0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
The Spontaneous Reactivation Function of the Herpes Simplex
Virus Type 1 LAT Gene Resides Completely within the First
1.5 Kilobases of the 8.3-Kilobase Primary Transcript
GUEY-CHUEN PERNG,1HOMAYON GHIASI,1,2SUSAN M. SLANINA,1
ANTHONY B. NESBURN,1,2ANDSTEVEN L. WECHSLER1,2*
Ophthalmology Research Laboratories, Cedars-Sinai Medical Center Research Institute,
Los Angeles, California 90048,1and Department of Ophthalmology,
UCLA School of Medicine, Los Angeles, California 900242
Received 21 August 1995/Accepted 29 October 1995
The herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) gene is essential for efficient spontaneous reactivation of HSV-1 from latency. We report here that although the LAT gene is 8.3 kb in length, the first 1.5 kb of the LAT gene alone is sufficient for wild-type levels of spontaneous reactivation. We began
with a LAT deletion mutant of HSV-1 strain McKrae in which the LAT promoter and the first 1.6 kb of the 5*
end of the LAT gene had been deleted from both copies of LAT (one in each viral long repeat). As we previously
reported, this mutant (dLAT2903) was significantly impaired for spontaneous reactivation (G. C. Perng, E. C.
Dunkel, P. A. Geary, S. M. Slanina, H. Ghiasi, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler, J. Virol. 68:8045– 8055, 1994). We then inserted the LAT promoter and the first 1.5 kb of the LAT gene into a location in the
unique long region ofdLAT2903 far removed from the normal location of LAT in the long repeats. This resulted
in a virus (LAT15a) whose capacity for transcribing LAT RNA was limited to the first 1.5 kb of the 8.3-kb LAT primary transcript. Rabbits were ocularly infected with this mutant, and spontaneous reactivation was mea-sured in comparison to those of the original LAT-negative mutant and its marker-rescued (wild-type) virus,
dLAT2903R. LAT15a had an in vivo spontaneous reactivation rate of 12%, compared with a rate of 11% for the
marker-rescued virus and 0% for the LAT-negative virus. Southern analysis confirmed that the spontaneously reactivated LAT15a virus retained the original deletions in both copies of LAT and the 1.5-kb LAT insertion in the unique long region. Thus, insertion of the first 1.5 kb of LAT (and its promoter) at a site distant from the normal LAT location appeared to completely restore in vivo spontaneous reactivation to wild-type levels, despite the remaining inability of the original LAT genes to transcribe any LAT RNA. The function of LAT involved in efficient spontaneous reactivation therefore appeared to map completely within the first 1.5 kb of the LAT gene.
A hallmark of herpes simplex virus type 1 (HSV-1) latent infection is the propensity of the latent virus to reactivate at various times and produce recurrent disease. Recurrent HSV-1 corneal infection, which can lead to blindness due to scarring of the cornea, is the most common cause of infectious blind-ness in the developed world (21). During latency, LAT is the only viral gene that is abundantly transcribed (28, 35). LAT is located in the long repeat region of the HSV-1 genome and is therefore present in two copies per genome. The primary LAT transcript is 8.3 kb and gives rise to a family of LAT RNAs (LATs), including very stable ones of 2 and 1.5 kb (6, 9, 28, 32, 34, 35, 40–44, 47). LAT is essential for efficient reactivation from sensory neurons, since LAT transcription-negative mu-tants have been shown to reactivate poorly by explant or in-duced reactivation in the mouse (1, 11–13, 18, 19, 29, 33, 39), by induced reactivation in the rabbit (2), and by spontaneous reactivation in the rabbit (24).
Despite the large body of work indicating that LAT is re-quired for efficient reactivation, the mechanism by which LAT functions remains unknown. No LAT-encoded protein has been detected during latency, nor has the functional region of the
primary 8.3-kb LAT transcript been mapped. It has been pro-posed that LAT may function via some type of antisense reg-ulation of the important ICP0 and/or ICP34.5 viral genes, both of which the primary 8.3-kb LAT transcript completely over-laps in an antisense direction (47) (see Fig. 1). Presumably such an antisense interaction would block ICP0 or ICP34.5 function, thereby leading to latency. Increased latency would then result in increased reactivation rates. Although this in-triguing hypothesis has received continued bolstering by the uniform inability to detect a LAT-encoded protein during la-tency in experimental animals, no direct supportive experimen-tal evidence has yet been presented.
Determining the region(s) of LAT required for LAT’s func-tion would be very helpful in determining if an antisense mech-anism may be involved. Unfortunately, the same overlap with ICP0 and ICP34.5 that makes antisense a possible mechanism severely complicates attempts to make LAT mutants that would address the likelihood of such an antisense mechanism. It is not possible to delete or mutate portions of LAT that are colinear with ICP0 or ICP34.5 without also altering the colinear gene. Likewise, it is not possible to alter ICP0 or ICP34.5 without altering LAT. If such mutants had an altered phenotype, it would be very difficult to determine if the phenotype was due to the alteration in LAT or to the alteration in the overlapping gene.
This concern would not apply to mapping that portion of the LAT gene (approximately 1.8 kb) prior to the regions of ICP0 * Corresponding author. Mailing address: Ophthalmology Research
Laboratories, Cedars-Sinai Medical Center Research Institute, Davis Bldg., Room 5072, 8700 Beverly Blvd., Los Angeles, CA 90048. Phone: (310) 855-6455. Fax: (310) 652-8411.
976
on November 9, 2019 by guest
http://jvi.asm.org/
and ICP34.5 overlap. One approach might be to insert a tran-scriptional stop site (i.e., a polyadenylation signal) prior to the region of ICP0 overlap. However, interpretation would be am-biguous because it would not be possible to unequivocally prove that the stop site blocked transcription with 100% effi-ciency. Even a very small, undetectable level of readthrough transcription could result in maintenance of a biological func-tion. We therefore took a novel approach. Starting with a LAT mutant (dLAT2903) that we had previously made and which lacks the LAT promoter and the first 1.6 kb of LAT (24), we introduced the first 1.5 kb of the LAT gene along with 1.7 kb of upstream promoter sequences into a distant location in the unique long region of the virus genome (see Fig. 1). This virus (LAT15a) can transcribe only the first 1.5 kb of the 8.3-kb LAT transcript from the inserted material. No LAT transcription should occur from either original promoter-negative LAT gene. In addition, it is not possible for the original LAT genes to be rescued by homologous recombination with the 1.5-kb LAT insert, because the 1.5-kb insert is 168 nucleotides (nt) shorter than the corresponding deletion in the original LAT genes.
Rabbits were infected in both eyes with 23 105 PFU of
LAT15a, its LAT-deficient parent dLAT2903, or the dLAT2903 marker-rescued virus dLAT2903R per eye. Although LAT15a is capable of making only the first 1.5 kb of the 8.3-kb LAT, it nonetheless reactivated spontaneously in a manner indistin-guishable from that of wild-type virus (spontaneous-reacti-vation rates were 12% for LAT15a, 11% for dLAT2903R, and 0% for dLAT2903). In addition, Southern analyses con-firmed that the reactivated LAT15a virus was identical to the input virus. Specifically, it retained the original deletion in both original copies of LAT. Thus, the function of LAT involved in spontaneous reactivation appeared to be completely contained within the first 1.5-kb portion of LAT. Since this region of LAT does not overlap any known HSV-1 gene, our findings formally eliminate an ICP0 or ICP34.5 antisense mechanism from being essential in the LAT function responsible for efficient sponta-neous reactivation.
