpes simplex virus infections in patients with the acquired immunodeficiency syndrome.N. Engl.J. Med. 320:293-296.
13. Field,H.J., and P.Wildy. 1978. Thepathogenicityofthymidine kinase-deficientmutants ofherpes simplexvirus in mice. J. Hyg. 81:267-277.
14. Fisher,F.B., and V. G. Preston. 1986.Isolation and characteriza-tion of HSV-1 mutants which fail to induce dUTPase activity. Virology 148:190-197.
15. Focher, F.,A.Verri, S.Verzeletti, P.Mazzarello, and S. Spadari. 1992.Uracil in OriS ofherpes simplex virus 1 alters itsspecific recognition by origin binding protein (OBP): doesvirus induced uracil-DNA glycosylase playa key role in viral reactivation and replication? Chromosoma102:S67-S71.
16. Gentry, G. A. 1992. Viral thymidine kinases and their relatives. Pharmacol.Ther. 54:319-355.
17. Glorioso, J.,W. F. Goins, and D. J. Fink. 1992. Herpes simplex virus-basedvectors.Semin. Virol. 3:265-276.
18. Hall, J. D., and R. E. Almy. 1982. Evidence for the control of herpes simplex virus mutagenesis by the viral DNApolymerase. Virology116:535-543.
19. Hall, J. D.,D. M.Coen, B. L.Fisher, M.Weisslitz, S. Randall, R.E.Almy, P. T. Gelep, and P. A. Schaffer. 1984.Generation of diversityinherpes simplex virus:anantimutatorphenotypemaps totheDNApolymeraselocus.Virology132:26-37.
20. Hall,J. D., P.A.Furman, M.H.St.Clair, and C. W.Knopf.1985. Reduced in vivo mutagenesis by mutant herpes simplex DNA polymerase involves improved nucleotide selection. Proc. Natl. Acad. Sci.USA 82:3889-3893.
21. Holliday, R. 1985. Aspects ofDNA repair and nucleotide pool imbalance, p. 453-460. In F. J. de Serres (ed.), Genetic conse-quencesofnucleotidepool imbalance. PlenumPress,NewYork. 22. Jamieson,A. T., G. A. Gentry, andJ. H. Subak-Sharpe. 1974. Induction ofboththymidine and deoxycytidine kinase activity by
herpesviruses.J.Gen. Virol.24:465-480.
23. Kit, S., and D. R. Dubbs. 1965. Properties ofdeoxythymidine kinase partially purified from noninfected and virus-infected mousefibroblastcells. Virology26:16-27.
24. Kornberg,A. 1980. DNAreplication,p. 52-64. W. H. Freeman & Co.,SanFrancisco.
25. Kunz,B.A.,andS. E. Kohalmi. 1991.Modulation ofmutagenesis by deoxynucleotidelevels.Annu. Rev.Genet. 25:339-359. 26. Larder,B.A.,Y.Cheng,andG.Darby.1983.Characterizationof
abnormal thymidinekinases induced bydrug-resistant strains of herpessimplexvirus type 1. J. Gen.Virol.64:523-532.
27. Lindahl,T. 1979. DNAglycosylases,endonucleases forapurinic/
apyrimidinic sites, and base excision repair. Prog. Nucleic Acid Res.22:135-192.
28. Lo, K.,and M.J. Bessman. 1976. Anantimutator deoxyribonucleic acidpolymerase.J.Biol.Chem.251:2480-2486.
29. McGeoch,D.J.,M. A.Dalrymple,A.J.Davison, A. Dolan, M. C. Frame,D.McNab,L.J.Perry, J. E. Scott, and P. Taylor. 1988. The complete sequenceofthelong unique region in the genome of herpes simplexvirustype 1. J.Gen. Virol. 69:1531-1574. 30. Mullaney, J.,H. W.Moss,and D.J. McGeoch.1989. Gene UL2 of
herpessimplex virustype 1 encodes a uracil-DNA glycosylase. J. Gen. Virol. 70:449-454.
31. Parris, D. S., andJ. E. Harrington. 1982. Herpes simplex virus variants resistant to high concentrations of acyclovir exist in clinicalisolates. Antimicrob. AgentsChemother. 22:71-77. 31a.Pyles,R.B.,etal.Unpublished data.
