0022-538X/10/$12.00 doi:10.1128/JVI.02200-09
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Hepatitis C Virus Hypervariable Region 1 Modulates Receptor
Interactions, Conceals the CD81 Binding Site, and
Protects Conserved Neutralizing Epitopes
䌤
§
Dorothea Bankwitz,
1† Eike Steinmann,
1† Julia Bitzegeio,
1Sandra Ciesek,
1,2Martina Friesland,
1Eva Herrmann,
3Mirjam B. Zeisel,
4Thomas F. Baumert,
4Zhen-yong Keck,
5Steven K. H. Foung,
5Eve-Isabelle Pe
´cheur,
6and Thomas Pietschmann
1*
Division of Experimental Virology, Twincore Centre for Experimental and Clinical Infection Research,‡Hannover, Germany1;
Department of Gastroenterology, Hepatology and Endocrinology, Medical School Hannover, Hannover, Germany2; Institute for
Biostatistics and Mathematical Modeling, Johann Wolfgang Goethe-University, Frankfurt, Germany3; INSERM U748,
Universite´ de Strasbourg, Strasbourg, France4; Department of Pathology, Stanford University, Stanford, California5;
and Institut de Biologie et Chimie des Prote´ines, UMR CNRS 5086, Universite´ Lyon, Lyon, France6
Received 19 October 2009/Accepted 16 March 2010
The variability of the hepatitis C virus (HCV), which likely contributes to immune escape, is most pro-nounced in hypervariable region 1 (HVR1) of viral envelope protein 2. This domain is the target for neutral-izing antibodies, and its deletion attenuates replicationin vivo. Here we characterized the relevance of HVR1 for virus replicationin vitro using cell culture-derived HCV. We show that HVR1 is dispensable for RNA replication. However, viruses lacking HVR1 (⌬HVR1) are less infectious, and separation by density gradients revealed that the population of⌬HVR1 virions comprises fewer particles with low density. Strikingly,⌬HVR1 particles with intermediate density (1.12 g/ml) are as infectious as wild-type virions, while those with low density (1.02 to 1.08 g/ml) are poorly infectious, despite quantities of RNA and core similar to those in wild-type particles. Moreover,⌬HVR1 particles exhibited impaired fusion, a defect that was partially restored by an E1 mutation (I347L), which also rescues infectivity and which was selected during long-term culture. Finally,
⌬HVR1 particles were no longer neutralized by SR-B1-specific immunoglobulins but were more prone to neutralization and precipitation by soluble CD81, E2-specific monoclonal antibodies, and patient sera. These results suggest that HVR1 influences the biophysical properties of released viruses and that this domain is particularly important for infectivity of low-density particles. Moreover, they indicate that HVR1 obstructs the viral CD81 binding site and conserved neutralizing epitopes. These functions likely optimize virus replication, facilitate immune escape, and thus foster establishment and maintenance of a chronic infection.
Hepatitis C virus (HCV) is a single-stranded positive-sense
RNA virus of the familyFlaviviridaethat has infected an
esti-mated 130 million people worldwide (1). Acute HCV infection is mostly asymptomatic; however, virus persistence can lead to severe liver disease, and within 20 years ca. 20% of chronically infected adults develop cirrhosis (46). In fact, morbidity asso-ciated with chronic HCV infection is the most common indi-cation for orthotopic liver transplantation (7). The mecha-nisms that permit the virus to establish chronic infection in ca. 55 to 85% of cases (24) despite vigorous immune responses are incompletely understood.
A number of studies have highlighted the pivotal role of strong, multispecific, and sustained T-cell responses for control of HCV infection (summarized in reference 53). Although
resolution of acute HCV infection can occur in the absence of antibodies (47), mounting evidence indicates that neutralizing antibodies also contribute to protective immunity (summarized in reference 62). Nevertheless, HCV often successfully evades cellular and humoral immune pressure likely at least in part via the constant generation of variants created by an error-prone RNA replication machinery. In line with this notion, a high degree of HCV sequence evolution is associated with chronic disease, while a comparatively static pool of variants correlates with resolution (13, 15, 43).
Virus isolates from patients are classified into at least 7 different genetic groups (genotypes [GTs]), which differ from each other by ca. 31 to 33% at the nucleotide level (20, 48). However, genetic variability is not equally distributed across the HCV genome, which encodes a large polyprotein of ca.
3,000 amino acids and contains 5⬘- and 3⬘-terminal
nontrans-lated regions (NTR) required for RNA replication. More
spe-cifically, the 5⬘NTR and the terminal 99 bases of the 3⬘NTR
are most conserved, while the N-terminal 27 amino acids of the envelope glycoprotein 2 (E2), called HVR1, are most diver-gent among HCV isolates (48). Notably, HVR1 contains epitopes which are recognized by patients’ antibodies (28, 29, 51, 59) and by antibodies that neutralize infection of chimpan-zees (14). Moreover, during an acute infection, sequence changes occur almost exclusively within this region, and these
* Corresponding author. Mailing address: Division of Experimental Virology, Twincore Center for Experimental and Clinical Infection Research, Feodor-Lynen-Strasse 7-9, 30625 Hannover, Germany. Phone: 49 511 220027 130. Fax: 49 511 220027 186. E-mail: thomas [email protected].