MATERIALS AND METHODS
Virus and cells.All mutants were derived from HSV-1 strain McKrae. The parental McKrae virus and all mutants were triple plaque purified and passaged only one or two times prior to use. Rabbit tear films were cultured on primary rabbit kidney cell monolayers to look for the presence of reactivated HSV-1. Rabbit skin (RS) cells were used for all other experiments, unless otherwise indicated. All cells were grown in Eagle’s minimal essential medium supple-mented with 10% fetal calf serum.
Construction of LAT15a, containing the first 1.5 kb of LAT in a novel location.
The parental virus for the LAT15a construct was dLAT2903, a mutant of HSV-1 strain McKrae containing a 1.8-kb EcoRV-HpaI) deletion in both copies of LAT that removed 0.2 kb of the LAT promoter and 1.6 kb of the 59end of the primary 8.3-kb LAT transcript (24). The previously cloned EcoRI A fragment from HSV-1 strain McKrae (25) was digested with BamHI, and the products were separated by agarose gel electrophoresis. A resulting 7.5-kb band containing the McKrae genomic region including UL37 and UL38 was isolated by electroelu-tion and cloned into the BamHI site of plasmid pEV-vrf3 (7, 25) to produce the plasmid pV375. pV375 was digested with AflII, the overhang was filled in by using the Klenow fragment, and the blunt ends were self ligated to create a unique PacI site in the plasmid between the sequences for UL37 and UL38. The resulting plasmid, designated pV375Pac, was amplified by transformation into Escherichia coli RR1lCI857 according to standard protocols.
A portion of the LAT gene was isolated and prepared for insertion into the PacI site of pV375Pac as follows. The previously cloned BamHI B restriction fragment of strain McKrae (25) was digested with HpaI to produce a 3.2-kb DNA fragment containing the first 1.5 kb of the 59end of the primary 8.3-kb LAT transcript and 1.7 kb of the immediately upstream region (containing the LAT promoter and known and potential upstream regulatory regions that might be essential for long-term LAT expression during neuronal latency) (see Fig. 1D). The band containing the LAT fragment was eluted from an agarose gel. A PacI linker (New England Biolabs, Beverly, Mass.) was added, and the fragment was digested with PacI to produce PacI ends. This fragment was then ligated into the
PacI site of the plasmid pV375Pac. The resulting plasmid was designated pV375LAT3.2. This plasmid contains the HSV-1 McKrae LAT fragment de-scribed above, bounded by UL37 and UL38. pV375LAT3.2 was then amplified in E. coli as described above.
LAT15a was generated by homologous recombination as we previously de-scribed (23, 24, 26). Briefly, pV375LAT3.2 was cotransfected with infectious dLAT2903 DNA (the LAT deletion mutant described above) by the calcium phosphate method. Viruses from the cotransfection were plated, and isolated plaques were picked and screened for insertion of the 3.2-kb LAT fragment between UL37 and UL38 by restriction digestion and Southern analysis. Selected plaques were triple plaque purified and reanalyzed by restriction digestion and Southern analysis to ensure that the 3.2-kb LAT fragment was present between UL37 and UL38 and that both long repeats retained the original 1.8-kb LAT deletion of the promoter and first 1.6 kb of the 59end of the primary LAT transcript (see Fig. 6). A final plaque was purified and designated LAT15a (LAT15 indicates 1.5 kb of the 59end of LAT; a indicates addition).
Replication of virus in tissue culture.Cell monolayers at approximately 70 to 80% confluency were infected with virus at 0.01 PFU per cell, and all monolayers were refed with exactly the same amount of minimal essential medium contain-ing 10% fetal calf serum. Virus was harvested for titration at various times by two cycles of freeze-thawing of the monolayers plus medium (2808C to room tem-perature). The numbers of PFU per milliliter were determined by standard plaque assays on RS cells.
Rabbits.Eight- to 10-week old New Zealand White female rabbits (Irish Farms) were used for all experiments. Rabbits were treated in accordance with the guidelines of the Association for Research in Vision and Ophthalmology, the American Association for Laboratory Animal Care, and the National Institutes of Health.
Rabbit model of ocular HSV-1 infection, latency, and spontaneous reactiva-tion. Rabbits were bilaterally infected without scarification or anesthesia by placing 23105PFU of HSV-1 per eye into the conjunctival cul-de-sac, closing the eye, and rubbing the lid gently against the eye for 30 s (28). At this dose of HSV-1 McKrae, virtually all of the surviving rabbits harbor a bilateral latent HSV infection in both trigeminal ganglia (TG), resulting in a high group rate of spontaneous reactivation with the McKrae strain of HSV-1. Latency is assumed to have been established by 28 days postinfection. Acute ocular infection of all eyes was confirmed by HSV-1-positive tear film cultures collected on days 3 and 4 postinfection.
Detection of spontaneous reactivation by ocular shedding.Beginning on day 30 postinfection, tear film specimens were collected daily from each eye for 26 days as previously described (22), using a nylon-tipped swab. The swab was then placed in 0.5 ml of tissue culture medium and squeezed, and the inoculated medium was used to infect primary rabbit kidney cell monolayers. These cell monolayers were observed in a masked fashion by phase-contrast microscopy for up to 30 days for HSV-1 cytopathic effects. All positive monolayers were blindly passaged onto fresh cells to confirm the presence of virus. DNA was purified from randomly selected positive cultures derived from latently infected rabbits and analyzed by restriction enzyme digestion and Southern blots to confirm that the cytopathic effect was due to reactivated HSV-1 and that the reactivated virus was identical to the input virus.
Reverse transcription-PCR (RT-PCR).RNA was isolated from individual TG from latently infected rabbits. Each TG was placed in 1 ml of lysis buffer (a 1:1:0.1 solution of water-saturated phenol, 4 M guanidinium thiocyanate in 25 mM sodium citrate [pH 7.0], and 2 M sodium acetate [pH 4.0]) supplemented with 7.2ml ofb-mercaptoethanol per ml, vortexed for 30 s, incubated at 558C for 1 h, and revortexed, and debris was removed by microcentrifugation for 2 min. The supernatants were transferred to fresh microcentrifuge tubes, and 0.1 vol-ume of chloroform-isoamylalcohol (24:1) was added. The mixture was vortexed until emulsified, incubated at 48C for 15 min, and centrifuged for 15 min at 48C, and the supernatant was collected and precipitated with 2.5 volumes of ethanol. The pellet was treated with DNase I (12 U, 378C, 30 min; Stratagene, La Jolla, Calif.), digested with proteinase K (200mg/ml, 558C, 1 h; Sigma), extracted with phenol-chloroform, and precipitated with 100% ethanol. Each RNA pellet was resuspended in diethyl pyrocarbonate-treated double-distilled water, and 5mg was subjected to first-strand cDNA synthesis with Superscript II (Gibco BRL, Grand Island, N.Y.) according to the manufacturer’s protocol. The primer for first-strand cDNA synthesis from the LAT RNA was 59-CTTTGTTGAACGA CACCGGGGCGCCCTCGA-39. The cDNA product was then amplified by PCR with the primer 59-CCACAACGGCCCGGCGCATGCGCTGTGGTT-39 and the first-strand primer. These primers generate a 160-bp product specific for LAT nucleotides 471 to 631. The PCRs were done in 100ml containing 5ml of the first-strand synthesis mixture, 10ml of 103buffer (Boehringer Mannheim Biochemicals, Indianapolis, Ind.), 2ml of 100 mM deoxynucleoside triphosphate mix (New England Biolabs), 1 ml of each of the two primers, 1ml of Taq polymerase (Boehringer Mannheim Biochemicals), and 80ml of double-distilled water. This mixture was overlaid with 100ml of mineral oil. Cycling reactions were performed on a thermal cycler (Appligene, Pleasanton, Calif.) as follows: (i) one cycle of denaturation at 958C for 4 min; (ii) 30 cycles of denaturation at 948C for 40 s, annealing at 608C for 30 s, and extension at 718C for 2 min; and (iii) one cycle of extension at 748C for 10 min. The amplified products were fraction-ated on a 4% NuSieve agarose gel running in Tris-borate-EDTA buffer, trans-ferred to a nylon membrane, and hybridized to the32
P-labeled internal probe
VOL. 70, 1996 LAT’S FUNCTION MAPS COMPLETELY WITHIN THE FIRST 1.5 kb 977
on November 9, 2019 by guest
http://jvi.asm.org/
59-TCTCCCCCCCCCCTTCTTCACCCCCAGTAC-39, corresponding to LAT nt 550 to 580.