32. Pyles,R. B., N. M. Sawtell, and R. L. Thompson. 1992. Herpes
simplex virus type 1 dUTPase mutants areattenuated for neuro-virulence, neuroinvasiveness, and reactivation from latency. J. Virol.66:6706-6713.
33. Pyles, R. B., and R. L. Thompson. Evidence that the herpes simplex virus type 1 uracil DNAglycosylaseisrequiredforefficient viral replication and latency in the murine nervous system. J. Virol.,in press.
34. Richards, R.G., 0. Brown, and W. D. Sedwick. 1985. Misincorpo-rationofdeoxyuridine inhuman cells:consequencesofantifolate exposure, p. 149-162. In F. J. de Serres (ed.), Genetic conse-quencesof nucleotidepoolimbalance. PlenumPress,New York. 35. Roizman, B., and A. E.Sears. 1990. Herpessimplex viruses and their replication, p. 1795-1841. In B. N. Fields et al. (ed.), Virology, 2nd ed. Raven Press, New York.
36. Schnipper, L. L., and C. S. Crumpacker. 1980. Resistance of herpes simplexvirus to acycloguanosine: role of viral thymidine kinase and DNA polymerase loci. Proc. Natl. Acad. Sci. USA 77:2270-2273.
37. Sedwick, W. D., 0. E. Brown, and B. W. Glickman. 1986. Deoxyuri-dinemisincorporation causessite-specific mutationallesions inthe lacI gene of Eschenichia coli. Mutat. Res. 162:7-20.
38. Sibrack, C. D., C. McLaren, and D. W.Barry. 1982.Diseaseand latency characteristics of clinical herpes simplex isolates after acyclovirtherapy. Am. J. Med. 37:372-375.
39. Smith, G. H. 1987. Insitu detection of transcription in transfected cells using biotin-labeled molecular probes. Methods Enzymol. 151:530-539.
40. Smith, K. 0. 1963. Some biological aspects ofherpesvirus-cell interactions in the presence of 5-iodo, 2-desoxyuridine (IDU). J. Immunol. 91:582-590.
41. Smith, K. O., W. L. Kennell, R. H. Poirier, and F. T. Lynd. 1980. In vitro and in vivo resistance of herpes simplex virus to 9-(2-hydroxyethoxymethyl)guanine (acycloguanosine). Antimicrob. Agents Chemother. 17:144-150.
42. Sonkin, P. L., K. H. Baratz, R. Frothingham, and L. M. Cobo. 1992.Acyclovir resistant herpes simplex virus keratouveitis after penetrating keratoplasty. Ophthalmology 99:1805-1808. 43. Southern, E. M. 1975. Detection of specific sequences among
DNAfragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517.
44. Speyer, J. F. 1965. Mutagenic DNA polymerase. Biochem. Bio-phys. Res. Commun. 21:6-8.
45. Tenser, R. B. 1991. Role of herpes simplex virus thymidine kinase expression in viral pathogenesis and latency. Intervirology 32:76-92.
46. Thompson, R. L., E. K. Wagner, and J. G. Stevens. 1983. Physical location of a herpes simplex virus type-1 gene function(s) specif-ically associated with a 10 million-fold increase in HSV neuroviru-lence. Virology 131:180-192.
47. Veerisetty, V., and G. A. Gentry. 1983. Alterations in substrate specificity and physicochemical properties of deoxythymidine ki-nase of a drug-resistant herpes simplex virus type 1 mutant. J. Virol. 46:901-908.
48. Verri, A., P. Mazzarello, G. Biamonti, S. Spadari, and F. Focher. 1990. The specific binding of nuclear proteins to the cAMP responsive element (CRE) sequence (TGACGTCA) is reduced by themisincorporation of U and increased by the deamination of C. Nucleic Acids Res. 18:5775-5780.
49. Verri, A., P. Mazzarello, S. Spadari, and F. Focher. 1992. Uracil-DNA glycosylasespreferentially excise mispaired uracil. Biochem. J. 287:1007-1010.
50. Williams, M. V. 1988. Herpes simplex virus-induced dUTPase: targetsite for antiviral chemotherapy. Virology 166:262-264.
J. VIROL.