† These authors contributed equally to this work.
‡ A joint venture between the Medical School Hannover (MHH) and the Helmholtz Centre for Infection Research (HZI).
§ Supplemental material for this article may be found at http://jvi .asm.org/.
䌤Published ahead of print on 31 March 2010.
5751
on November 8, 2019 by guest
http://jvi.asm.org/
HCV mutant lacking HVR1 (⌬HVR1) was infectious for chim-panzees but clearly attenuated (17). Interestingly, an increase
in titers of the⌬HVR1 virus coincided with emergence of two
mutations in the ectodomain of E2, suggesting that these changes may have compensated for a putative functional im-pairment of the mutant (17).
The development of retroviral particles which carry HCV gly-coproteins on their surfaces (HCV pseudoparticles [HCVpp]) and, more recently, cell culture-derived HCV (HCVcc) based on the JFH1 strain provides robust models for dissecting the
mech-anisms of HCV entryin vitro(3, 25, 35, 58, 64). By means of these
systems, the tetraspanin CD81, the lipoprotein receptor SR-BI, and tight junction proteins claudin-1 and occludin were identified as essential host factors for HCV infection (3, 5, 12, 25, 41, 42, 45). Moreover, it was recognized that there is a complex in-terplay between HCV and lipoproteins. Specifically, high-density lipoprotein (HDL) and oxidized low-high-density lipopro-tein (oxLDL), both ligands of SR-BI, modulate HCVpp infection in an SR-BI-dependent fashion (4, 56, 57). Of note, HVR1 seems to be involved in SR-BI-mediated entry of HCVpp (5), since deletion of this domain ablated stim-ulation of HCVpp infection by HDL and rendered the virus resistant to inhibition by SR-BI-specific antibodies (4, 5), which prevent infection of HCVpp carrying wild-type HCV glycoproteins. Finally, HCVpp lacking HVR1 are more sus-ceptible to neutralization by patient serum-derived immu-noglobulins (4). Thus, altogether these results indicated an important role for HVR1 in viral fitness, likely due to an involvement in HCV entry via SR-BI, and in the interaction of HCV with the humoral immune system. Despite these important observations in the HCVpp system, the role of HVR1 in infection by authentic HCV particles was not de-fined. In addition, it was unclear if HCVpp produced in 293T cells that are unable to produce lipoproteins reflect natural HCV particles with regard to HVR1 function.
Therefore, to better understand the role of HVR1 for virus replication and immune evasion, in this study we analyzed the importance of HVR1 for virus replication and neutralization using authentic, cell culture-derived HCV. We dissected the influence of this domain on HCV receptor interactions and membrane fusion and investigated compensatory mechanisms that permit the virus to regain fitness after deletion of HVR1.
MATERIALS AND METHODS
Ethics statement.The study was approved by ethics committee of the Han-nover Medical School. Patients provided written informed consent.
Plasmids.The plasmids pFK-Jc1 and pFK-Luc-Jc1, encoding the Jc1 chimera with or without the firefly luciferase reporter gene, were described recently (34, 40). Jc1/⌬HVR1 and Jc1/⌬HVR1/I347L were constructed by standard PCR-based techniques and verified by sequencing. pFK-Jc1/⌬HVR was generated by
DHVR/I347L was created with an analogous strategy using pFK-Jc1/⌬HVR as the template and S-J6/I347L (CATTATAGACCTCATTAGCGGGGCTCATT GGGGC), A-J6/I347L (CCCCGCTAATGAGGTCTATAATGACCTCGGGG AC), A/2A/3089 (CTTATCAGAGCGTGAGCTCTGACG), and S-J6/1291 (CA TGGCATGGGACATGATGAT) as primers. Detailed sequence information is available upon request. The constructs pcDNA3⌬cE1E2/⌬HVR1-J6 and pcDNA3⌬cE1E2/⌬HVR1/I347L-J6 were created by transferring a BsiWI-BsmBI restriction fragment derived from pFK-Jc1/⌬HVR or pFK-Jc1/⌬HVR/I347L into pcDNA3⌬cE1E2-J6.
Cell culture and infectivity assay.The Huh-7 cell-derived clone Huh-7.5 is highly permissive for HCV replication (6) and was used for transfections, for passaging of the Jc1/⌬HVR1 virus, and for virus titrations with the limiting dilution assay. Luciferase reporter virus infection assays were conducted in Huh7-Lunet N hCD81 cells, a derivative of Huh7-Lunet cells (18) transduced by lentiviral gene transfer to express high levels of human CD81. Details about the generation of these cells, which were used to provide optimal sensitivity of the assay, will be described elsewhere (J. Bitzegeio and T. Pietschmann, unpublished data). Luciferase reporter virus-associated infectivity was determined as de-scribed previously (34). Authentic viruses were titrated by using a limiting dilu-tion assay (35) with a few minor modificadilu-tions (34).