Statistical analyses.Statistical analyses were performed by using Instat, a personal-computer software program. For analyses with either the Student t test, the Mann-Whitney rank sum test, the chi-square test, or the Fisher exact test, results were considered statistically significant when the P value was,0.05.
RESULTS
Structure of the LAT15a virus.The McKrae strain was used
as the original parental virus for all mutants because its high spontaneous-reactivation rate in rabbits allows changes in the spontaneous-reactivation rates of mutants to be detected and analyzed statistically. The genomic structures of wild-type HSV-1 McKrae and dLAT2903R, a marker-rescued virus of the LAT deletion mutant dLAT2903 (24), are shown schematically in Fig. 1A. The HSV-1 genome contains a unique long region and a unique short region, both of which are flanked by inverted
repeats (long terminal and internal repeats and short terminal and internal repeats). The long repeats are expanded in Fig. 1A to show the relative locations and statuses of the LAT, ICP0, and ICP34.5 genes. The primary 8.3-kb LAT transcript is unstable and difficult to detect. It gives rise to the very stable and easily detected 2-kb LAT, the location of which is shown in Fig. 1A; the location of the LAT promoter TATA box is also indicated. Transcription of the 8.3-kb LAT (11) starts 28 nt downstream of the TATA box (47).
The LAT promoter mutant dLAT2903, which we have pre-viously described (24) is shown schematically in Fig. 1B.
dLAT2903 contains a 1.8-kb deletion in both copies of the
LAT gene (one in each long repeat). This deletion consists of 0.2 kb of the LAT promoter and the portion of the LAT gene encoding the first 1.6 kb of the 8.3-kb primary LAT transcript. As indicated in Fig. 1B, the deletion extends to LAT nt 1667. As we previously described (24), dLAT2903 was marker
res-FIG. 1. Construction and structure of LAT15a. (A) Schematic representation of marker-rescued dLAT2903R virus, which is identical to wild-type HSV-1 McKrae. The prototypic orientation of HSV-1 shown here contains a unique long region and a unique short region, each bounded by inverted repeats. The unique regions are shown as solid lines, and the repeats are shown as open rectangles. UL, unique long region; US, unique short region; TRL, long terminal repeat; IRL, long internal repeat; TRS, short terminal repeat; IRS, short internal repeat. The lines with arrows under the genome indicate the locations and directions of the LAT, ICP34.5, and ICP0 transcripts. The solid rectangle within the primary 8.3-kb LAT transcript indicates the location of the stable 2-kb LAT. TATA indicates the location (in the genomic DNA) of the LAT promoter TATA box. (B) The previously described (24) LAT deletion mutant dLAT2903. The dashed rectangle and the preceding dashed line represent the portion of LAT deleted from both long repeats. (C) Enlargement showing the location of the 3.2-kb LAT fragment inserted between genes UL37 and UL38 in the unique long region of the LAT deletion mutant dLAT2903 to generate the virus LAT15a. The insertion site is outside the domains of the UL37 and UL38 promoters (see Materials and Methods for additional details). (D) Enlargement of the insert, which consists of 1.7 kb of the LAT promoter and upstream sequences and 1.5 kb of the structural portion of LAT. (E) Further enlargement of the 1.5-kb region of the LAT insertion. This 1,499-nt region consists of 662 nt from the TATA box to the start of the stable 2-kb LAT and the first 837 nt of the 2-kb LAT. LAT15a retains the 1.8-kb deletion in both original copies of LAT.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.66.546.68.444.2]cued back to wild type (dLAT2903R) by homologous recom-bination. The double-headed arrow between panels A and B of Fig. 1 indicates that dLAT2903 was derived from wild-type McKrae and that dLAT2903R, which is identical to wild-type McKrae, was derived from dLAT2903.
LAT15a was derived from dLAT2903 by the insertion of the LAT promoter and the DNA encoding the first 1.5 kb of the 8.3-kb primary LAT transcript into the unique long region (Fig. 1C to E). The insertion was at an AflII site between the 59 ends of UL37 and UL38 (genomic nt 84252). UL37 and UL38 are both gamma genes that require only the first 29 to 34 nt upstream of the mRNA cap site for transcriptional activity (8, 14, 30, 31). The AflII site that we converted to a PacI site and into which we then cloned the LAT insert is located 36 nt upstream of the UL37 cap site and 126 nt upstream of the UL38 cap site. Thus, neither the UL37 nor the UL38 promoter is disrupted in this construct. The complete insert is 3.2 kb and consists of the LAT promoter and associated upstream se-quences, totaling 1.7 kb, and the first 1.5 kb of the structural portion of the 8.3-kb LAT gene. Further details of the con-struction of LAT15a are given in Materials and Methods. LAT15a retains the dLAT2903 deletion in both copies of LAT and differs from dLAT2903 only in that it contains a small portion of LAT introduced into the viral unique long region rather than the normal LAT location. As indicated in Fig. 1E, the 1.5-kb structural portion of the insert contains the 662-nucleotide region between the LAT TATA box and the start of the 2-kb LAT along with the first 837 nucleotides of the 2-kb LAT. The structure of LAT15a was confirmed by Southern analysis (see Fig. 6).
Replication of LAT15a in tissue culture. To ensure that
LAT15a was capable of wild-type replication in tissue culture, RS cells were infected with 0.01 PFU of LAT15a, dLAT2903, or wild-type McKrae per cell. All three viruses grew to the same final titer of approximately 23108PFU per ml (Fig. 2).
At each of the time points, the titers of all three viruses ap-peared to be similar. Thus, as we previously showed (24), deletion of the LAT promoter and the beginning of LAT did not affect replication of the virus in tissue culture. Further-more, insertion of the 3.2-kb LAT fragment between UL37 and UL38 did not adversely affect viral replication.
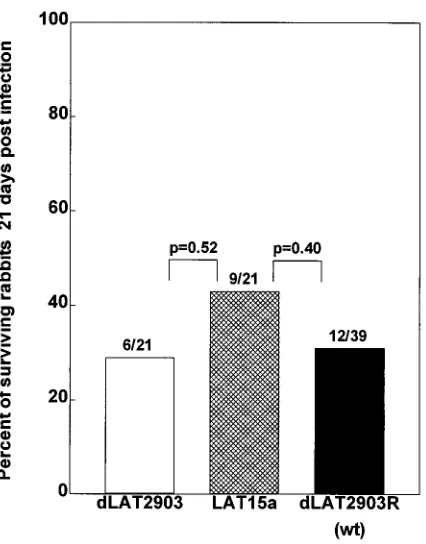
Virulence of LAT15a in rabbits.Groups of 21 or 39 rabbits
were infected with 23105PFU of LAT-negative dLAT2903,
marker-rescued wild-type dLAT2903R, or LAT15a per eye. Survival was determined at 21 days postinfection (Fig. 3). All deaths were attributable to HSV-1-induced encephalitis. There was no difference in survival between rabbits ocularly infected with LAT15a and those infected with dLAT2903 (P50.52) or between rabbits ocularly infected with LAT15a and those in-fected with dLAT2903R (P 5 0.40). Thus, insertion of the 3.2-kb LAT fragment between UL37 and UL38 did not de-crease the virulence of LAT15a.
Transcription of LAT in TG of rabbits latently infected with
LAT15a. Rabbits were infected ocularly, and 60 days later
(.30 days after latency had been established), TG were re-moved, total RNA was isolated from individual TG, and RT-PCR was performed to detect LAT transcription as described in Materials and Methods. The primers used generate a 160-bp product specific for LAT nt 471 to 631 (the 2-kb LAT starts at nt 662 [Fig. 1E]). The RT-PCR products were subjected to Southern analysis with an internal32P-labeled probe (LAT nt
550 to 580) (Fig. 4). Each lane wt in Fig. 4 shows the result from one randomly selected TG, each from a different rabbit latently infected with wild-type McKrae virus. As expected, a single band migrating with an apparent size of 160 bp is seen in each case. Lanes Dshow the results for two randomly
se-lected rabbits latently infected with the LAT deletion mutant
dLAT2903. No RT-PCR product is seen, confirming that this
mutant does not transcribe this region of LAT during latency. Negative results were also seen with RNA from TG of unin-fected rabbits (not shown). Lanes 15a show the results from two rabbits latently infected with LAT15a. Each lane shows a single band identical to that seen in lanes wt. Identical RT-PCRs were also done with a primer pair that produces a prod-uct corresponding to LAT nt 960 to 1130, within the 2-kb LAT region. As before, Southern analysis with an internal probe detected the expected RT-PCR product in both rabbits latently infected with LAT15a (data not shown). Thus, the LAT insert in LAT15a appeared to rescue the ability of the original
dLAT2903 to transcribe this region of LAT.
Spontaneous reactivation of LAT15a.We recently showed
that the LAT deletion mutant dLAT2903, which has the first 1.6 kb of LAT deleted and is incapable of making any LAT RNA, had significantly reduced spontaneous reactivation in latently infected rabbits (24). To determine if restoration of the first 1.5 kb of LAT in a distant location (along with upstream elements) could restore the ability of the virus to reactivate spontaneously, we examined the ability of LAT15a to reacti-vate spontaneously. Rabbit eyes were infected with 2 3 105
PFU of LAT15a, dLAT2903, or marker-rescued dLAT2903R. Beginning 30 days postinfection (at which time latency had already been established), all eyes were swabbed once a day to collect tear films for analysis of spontaneously reactivated virus as described in Materials and Methods. The cumulative num-ber of virus-positive tear film cultures per eye during 26 days
FIG. 2. Growth kinetics of LAT15a in tissue culture. Semiconfluent mono-layers of RS cells were infected with 0.01 PFU of LAT15a, dLAT2903, or wild-type McKrae per cell. At the indicated times, the infected-cell monolayers were harvested by freeze-thawing, and the amount of virus was determined by plaque assay on RS cells. Each datum point is the average of two determinations.
VOL. 70, 1996 LAT’S FUNCTION MAPS COMPLETELY WITHIN THE FIRST 1.5 kb 979
on November 9, 2019 by guest
http://jvi.asm.org/
for each virus is shown in Fig. 5. Consistent with the very low spontaneous-reactivation rate that we previously reported for the LAT-negative mutant dLAT2903 (24), during the time period observed, no spontaneous reactivations were detected in any of the dLAT2903-infected eyes (Fig. 5). In contrast, rabbits latently infected with LAT15a or wild-type (marker-rescued dLAT2903R) virus had virtually identical cumulative spontaneous-reactivation rates of approximately three positive cultures per eye (Fig. 5).
A statistical analysis of positive (spontaneously reactivated) cultures versus negative cultures is shown in Table 1. Almost 12% (55 of 468) of the tear films from rabbits latently infected with LAT15a contained spontaneously reactivated virus. This was significantly higher than the 0% (0 of 312) positive tear film cultures from the rabbits latently infected with
LAT-neg-ative dLAT2903 (P,0.0001 by the Fisher exact test) and was not different from the 11% (70 of 624) positive cultures from rabbits latently infected with dLAT2903R marker-rescued wild-type virus (P 5 0.85). Thus, although LAT15a contains only a single copy of the first 1.5 kb of the 8.3-kb LAT, its spontaneous-reactivation rate appeared to have been com-pletely restored back to wild-type levels.
Because the analyses described above do not take into ac-count the number of eyes in each of the groups, the data were analyzed by additional methods. The fraction of virus-positive cultures for each eye in each group (i.e., the fraction of time that each eye was virus positive) was calculated, and these fractions were analyzed (Table 1). Spontaneous reactivation with the LAT15a virus was significantly higher than that with the LAT-negative dLAT2903 virus (P5 0.001 by the Mann-Whitney rank sum test) and was similar to that with wild-type virus (P50.66).
[image:5.612.73.286.68.340.2]The number of eyes in each group that had at least one spontaneous reactivation was also analyzed (Table 1). Again, the result for the LAT15a virus was significantly higher than the result for the LAT-negative dLAT2903 virus (72 versus 0%; P, 0.0001 by the Fisher exact test) and similar to the wild-type results (72 versus 75%; P51.0). Likewise, the num-ber of rabbits that had at least one spontaneous reactivation (Table 1) was significantly higher for the rabbits latently in-fected with the LAT15a virus than for those latently inin-fected with dLAT2903 (89 versus 0%; P50.001 by the Fisher exact test) and was similar to that of the wild-type-infected group (83%; P51.0).
[image:5.612.314.554.71.335.2]FIG. 3. Virulence of LAT15a. Rabbits were infected ocularly as described in Materials and Methods. The percentage of rabbits surviving until day 21 postin-fection is shown by the bars. The numbers above each bar show the number of surviving rabbits/total number of infected rabbits for that group. P values com-pare adjacent bars and were determined by the Fisher exact test. wt, wild type.
FIG. 4. LAT transcription in rabbits latently infected with LAT15a. RT-PCR of total RNA isolated from individual TG from latently infected rabbits was done as described in Materials and Methods with primers corresponding to LAT nt 471 to 500 and 602 to 631. Southern analysis of the RT-PCR products was done by using a32
P-labeled probe corresponding to LAT nt 550 to 580. Each lane shows the RT-PCR product from one TG. Duplicate lanes are from different rabbits. Lanes: wt, dLAT2903 marker-rescued wild-type virus;D, dLAT2903 LAT-negative virus; 15a, LAT15a virus.