Preparation of HCV pseudoparticles.Murine leukemia virus (MLV)-based pseudotypes bearing vesicular stomatitis virus glycoproteins (VSV-G) or wild-type as well as mutant HCV J6CF-derived E1 and E2 proteins were generated by cotransfection of 293T cells. Briefly, 1.5⫻106293T cells were
seeded into 6-cm-diameter plates 1 day before transfection with 2.6g of the envelope protein expression the construct pczVSV-G, pcDNA3⌬cE1E2-J6, pcDNA3⌬cE1E2/⌬HVR1-J6, or pcDNA3⌬cE1E2/⌬HVR1/I347L-J6 or empty vector control, 2.6g MLV Gag-Pol expression construct pHIT60, and 2.6g firefly luciferase transducing vector by using Lipofectamine 2000 (Invitrogen). The medium was replaced 6 h after transfection, and the supernatants containing the pseudoparticles were harvested 48 h later. The supernatants were cleared from cells by passage through a 0.45-m-pore-size filter and used to infect Huh-7.5, HuH6, or 293T cells. The efficiency of pseudotype virus infection was evaluated by luciferase assays 72 h postinfection.
Metabolic radiolabeling of Huh-7.5 cells and immunoprecipitation. Twenty-four hours after electroporation of Huh-7.5 cells with the various viral RNAs, cells were washed with phosphate-buffered saline (PBS), starved in methionine-and cysteine-free medium for 1 h, methionine-and incubated for 24 h in methionine- methionine-and cysteine-free Dulbecco’s modified Eagle medium supplemented with 100Ci/ml of Express protein labeling mix (Perkin Elmer, Rodgau-Ju¨gesheim, Germany). Cell lysates were prepared by using 1 ml of ice-cold sodium phosphate buffer per well of a 6-well plate (50 mM Tris-Cl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) supple-mented with a complete protease inhibitor cocktail, as recommended by the manufacturer (Roche). Lysates were cleared by centrifugation at 13,800⫻gfor 15 min at 4°C. The cleared lysates were used for immunoprecipitation using the E2-specific antibody AP33 (9), the R1233 polyclonal rabbit serum specific to E1 (60). For deglycosylation studies, we treated the immunocomplexes with 2,000 U endoglycosidase H (New England Biolabs) as recommended by the manufac-turer and stopped the deglycosylation by adding an equal volume of protein sample buffer. Immune complexes were resolved by denaturing SDS-polyacryl-amide gel electrophoresis (PAGE) and detected by autoradiography.
In vitrotranscription and electroporation.The methods used for transcription and electroporation are described elsewhere (34, 58).
Quantitative detection of HCV core protein by ELISA.HCV core protein was quantified using an enzyme-linked immunosorbent assay (ELISA) according to the instructions of the manufacturer (Wako Chemicals, Neuss, Germany).
Quantitative detection of HCV RNA.Viral RNA was isolated from gradient fractions or agarose beads using a Nucleo Spin RNAII kit (Macherey-Nagel, Du¨ren, Germany) as recommended by the manufacturer. Two microliters of the
on November 8, 2019 by guest
http://jvi.asm.org/
RNA sample was used for quantitative reverse transcription-PCR (qRT-PCR) analysis using a Light Cycler 480 device (Roche, Mannheim, Germany). HCV-specific RT-PCRs were conducted in duplicate utilizing a one-step RT-PCR LightCycler 480 RNA master hydrolysis probes kit (Roche, Mannheim, Ger-many) and the following HCV-specific probe (TIB Molbiol, Berlin, GerGer-many) and primers (MWG-Biotech, Martinsried, Germany): A-195, 5⬘-6FAM (6-car-boxy-fluorescein)-AAA GGA CCC AGT CTT CCC GGC AAT T-TAMRA (tetra-chloro-6-carboxy-fluorescein)-3⬘; S-146, 5⬘-TCT GCG GAA CCG GTG AGT A-3⬘; and A-219, 5⬘-GGG CAT AGA GTG GGT TTA TCC A-3⬘. Reac-tions were performed in three stages by using the following condiReac-tions: stage 1, 3 min at 63°C (reverse transcription); stage 2, 30 s at 95°C (initial denaturation); and stage 3, 35 cycles of 15 s at 95°C and 30 s at 60°C (amplification). The amount of HCV RNA was calculated by comparison to serially dilutedin vitrotranscripts. Indirect immunofluorescence.The method used for indirect immunofluores-cence has been described (34). HCV-infected cells were detected using the NS5A-specific monoclonal antibody 9E10 at a dilution of 1:2,000. Bound primary antibodies were detected using goat anti-mouse IgG-specific secondary antibod-ies conjugated to Alexa Fluor 488 (Sigma, Steinheim, Germany) at a dilution of 1:1,000. Nuclear DNA was stained using DAPI at a dilution of 1:3,000.