FIG. 5. Spontaneous reactivation in rabbits latently infected with LAT15a. Rabbits were ocularly infected with LAT15a, dLAT2903, or dLAT2903R (mark-er-rescued wild type). Beginning on day 30 postinfection (day 0), at which time latency had been established, tear films were collected daily, plated on primary rabbit kidney cells, and observed for up to 30 days for the presence of cytopathic effects. All positive cultures were confirmed by passage and Southern analysis (see Fig. 6). The y axis represents the cumulative number of HSV-1 positive cultures for each virus group divided by the number of eyes in the group.
Statistical analyses are shown in Table 1.
on November 9, 2019 by guest
http://jvi.asm.org/
Another method of examining the effect of the 1.5-kb LAT insert on spontaneous reactivation is to analyze the number of times spontaneous reactivation is detected in each eye, regard-less of the length of time that virus is present. This is equivalent to the number of episodes in which reactivated virus is detect-ed in the tears, with consecutive days of positive cultures being treated as a single event. Thus, an eye that sheds virus for a sin-gle day and an eye that sheds virus for 5 consecutive days would each constitute one episode of spontaneous reactivation. The average number of spontaneous reactivations per eye in each group calculated in this manner are shown in Table 1. The num-ber of episodes of spontaneous reactivation in LAT15a-infect-ed eyes was much higher than that in dLAT2903-infectLAT15a-infect-ed eyes (1.4 versus 0; P5 0.001) and similar to that in wild-type-in-fected eyes (1.6; P50.68). All of the analyses described above indicate that LAT15a reactivated spontaneously at a rate in-distinguishable from that of the marker-rescued dLAT2903R (wild-type) virus.
Southern analysis of spontaneously reactivated virus. To
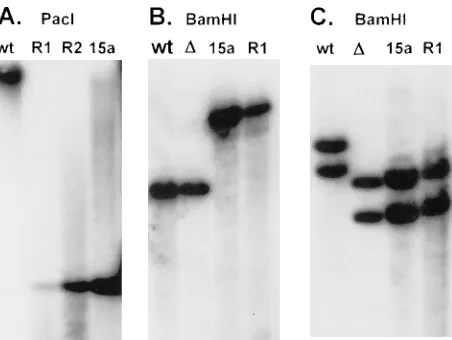
eliminate the unlikely possibility that the spontaneously reac-tivated virus recovered from eyes of rabbits latently infected with the LAT15a virus was contaminating wild-type virus or LAT15a virus that had somehow reverted back to wild-type virus, Southern analyses were done on spontaneously reacti-vated virus recovered from all eyes that had been infected with LAT15a. Examples of some of these Southern analyses are shown in Fig. 6. Purified LAT15a viral DNA was obtained prior to infection (lanes 15a) and from LAT15a virus in tears after spontaneous reactivation (lanes R1 and R2). Figure 6A shows a Southern analysis of DNAs that were individually digested with PacI and probed with a32P-labeled restriction
fragment that is completely within the LAT region deleted in
dLAT2903 and that therefore can hybridize only with LAT
sequences in the LAT15a insert. The wild-type virus (lane wt) produced a single large band of genome size, as expected since
PacI does not cut wild-type HSV-1. In contrast, PacI should cut
the PacI sites flanking the 3.2-kb LAT fragment (LAT nt 1 to 1499 plus 1.7 kb of upstream DNA) inserted between UL37 and UL38 in LAT15a. This should produce a single band of 3.2 kb that hybridizes to the probe, as is seen in lane 15a. The spontaneously reactivated LAT15a DNAs from two different rabbits (R1 and R2) each produced the same 3.2-kb band, indicating that the spontaneously reactivated LAT15a virus retained the LAT insert.
The viral DNAs in Fig. 6B were digested with BamHI and probed with the BamHI H restriction fragment that hybridizes to the UL37-UL38 region but not to any of the LAT
se-quences. Both the wild type (lane wt) and dLAT2903 (laneD) show a single band of 7.5 kb, corresponding in size to the
BamHI H restriction fragment containing UL37 and UL38.
Lanes 15a and R1 both contain only a more slowly migrating band of 10.7 kb, corresponding to BamHI-H plus the 3.2-kb LAT insert. This again indicates that the spontaneously reac-tivated LAT15a virus retained the LAT insert between UL37 and UL38.
[image:6.612.58.554.84.140.2]The viral DNAs in Fig. 6C were also digested with BamHI but were hybridized to a probe specific for a region of LAT that is present in the wild type and dLAT2903 but not in the LAT insert of LAT15a (a cloned HpaI-MluI restriction frag-ment corresponding to LAT nt 1667 to 2850). This probe should hybridize only to the BamHI B and BamHI E restric-tion fragments generated from the long repeats (one from each repeat) that contain the HSV-1 sequence corresponding to the probe. As seen in Fig. 6C, lane wt, these two bands are of different sizes because in both restriction fragments one TABLE 1. Spontaneous reactivation of HSV-1 in rabbits latently infected with LAT15aa
Virus (no. of eyes)
No. of positive tear film
cultures/total (%)b Fraction of
posi-tive cultures/eyec No. of eyes that
reacti-vated/total (%)
No. of rabbits that reactivated/total (%)
Avg no. of reactiva-tion episodes/eyed
LAT15a (18) 55/468 (11.8) 0.12 13/18 (72) 8/9 (89) 1.4
dLAT2903 (12) 0/312 (0) (P,0.0001e) 0.0 (P50.001f) 0/12 (0) (P,0.0001e) 0/6 (0) (P50.001e) 0 (P50.001f)
dLAT2903R (wt) (24)g 70/624 (11.2) (P50.85e) 0.11 (P50.66f) 18/24 (75) (P51.0e) 10/12 (83) (P51.0e) 1.6 (P50.68f)
aStatistical analysis of the data presented in Fig. 5. Rabbits were bilaterally ocularly infected with dLAT2903 (virus with the LAT promoter and the first 1.6 kb of
LAT deleted), dLAT2903R (marker-rescued wild-type virus), or LAT15a (dLAT2903 containing the LAT promoter and the first 1.5 kb of LAT at a distant location) as described in Materials and Methods.
bSpontaneous reactivation was assessed beginning on day 30 postinfection by culturing tear films collected daily for 26 days as described in the text.
cFor each eye within each group, the fraction of days on which positive cultures were obtained was calculated (total HSV-1-positive cultures for eye/total cultures
for eye). The average is shown.
dThe total number of reactivations in each group, assuming that consecutive days of HSV-1-positive tear film cultures from a single eye are the result of a single
reactivation event or episode.
eVersus LAT15a; determined by a two-sided Fisher exact test. The groups are considered significantly different if P is,0.05.
fVersus LAT15a; determined by a two-sided Mann-Whitney rank sum test. The groups are considered significantly different if P is,0.05. gWild-type (wt) virus. This marker-rescued virus is indistinguishable from the original parental McKrae virus.