Iodixanol density-gradient fractionation.Density gradient centrifugation was performed as described recently (22). Briefly, viruses were separated by over-night centrifugation through a 0% to 40% iodixanol step gradient at 154,000⫻
gin a TH-641 swing-out rotor at 4°C using a Sorvall Ultra WX80 centrifuge. Ten fractions of 1 ml were collected from the bottom and analyzed for virus infec-tivity, core protein levels, and viral RNA copies.
Monoclonal antibody- and patient serum-mediated neutralization of HCV infection.For inhibition of HCV infection, 200l of an Huh7-Lunet N hCD81 cell suspension (3.5⫻104cells per ml) was seeded into each well of a 96-well
plate 24 h prior to inoculation. Luciferase reporter viruses were mixed with serial dilutions of antibodies and were used to inoculate cells. Neutralization assays were performed using the human E2-specific MAbs CBH4D, CBH4G, CBH5, CBH7, CBH23, HC-1, HC-11, and R04 (21, 31). In addition, mouse monoclonal antibodies specific to E2 (AP33 [37] or CD81 [JS81 {Becton Dickinson, berg, Germany}, 1.3.3.22 {Ancell, Bayport, MN}, or 5A6 {Santa Cruz, Heidel-berg, Germany}]) or specific to CD13 (Becton Dickinson, HeidelHeidel-berg, Germany) were utilized. Polyclonal rat immunoglobulins specific to SR-B1 were generated by immunization of a Wistar rat with a plasmid expressing the full-length SR-B1 cDNA (63). Immunoglobulins from this rat (SR-BI IgG) and a control rat (control IgG) that was not immunized were purified from sera using a MAb Trap kit (GE Healthcare) according to the instructions of the manufacturer. Human sera were obtained from healthy donors or patients chronically infected with HCV of genotype 1, 2, 3, or 4.
Precipitation of HCV particles with antibodies or soluble CD81-LEL.Equal quantities of HCV particles (normalized for HCV RNA copy numbers) were subjected to immunoprecipitation with 1g anti-E2 antibodies or an isotype-matched control antibody R04 (anti-CMV). Antibodies and HCV RNA-contain-ing particles recognized by the antibodies were incubated for 2 h at 4°C and were then precipitated by the addition of 25l protein G-agarose. For the CD81 capture assay, 10g glutathioneS-transferase (GST)-CD81-LEL or GST was preincubated with 50l glutathione agarose (Sigma) for 2 h at 4°C. The agarose beads were incubated with the samples overnight at 4°C with rotation. Immu-noprecipitated material was washed three times with PBS. Finally, precipitated viral RNA was quantified using qRT-PCR as described above.
Production and purification of recombinant hCD81-LEL and GST.CD81 fused to glutathione-S-transferase was prepared as described previously (16). Briefly, transformedE. coliRosetta-gami cells were induced to express the hCD81 LEL-GST and GST by addition of isopropyl-1-thio- -D-galactopyrano-side (IPTG; Sigma) to a final concentration of 100M overnight at room temperature. Bacteria were lysed with 0.5% NP-40 (Roth, Karlsruhe, Germany), lysozyme, and Benzonase (Merck, Darmstadt, Germany) in PBS. The proteins were further purified using glutathione agarose followed by elution with 100 mM L-glutathione (Sigma) in PBS and dialysis in PBS.
Fusion assay.The fusion assay was described recently (22). Briefly, octadecyl rhodamine B chloride (R18)-labeled liposomes composed of phosphatidylcho-line (PC) and cholesterol (final lipid concentration, 15M) were mixed with the virus followed by acidification with HCl to initiate fusion (final HCl concentra-tion, 0.1 M). Lipid mixing was monitored as the dequenching of R18and was
recorded using a dual-channel PicoFluor hand-held fluorimeter (Turner Biosys-tems, CA) operated under the rhodamine channel (excitation and emission wavelengths, 540⫾20 and⬎570 nm, respectively). Maximal dequenching of R18
was determined after 0.1% Triton X-100 was added to the liposomes. Statistical data analysis.Data are presented as means⫾standard deviations or standard errors, as appropriate. The 50% tissue culture infectious dose
(TCID50) was calculated based on methods described by Ka¨rber and by
Spear-man (27, 49). Statistical comparison of TCID50between groups was based on the
asymptotic Gauss distribution. If different experiments were performed, a linear mixed-effect model was used to compare infectivity between groups. Further-more, weighted linear or nonlinear regression analysis was used to assess dy-namics of infectivity. In the case of multiple tests, the Bonferroni correction was used. Statistical tests were two-sided, andPvalues below 5% are considered significant.
RESULTS
Deletion of HVR1 selectively impedes HCV infectivity. To analyze the role of HVR1 in replication, assembly, and infec-tivity of HCV, we used the Jc1 chimera, which grows to high virus titers in tissue culture (40), and compared RNA replica-tion, virus producreplica-tion, and infectivity of released particles be-tween Jc1 and a Jc1 mutant lacking HVR1, i.e., the 27 amino-terminal residues of E2. Removal of HVR1 did not affect RNA replication or release of HCV particles, since accumulation of core in transfected Huh-7.5 cells and in the culture fluid was similar between viruses (Fig. 1A) and as replication kinetics determined with luciferase reporter virus constructs were com-parable (data not shown). However, when infectivity of parti-cles normalized for equal quantity of core protein was
deter-mined, infectivity of Jc1/⌬HVR1 was reduced about 6-fold
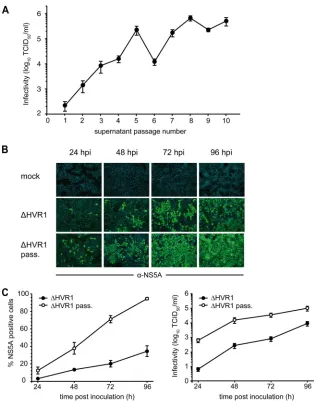
FIG. 1. HCV particles devoid of HVR1 have reduced infectivity. RNA transcripts of Jc1 and a Jc1 derivative lacking HVR1 (⌬HVR1) were transfected into Huh-7.5 cells. (A) Forty-eight hours later, intra-and extracellular core protein levels were determined using a core-specific ELISA as an indicator for efficiency of RNA replication and virus production. (B) In parallel, virus infectivity normalized for equal quantity of core was determined by inoculation of naïve Huh-7.5 cells using a limiting dilution assay. Error bars show standard errors. (C) Representative microscopic views of Huh-7.5 cells inoculated with Jc1 and⌬HVR1 particles. Cells were stained with NS5A-specific an-tibodies by immunohistochemistry. Results of one representative ex-periment out of three independent repetitions are shown.
on November 8, 2019 by guest
http://jvi.asm.org/
(3.1- to 11.6-fold based on the corresponding 95% confidence interval) compared to the parental virus (Fig. 1B), and fewer infected cells were detected 72 h postinoculation (Fig. 1C). Accordingly, deletion of HVR1 does not affect HCV RNA replication or virus release but impairs virus entry.
Impaired virus infectivity due to deleted HVR1 is compen-sated for by a single mutation in E1.To assess if impaired virus
entry of Jc1/⌬HVR1 can be overcome by compensatory
muta-tions, we serially passaged Jc1/⌬HVR1 in highly permissive
Huh-7.5 cells. In the course of 10 consecutive supernatant
passages, we observed a ⬎1,000-fold increase in viral titers
with a mean increasing rate of 0.33/passage (Fig. 2A). To analyze if the passaged virus had regained fitness, we
inocu-lated Huh-7.5 cells with an equally low dose of Jc1/⌬HVR1
viruses derived 72 h posttransfection and the Jc1/⌬HVR1 virus
[image:4.585.134.455.65.466.2]population present after 10 passages. Notably, the passaged viruses spread through the cell culture much more rapidly (Fig. 2B and C) and produced ca. 100-fold more progeny 24 and 48 h postinoculation (Fig. 2C, right). Sequencing the viral quasispe-cies after 10 passages, we identified four changes. Two of these were silent mutations in NS5A and NS5B. In addition, an amino acid change in the vicinity of the transmembrane do-main of E1 (I347L) and a mutation within NS5B (S2508L) were identified. It is interesting that residue I347 resides in the membrane-proximal heptad repeat region of E1 that has pre-viously been shown to be important for infectivity of genotype 1a HCVpp (11). To analyze if these amino acid changes com-pensated for the impairment caused by deletion of HVR1, we
FIG. 2. Serial cell culture passage of an⌬HVR1 mutant yields viruses with increased fitness. (A) Huh-7.5 cells were transfected with the
⌬HVR1 mutant RNA. Forty-eight hours later, the cell culture fluid of these cells was harvested and used to infect naïve Huh-7.5 cells. Subsequently, cell-free culture fluids were passaged in naive Huh-7.5 cells every two to three days in total for 10 serial rounds of passaging. Infectivity of the respective culture fluid at the time of passaging was determined using the limiting dilution assay. (B) Huh-7.5 cells were inoculated with⌬HVR1 virus harvested 72 h posttransfection or with⌬HVR1 viruses collected after 10 consecutive passages at a multiplicity of infection of 0.1. Inoculated cells were fixed at the indicated time points and stained for NS5A expression by indirect immunofluorescence (green). Nuclear DNA was stained with DAPI (blue). (C) The percentage of infected cells at the respective time points was calculated by counting total cells (DAPI stain) and infected cells (NS5A expression) in five arbitrarily chosen microscopic views, respectively (left). Data are means and standard errors of the means. Production of viral progeny was quantified by inoculation of naïve Huh-7.5 cells (right).