FIG. 6. Southern analysis of spontaneously reactivating LAT15a. DNA was isolated from the tears of rabbits latently infected with LAT15a virus and com-pared with DNA from tissue culture-grown virus by restriction digestion and Southern analysis. (A) The viral DNAs were cut with PacI and hybridized to a 32
P-labeled probe corresponding to part of the LAT region deleted in dLAT2903 but present in the LAT insert in LAT15a (see text). (B) DNAs were cut with BamHI. The probe was specific for the UL37-UL38 region. (C) DNAs were cut with BamHI. The probe was specific for a region of LAT near the deletion in dLAT2903 but outside the region inserted into LAT15a. Lanes: wt, dLAT2903R marker-rescued wild-type virus; 15a, LAT15a;D, dLAT2903; R1 and R2, spon-taneously reactivated LAT15a from two different rabbits.
VOL. 70, 1996 LAT’S FUNCTION MAPS COMPLETELY WITHIN THE FIRST 1.5 kb 981
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.320.546.463.633.2]BamHI cut is in the repeat and one is in the adjacent unique
long region. The larger band contains LAT from the internal repeat, while the smaller band is from the terminal repeat. Both of these bands are smaller in the original LAT deletion
dLAT2903 (laneD) and LAT15a (lane 15a), indicative of the
1.8-kb LAT deletion in each long repeat in these viruses. Lane R1 (containing a spontaneously reactivated LAT15a virus) contains bands identical to those seen in lanes D and 15a, confirming that the original LAT deletion was retained in both original copies of LAT in the LAT15a spontaneously reacti-vated virus.
These Southern analyses confirm the structure of the LAT15a virus and demonstrate that the spontaneously reacti-vated LAT15a virus retained the 1.8-kb LAT deletion in both repeats as well as the 3.2-kb LAT insert in the unique long region. This was found to be the case for all of the spontane-ously reactivated LAT15a virus examined. Thus, the ability of LAT15a to reactivate spontaneously was due to inherent prop-erties of the virus and not to rescue of the original LAT deletions by homologous recombination with LAT gene se-quences inserted into the unique long region.
DISCUSSION
The function of LAT involved in spontaneous reactivation maps to within the first 1.5 kb of the primary LAT transcript.
As we previously described, the 1.8-kb deletion in dLAT2903 removed both the authentic LAT promoter and a hypothetical cryptic promoter from both copies of LAT, thereby rendering the mutant unable to express any LAT-related RNA (24). This virus is severely impaired for spontaneous reactivation in the rabbit ocular model. In this study, we have restored spontane-ous reactivation in this mutant to wild-type levels by insertion of the first 1.5 kb of LAT (and 1.7 kb of upstream sequences) into a distant location (LAT15a). Since the 39 extent of the original LAT deletion (LAT nt 1667) exceeded that of the 1.5-kb insert (LAT nt 1499) (Fig. 1), it was not possible for the original LAT region to be rescued by homologous recombina-tion. This was confirmed by Southern analysis, which demon-strated that all of the spontaneously reactivated virus retained the original deletion in both copies of LAT. These results therefore mapped the function of LAT involved in spontane-ous reactivation to within the first 1.5 kb of the LAT transcript.
Establishment of latency.In situ hybridization to estimate
the percentage of neurons containing the 2-kb LAT RNA and PCR to estimate the HSV-1 genomic copy number in ganglia have both been used to estimate the relative amount of latency in animals latently infected with HSV-1 mutants (15, 27, 36). For mutants such as LAT2903 that do not express the 2-kb LAT RNA, in situ hybridization is not a feasible method. In this situation, PCR analysis is the most sensitive technique currently available. Using this approach, we have previously observed that the amount of HSV-1 DNA detected in TG of rabbits latently infected with dLAT2903 (the LAT mutant res-cued in this study to produce LAT15a) and marker-resres-cued
dLAT2903R appeared to be identical (24). Since this method
was unable to distinguish between establishment of latency by wild-type virus and by the LAT-negative dLAT2903 virus, there was no purpose in using this method with LAT15a. Therefore, although we have not specifically determined in this study that LAT15a established latency as well as wild-type virus, our prior results with dLAT2903 and dLAT2903R indi-cate that at the level of sensitivity of competitive PCR analysis, this must be the case. Assuming that these PCR analyses ac-curately reflect the relative levels of latency established by LAT-negative and LAT-positive viruses, the LAT function that
in this study was mapped to the first 1.5 kb of LAT must be involved in spontaneous reactivation, not establishment of la-tency. The similarity in intensity of hybridization to the LAT RT-PCR products from rabbits latently infected with wild-type virus and LAT15a virus also suggests a wild-type level of la-tency with LAT15a, further suggesting a role for the first 1.5 kb of LAT in reactivation.
The stable 2-kb LAT is not essential for wild-type levels of
spontaneous reactivation. The 2-kb LAT is extremely stable
compared with the remainder of the 8.3-kb LAT RNA (47). This stability makes the 2-kb LAT relatively easy to detect during latency compared with the rest of the LAT transcript. Thus, for several years after the initial reports of LAT (28, 35), it was thought that the 2-kb LAT (and a 1.5-kb LAT derived from the 2-kb LAT) constituted the entire LAT RNA. Because of its abundance during latency, it is still often assumed that the 2-kb LAT is the functionally important portion of LAT as regards reactivation from latency. The LAT15a mutant showed that this is not the case, as it is not capable of transcribing the entire 2-kb LAT. The 1.5-kb LAT insert contains only the first 837 nt of the 2-kb LAT, and hence only about 40% of the 2-kb LAT can be transcribed. Since LAT15a reactivated like wild-type virus, it appears that the presence of the entire 2-kb LAT is not essential for efficient spontaneous reactivation.
A possible role for the remaining 6.8 kb of the 8.3-kb LAT.
The primary LAT transcript is 8.3 kb in length and appears to be well conserved in various HSV-1 and HSV-2 strains. From an evolutionary standpoint this argues strongly that there has been continued selective pressure to maintain the entire 8.3-kb LAT gene. This further argues that regions of LAT in addition to the first 1.5 kb of LAT may have a function. There are at least two possibilities. First, it is possible that the remaining region of LAT has a function unrelated to latency and that this function, although useful in nature, is not essential for repli-cation in tissue culture or in laboratory animals. Second, it is possible that the remaining region of LAT has a function in the latency cycle but that this function is not easily observed in laboratory animals. Thus, different regions of LAT may con-tribute to LAT’s function, with the LAT15a mutant having wild-type function in laboratory animals but possibly being impaired in nature. This would be similar to the situation for numerous HSV-1 genes that are designated nonessential. These genes are ‘‘nonessential’’ because they are not required for efficient tissue culture replication. However, it is obvious that such genes must be useful in nature and provide the virus some selective advantage; otherwise, they would not be con-served.
Recently, an HSV-1 RNA that is colinear and coterminal with approximately the last 25% of the primary 8.3-kb LAT has been described (3, 16, 17, 46). This RNA is transcribed inde-pendently of LAT, contains a presumptive open reading frame (ORF P), and appears to be made in tissue culture under some circumstances. Although the function of this RNA during acute or latent infection is unknown, it is possible that the sequence of this region of the HSV-1 genome has been con-served because of selective pressure to retain this newly dis-covered RNA (or the ICP34.5 gene, which is almost completely overlapped in an antisense direction by this transcript). How-ever, this would not explain why the primary LAT has been conserved as an 8.3-kb transcript.
Sequences upstream of the LAT promoter.In addition to the
first 1.5 kb of the primary LAT transcript included in the LAT insert in LAT15a, we also included 1,792 nt upstream of the start of LAT transcription. This was done to try to ensure that both mapped and unmapped important LAT promoter se-quences would be present. The inclusion of this large upstream
on November 9, 2019 by guest
http://jvi.asm.org/
region makes it formally possible that it is this region rather than the region corresponding to the first 1.5 kb of the LAT transcript to which the spontaneous reactivation function maps. In fact, if the LAT promoter was bidirectional, transcrip-tion might occur in the upstream region during latency. A small amount of such transcription in this region has been reported to occur in latently infected mice (20); however, the direction of transcription was not determined. Our computer analysis of the sequence in this region revealed three open reading frames, each beginning with an ATG and ending in a typical termination codon. All three have a very high Fickett test code probability of.0.92, indicating that codon usage is typical of a protein (IBI Pustell Sequence Analysis Program). Thus, the region upstream of the LAT promoter warrants additional study.
Overlapping genes.The 8.3-kb LAT gene completely
over-laps in an antisense direction two important HSV-1 genes, ICP0 and ICP34.5. ICP0 is an immediate-early gene involved in the early stages of the viral life cycle. ICP0 is particularly important for low-multiplicity infections, such as would be expected to occur in the initial stages of HSV-1 reactivation. Deletions in ICP0 (which of course also alter the correspond-ing region of LAT) have been shown to reduce explant reac-tivation in the mouse (10, 18). ICP34.5 is a neurovirulence gene (5, 37, 38, 45) that appears to be extremely important for viral replication in neurons, a function that would be expected to be essential for reactivation of HSV-1 neuronal latency. Some ICP34.5 mutations (which of course also alter the cor-responding region of LAT) have been shown to reduce reac-tivation, both in the mouse (4, 5, 26, 45) and in the rabbit (2, 26). It is therefore apparent that genetic mapping of the func-tional portion(s) of the 8.3-kb LAT gene is complicated by the colinearity of more than half of the LAT gene with the genes for ICP0 and ICP34.5.
The approach we used here should allow functional genetic mapping of the LAT region without regard to the overlapping genes. In this study we rescued LAT function in a LAT pro-moter-negative mutant containing unaltered ICP0 and ICP34.5 genes, by reintroducing the LAT promoter and part of LAT into a novel location to produce a virus that transcribed only the first 1.5 kb of LAT. It should be possible to extend this approach to include almost any length of LAT in the insertion. Mutations could then be introduced anywhere within this en-tire LAT gene without altering either of the original ICP0 and ICP34.5 genes. Alterations in phenotype associated with such mutations in LAT could then be assigned directly to LAT. Although our results reported here indicate that the first 1.5 kb of the 8.3-kb LAT alone is capable of producing wild-type levels of spontaneous reactivation, it is possible, as discussed above, that other regions of the 8.3-kb LAT have additional, as-yet-undetermined functions or that other regions of the 8.3-kb LAT enhance the function of the first 1.5 kb of LAT. We expect that the approach we have presented here will be invaluable in mapping such additional LAT functions.
ACKNOWLEDGMENTS
We thank Anita Avery for excellent technical assistance.
This work was supported by Public Health Service grants EY07566 and EY10243, the Discovery Fund for Eye Research, and the Skirball Program in Molecular Ophthalmology.
REFERENCES
1. Block, T. M., J. G. Spivack, I. Steiner, S. Deshmane, M. T. McIntosh, R. P.
Lirette, and N. W. Fraser. 1990. A herpes simplex virus type 1 latency-associated transcript mutant reactivates with normal kinetics from latent infection. J. Virol. 64:3417–3426.
2. Bloom, D. C., G. B. Devi-Rao, J. M. Hill, J. G. Stevens, and E. K. Wagner.
1994. Molecular analysis of herpes simplex virus type 1 during epinephrine-induced reactivation of latently infected rabbits in vivo. J. Virol. 68:1283– 1292.
3. Bohenzky, R. A., M. Lagunoff, B. Roizman, E. K. Wagner, and S. Silverstein. 1995. Two overlapping transcription units which extend across the L-S junc-tion of herpes simplex virus type 1. J. Virol. 69:2889–2897.
4. Bolovan, C. A., N. M. Sawtell, and R. L. Thompson. 1994. ICP34.5 mutants in herpes simplex virus type 1 strain 17 syn1are attenuated for neuroviru-lence in mice and for replication in confluent primary mouse embryo cells. J. Virol. 68:48–55.
5. Chou, J., E. R. Kern, R. J. Whitley, and B. Roizman. 1990. Mapping of herpes simplex virus-neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1265.
6. Croen, K. D., J. M. Ostrove, L. J. Dragovic, J. E. Smialek, and S. E. Straus. 1987. Latent herpes simplex virus in human trigeminal ganglia: detection of an immediate early gene ‘‘antisense’’ transcript by in situ hybridization. N. Engl. J. Med. 317:1427–1432.
7. Crowl, R., C. Seamens, P. Lomedico, and S. McAndrew. 1985. Versatile expression vectors for high-level synthesis of cloned gene products in E. coli. Gene 38:31–38.
8. Fanagan, W. M., A. G. Papavassiliou, M. Rice, L. B. Hect, S. Silverstein, and
E. K. Wagner.1991. Analysis of the herpes simplex virus type 1 promoter controlling the expression of UL38, a true late gene involved in capsid assembly. J. Virol. 65:769–786.
9. Gordon, Y. J., B. Johnson, E. Romanowski, and T. Araullo-Cruz. 1988. RNA complementary to herpes simplex virus type 1 ICP0 gene demonstrated in neurons of human trigeminal ganglia. J. Virol. 62:1832–1835.
10. Gordon, Y. J., J. L. C. McKnight, J. M. Ostrove, E. Romanowski, and T.
Araullo-Cruz.1990. Host species and strain differences affect the ability of an HSV-1 ICP0 deletion mutant to establish latency and spontaneously reacti-vate in vivo. Virology 178:469–477.
11. Hill, J. M., F. Sederati, R. T. Javier, E. K. Wagner, and J. G. Stevens. 1990. Herpes simplex virus latent phase transcription facilitates in vivo reactiva-tion. Virology 174:117–125.
12. Ho, D. Y., and E. S. Mocarski. 1989. Herpes simplex virus latent RNA (LAT) is not required for latent infection in the mouse. Proc. Natl. Acad. Sci. USA
86:7596–7600.
13. Javier, R. T., J. G. Stevens, V. B. Dissette, and E. K. Wagner. 1988. A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology 166:254–257.
14. Johnson, P. A., and R. D. Everett. 1986. The control of herpes simplex virus type 1 late gene transcription: a TATA box/cap site region is sufficient for fully efficient regulated activity. Nucleic Acids Res. 14:8247–8264. 15. Katz, J. P., E. T. Bodin, and D. M. Coen. 1990. Quantitative polymerase
chain reaction analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J. Virol. 64:4288–4295. 16. Lagunoff, M., and B. Roizman. 1994. Expression of a herpes simplex virus 1
open reading frame antisense to theg134.5 gene and transcribed by an RNA 39terminal with the unspliced latency-associated transcript. J. Virol. 68: 6021–6028.
17. Lagunoff, M., and B. Roizman. 1995. The regulation of synthesis and prop-erties of the protein product of open reading frame P of the herpes simplex virus 1 genome. J. Virol. 69:3615–3623.