on November 8, 2019 by guest
http://jvi.asm.org/
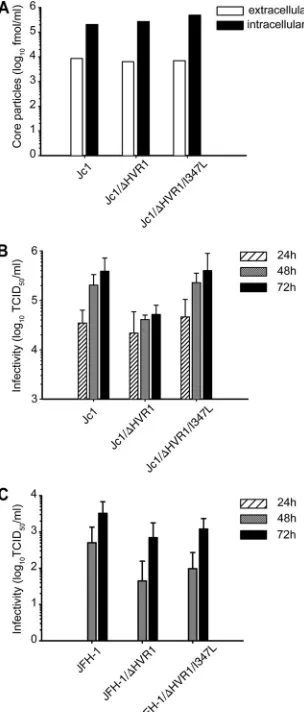
cloned them individually into the backbone of Jc1/⌬HVR1. Insertion of the S2508L mutation did not change RNA repli-cation, production of particles, or their infectivity (data not shown). Similarly, mutation I347L did not affect RNA
replica-tion or release of HCV particles, since Jc1 and Jc1/⌬HVR1
with or without the I347L exchange accumulated comparable quantities of intra- and extracellular core protein 48 h post-transfection (Fig. 3A). In contrast, the I347L mutation com-pensated for the entry defect caused by deletion of HVR1,
since insertion of this mutation into Jc1/⌬HVR1 yielded
infec-tivity similar to that of Jc1 and significantly higher than that of
Jc1/⌬HVR1. Statistical significance was confirmed using a
mixed linear effect model, which assesses the overall
differ-ences between these viruses for all three time points (P⬍0.001
for the comparison of Jc1 versus Jc1/⌬HVR1,P⬍0.001 for the
comparison of Jc1/⌬HVR1 versus Jc1/⌬HVR1/I347L, andP⬎
0.2 for the comparison of Jc1 versus Jc1/⌬HVR1/I347L) (Fig.
3B). Importantly, deletion of HVR1 also reduced infectivity of
JFH1 particles. However, in the context of JFH1/⌬HVR1, the
I347L mutation did not significantly enhance infectivity,
indi-cating that suppression of the⌬HVR1-specific entry defect by
this mutation may be specific to the JFH1-J6 context of the Jc1 chimera or may not be visible in a viral context that produces virus particles with lower efficiency, like JFH1 (Fig. 3C). Taken together, these results argue that loss of HVR1 and the asso-ciated entry defect can be compensated for by a single point mutation close to the transmembrane domain of E1.
Deletion of HVR1 heavily impairs HCVpp infectivity but has no influence on E1/E2 glycosylation. To investigate possible mechanisms for impaired infectivity of HCVcc lacking HVR1 and for the compensatory effect of the I347L mutation, we first inves-tigated the influence of this mutation on the glycosylation of E1/E2 proteins in virus-producing Huh-7.5 cells. To this end,
Huh-7.5 cells were transfected with Jc1, Jc1⌬HVR1, Jc1/⌬HVR1
I347L, or a subgenomic JFH1 replicon (SG) lacking the E1-E2 coding region. After metabolic labeling, E1/E2 complexes were precipitated with an E2-specific monoclonal antibody (AP33), or
a polyclonal E1-specific antiserum and subjected to endo-
[image:5.585.86.238.70.426.2]-N-acetylglucosaminidase H (Endo H) digestion. As is depicted in Fig. 4, deletion of HVR1 caused a subtle shift in the electro-phoretic mobility of the E2 protein which was evident both for the glycosylated and the deglycosylated protein, indicating that dif-ferential mobility is likely due to the deletion of 27 amino acids rather than divergent glycosylation. Moreover, we did not observe
an overt difference between Jc1/⌬HVR1 and the Jc1/⌬HVR1
FIG. 3. The suppressor mutation I347L restores infectivity of
⌬HVR1 viruses. (A) RNA transcripts of Jc1, Jc1/⌬HVR1, and Jc1/
[image:5.585.301.539.71.263.2]⌬HVR1/I347L were transfected into Huh-7.5 cells. (A) Forty-eight hours later, intra- and extracellular core protein levels were deter-mined using a core-specific ELISA. Results from a representative of two independent repetitions are shown. (B) Cell supernatants were harvested at the indicated time points and used for infection of naïve Huh-7.5 cells. The 50% tissue culture infectious dose of the samples was determined by a limiting dilution assay. Data are means from five independent repetitions. (C) RNA transcripts of JFH1, JFH1/⌬HVR1, and JFH1/⌬HVR1/I347L were transfected as for panel A, and infec-tivity released was quantified as for panel B. Data are means from three independent experiments.
FIG. 4. Deletion of HVR1 and the I347L mutation does not affect glycosylation of HCV E1-E2 complexes. Huh-7.5 cells were transfected with the indicated viral RNAs and metabolically labeled. Cells were then lysed under nondenaturing conditions, and E1/E2 complexes were precipitated using the E2-specific mouse monoclonal antibody AP33 or an E1-specific polyclonal serum. After extensive washing, proteins were deglycosylated with Endo-H or left untreated. Finally, immune complexes were resolved by denaturing SDS-PAGE and de-tected by autoradiography. Deglycosylated protein species are marked with asterisks.
on November 8, 2019 by guest
http://jvi.asm.org/
I347L mutant, suggesting that the I347L mutation does not grossly affect E1/E2 glycosylation.