18. Leib, D. A., C. L. Bogard, M. Kosz-Vnenchak, K. A. Hicks, D. M. Coen, D. M.
Knipe, and P. A. Schaffer.1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 63:2893–2900.
19. Leib, D. A., K. C. Nadeau, K. C. Rundle, S. A. Rundle, and P. A. Schaffer. 1991. Promoter of the latency-associated transcripts of herpes simplex virus type-1 contains a functional cAMP-response element: role of the latency associated transcripts and cAMP in reactivation of viral latency. Proc. Natl. Acad. Sci. USA 88:48–52.
20. Mitchell, W. J., R. P. Lirette, and N. W. Fraser. 1990. Mapping of low-abundance latency-associated RNA in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. Virology 71:125–132.
21. Nesburn, A. B. 1983. In A. B. Nesburn (ed.), Report of the corneal disease panel: vision research: a national plan 1983–1987, vol II, part III. The C.V. Mosby Co., St. Louis.
22. Nesburn, A. B., H. Ghiasi, and S. L. Wechsler. 1990. Ocular safety and efficacy of an HSV-1 gD vaccine during primary and latent infection. Invest. Ophthal. Vis. Sci. 31:77–82.
23. Perng, G.-C., K. Chokephaibulkit, R. L. Thompson, N. M. Sawtell, S. M.
Slanina, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.1996. The region of the herpes simplex virus type 1 LAT gene that is colinear with the ICP34.5 gene is not involved in spontaneous reactivation. J. Virol. 70:282–291. 24. Perng, G. C., E. C. Dunkel, P. A. Geary, S. M. Slanina, H. Ghiasi, R. Kaiwar,
A. B. Nesburn, and S. L. Wechsler.1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol. 68:8045–8055. 25. Perng, G. C., H. Ghiasi, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler. 1994.
An improved method for cloning portions of the repeat regions of herpes simplex virus type 1. J. Virol. Methods 46:111–116.
VOL. 70, 1996 LAT’S FUNCTION MAPS COMPLETELY WITHIN THE FIRST 1.5 kb 983
on November 9, 2019 by guest
http://jvi.asm.org/
26. Perng, G. C., R. L. Thompson, N. M. Sawtell, W. E. Taylor, S. M. Slanina,
H. Ghiasi, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler.1995. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol. 69:3033–3041.
27. Ramakrishnan, R., D. J. Fink, G. Jiang, P. Desai, J. C. Glorioso, and M.
Levine.1994. Competitive quantitative PCR analysis of herpes simplex virus type 1 DNA and latency-associated transcript RNA in latently infected cells of the rat brain. J. Virol. 68:1864–1873.
28. Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L. Lewis, J. R. Lokensgard,
and S. L. Wechsler.1987. Detection of latency-related viral RNAs in tri-geminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol. 61:3820–3826.
29. Sawtell, N. M., and R. L. Thompson. 1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol. 66:2157–2169. 30. Shapira, M., F. L. Homa, J. C. Glorioso, and M. Levine. 1987. Regulation of
the herpes simplex virus type 1 late (gamma 2) glycoprotein C gene: se-quences between base pairs234 to129 control transient expression and responsiveness to transactivation by the products of the immediate early alpha 4 and 0 genes. Nucleic Acids Res. 15:3097–3111.
31. Shelton, L. S., M. N. Pensiero, and F. J. Jenkins. 1990. Identification and characterization of the herpes simplex virus type 1 protein encoded by the UL37 open reading frame. J. Virol. 64:6101–6109.
32. Spivack, J. G., and N. W. Fraser. 1988. Expression of herpes simplex virus type 1 latency-associated transcripts in the trigeminal ganglia of mice during acute infection and reactivation of latent infection. J. Virol. 62:1479–1485. 33. Steiner, I., J. G. Spivack, R. P. Lirette, S. M. Brown, A. R. MacLean, J. H.
Subak-Sharpe, and N. W. Fraser.1989. Herpes simplex virus type 1 latency associated transcripts are evidently not essential for latent infection. EMBO J. 8:505–511.
34. Steiner, I., J. G. Spivack, D. R. O’Boyle II, E. Lavi, and N. W. Fraser. 1988. Latent herpes simplex virus type 1 transcription in human trigeminal ganglia. J. Virol. 62:3493–3496.
35. Stevens, J. G., E. K. Wagner, G. B. Devi-Rao, M. L. Cook, and L. T. Feldman. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominant in latently infected neurons. Science 235:1056–1059.
36. Takasu, T., Y. Furuta, K. C. Sato, S. Fukuda, Y. Inuyama, and K.
Na-gashima.1992. Detection of latent herpes simplex virus DNA and RNA in human geniculate ganglia by the polymerase chain reaction. Acta Otolaryn-gol. 112:1004–1011.
37. Thompson, R. L., G. V. Devi-Rao, J. G. Stevens, and E. K. Wagner. 1985.
Rescue of a herpes simplex virus type 1 neurovirulence function with a cloned DNA fragment. J. Virol. 55:504–508.
38. Thompson, R. L., E. K. Wagner, and J. G. Stevens. 1983. Physical location of a herpes simplex virus type 1 gene function(s) specifically associated with a 10 million fold increase in HSV neurovirulence. Virology 131:180–192. 39. Trousdale, M. D., I. Steiner, J. G. Spivack, S. L. Deshmane, S. M. Brown,
A. R. MacLean, J. H. Subak-Sharpe, and N. W. Fraser.1991. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-asso-ciated transcript variant in a rabbit eye model. J. Virol. 65:6989–6993. 40. Wagner, E. K., G. Devi-Rao, L. T. Feldman, A. T. Dobson, Y. Zhang, W. M.
Flanagan, and J. G. Stevens.1988. Physical characterization of the herpes simplex virus latency-associated transcript in neurons. J. Virol. 62:1194– 1202.
41. Wagner, E. K., W. M. Flanagan, G. Devi-Rao, Y. Zhang, J. M. Hill, K. P.
Anderson, and J. G. Stevens.1988. The herpes simplex virus latency-associ-ated transcript is spliced during the latent phase of infection. J. Virol. 62: 4577–4585.
42. Wechsler, S. L., A. B. Nesburn, R. J. Watson, S. Slanina, and H. Ghiasi. 1988. Fine mapping of the major latency related-RNA of herpes simplex virus type 1 in humans. J. Gen. Virol. 69:3101–3106.
43. Wechsler, S. L., A. B. Nesburn, R. J. Watson, S. M. Slanina, and H. Ghiasi. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J. Virol. 62:4051–4058.
44. Wechsler, S. L., A. B. Nesburn, J. C. Zwaagstra, and H. Ghiasi. 1989. Sequence of the latency related gene of herpes simplex virus type 1. Virology
168:168–172.
45. Whitley, R. J., E. R. Kern, S. Chatterjee, J. Chou, and B. Roizman. 1993. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Invest. 91:2837–2843.
46. Yeh, L., and P. A. Schaffer. 1993. A novel class of transcripts expressed with late kinetics in the absence of ICP4 spans the junction between the long and short segments of the herpes simplex virus type 1 genome. J. Virol. 67: 7373–7382.
47. Zwaagstra, J. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, S. C. Wheatley, K.
Lillycrop, J. Wood, D. S. Latchman, K. Patel, and S. L. Wechsler.1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J. Virol. 64:5019–5028.