Next we assessed if the adaptive mutation I347L selected in
the context of Jc1⌬HVR1 virus rescues impaired infectivity of
HCVpp carrying J6 glycoproteins devoid of HVR1. Interest-ingly, in the context of HCVpp, deletion of HVR1 caused a
⬎100-fold decrease of infectivity compared to HCVpp coated
with wild-type J6 glycoproteins (Fig. 5). Moreover, unlike for HCVcc, the I347L mutation did not restore infectivity of HCVpp. These results suggest that there are differences with regard to the degree of HVR dependence between HCVpp and HCVcc carrying J6 glycoproteins. Moreover, the mecha-nism or requirements for restoration of an entry defect caused by deletion of the J6-derived HVR1 seem to differ between HCVpp and HCVcc.
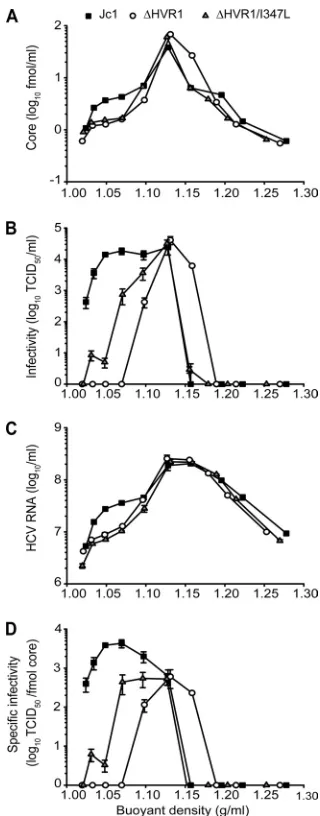
Deletion of HVR1 reduces abundance of HCV particles with very low buoyant density and ablates their infectivity. Cell culture- and serum-derived HCV associates with lipoproteins, and this interaction is important for infection. To assess if deletion of HVR1 modulates the relation of HCV with lipids or lipoproteins, an interplay that may not be fully recapitulated by HCVpp which are created in 293T cells, a human embryonic kidney cell line which is unlikely to produce lipoproteins, we
compared buoyant density between Jc1, the⌬HVR1 mutant,
and a⌬HVR1 virus carrying the I347L mutation (Jc1/⌬HVR1/
I347L). Both viruses lacking HVR1 exhibited a spectrum of core- and RNA-containing structures very similar to those in Jc1 wild-type particles, with peaks of RNA and core at a den-sity of ca. 1.12 g/ml (Fig. 6A and B). However, the virus pop-ulations produced from genomes lacking HVR1 were both characterized by a moderate yet reproducible decrease of par-ticles with low buoyant density ranging between 1.02 to 1.10 g/ml. In addition, at least based on core protein (Fig. 6A) and
infectivity (Fig. 6B) measurement Jc1/⌬HVR1 produced some
more particles with a density of ca. 1.16 compared to Jc1 and
Jc1/⌬HVR1/I347L. Strikingly,⌬HVR1 particles with a density
between 1.02 and 1.08 were not infectious at all, and the I347L
mutation partially restored infectivity of⌬HVR1 viruses of this
specific density (Fig. 6C). As a consequence, the specific infec-tivity of HCV particles, i.e., the infecinfec-tivity associated with a defined quantity of core, significantly differed among all three viruses in this density range but was indistinguishable at 1.12 g/ml (Fig. 6D). Therefore, the moderate reduction of
infectiv-ity of total Jc1/⌬HVR1 particles (Fig. 3) likely reflects a strong
impairment of those particles with a density between 1.02 and 1.08, which represent a minor fraction of total particles (less than 10% based on the detection of viral RNA and core pro-tein). As a consequence, most of the detectable infectivity of
⌬HVR1 particles likely reflects the virus particle type with
FIG. 5. The suppressor mutation I347L does not restore infectivity of HCVpp with the⌬HVR1 mutation. Murine leukemia virus (MLV)-based pseudotypes bearing vesicular stomatitis virus glycoproteins (VSV-G), HCV J6 wild-type (J6), J6/⌬HVR1 (⌬HVR1), or J6/
[image:6.585.340.500.66.474.2]⌬HVR1/I347L (⌬HVR1/I347L) glycoproteins were prepared by trans-fection of 293T cells. Preparations created in the absence of a viral envelope protein expression construct (pcDNA) served as controls. Infectivity of the pseudoparticles transducing a luciferase transgene was determined by inoculation of Huh-7.5 cells. Data are the means of quadruple measurements and are from a representative of three inde-pendent repetitions.
FIG. 6. Buoyant density and specific infectivity of Jc1, Jc1/⌬HVR1, and Jc1/⌬HVR1/I347L viruses. Individual viruses were harvested 48 h after transfection of Huh-7.5 cells and were resolved using an iodixanol step gradient. Ten fractions were harvested from the bottom, and HCV core protein (A), HCV RNA (B), and infectivity (C) were determined for each fraction. Values are plotted against the density of the respec-tive fraction measured by refractometry. (D) Specific infectivity of viruses contained in individual fractions is expressed as TCID50per fmol core protein. The results are from a representative of five inde-pendent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
intermediate density. Interestingly, the I347L mutation
par-tially, but not completely, restored infectivity of⌬HVR1 with
very low density, suggesting that this mutation is able to par-tially compensate for the defect of this specific virus particle type. The reason why this mutation, unlike the bulk virus prep-aration analyzed in Fig. 3, did not fully restore infectivity after iodixanol gradient centrifugation is currently unknown but may be related to the experimental procedure of gradient centrif-ugation. Taken together, these results indicate that deletion of HVR1 results in production of decreased numbers of virions with low density. In addition, removal of this domain selec-tively ablates infectivity of this particle type but does not affect the infectivity of particles with a density of 1.12 g/ml, the predominant virus species produced in tissue culture. There-fore, these data suggest that HVR1 is particularly important for infectivity of particles with low density.
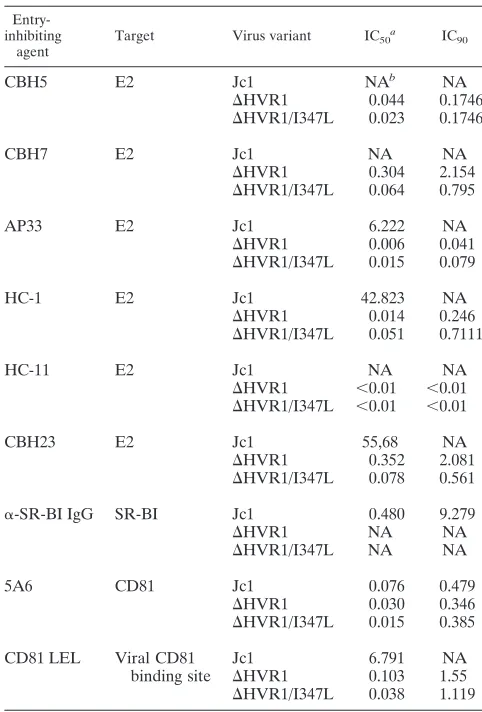
Deletion of HVR1 ablates neutralization by polyclonal SR-BI-specific immunoglobulins and exposes the viral CD81 bind-ing site.Previous reports have indicated that recombinant sol-uble E2 devoid of HVR1 exhibits increased CD81 binding yet decreased interaction with SR-BI compared to E2 carrying this domain (44, 45). In addition, entry of HCVpp lacking HVR1 was no longer inhibited by an SR-BI-specific antiserum, sug-gesting that deletion of this domain may have ablated viral dependence on this host factor (5). To investigate the impor-tance of HVR1 for interaction of authentic HCV particles with these two crucial entry factors, we performed a series of re-ceptor competition assays employing HCV particles with or without HVR1 and with the I347L mutation, using antibodies against SR-BI and CD81 as well as soluble human CD81. As target cells we used Huh7-Lunet CD81-high cells, which are highly permissive to HCV infection, thus providing optimal assay sensitivity. Polyclonal immunoglobulins specific to SR-BI which were prepared from a rat genetically immunized with an SR-BI expression construct (63) neutralized Jc1 in a dose-dependent fashion (Fig. 7 and Table 1). In contrast both Jc1 viruses lacking HVR1 were not neutralized by these immuno-globulins. These results are in line with previous findings ob-tained with HCVpp (5) and suggest that deletion of HVR1 has ablated or strongly reduced dependence on SR-BI.
CD81 usage was compared between these viruses using two complementary approaches employing the CD81-specific mono-clonal antibody 5A6 or soluble CD81-LEL GST fusion
[image:7.585.137.449.67.189.2]pro-teins (hCD81-LEL), which comprise the viral receptor binding site (41). While the former assay primarily analyzes the depen-dence of HCV entry on the availability of surface-resident CD81 molecules, the latter mainly probes the exposure of the
FIG. 7. HCV particles lacking HVR1 are more resistant to neutralization by SR-BI-specific sera. Huh7-Lunet N hCD81 cells were infected with luciferase reporter viruses normalized to equal quantities of HCV core and mixed with increasing doses of SR-BI-specific immunoglobulins (left) or antibodies purified from a control rat (right) for 72 h at 37°C. Luciferase reporter activity was determined and is expressed relative to that in infections performed in the absence of rat serum. Data are means and standard deviations of triplicate measurements. Two independent experiments were conducted.
TABLE 1. Inhibitory concentrations of various entry inhibitors used in this study
Entry-inhibiting
agent
Target Virus variant IC50 a
IC90
CBH5 E2 Jc1 NAb NA
⌬HVR1 0.044 0.1746
⌬HVR1/I347L 0.023 0.1746
CBH7 E2 Jc1 NA NA
⌬HVR1 0.304 2.154
⌬HVR1/I347L 0.064 0.795
AP33 E2 Jc1 6.222 NA
⌬HVR1 0.006 0.041
⌬HVR1/I347L 0.015 0.079
HC-1 E2 Jc1 42.823 NA
⌬HVR1 0.014 0.246
⌬HVR1/I347L 0.051 0.7111
HC-11 E2 Jc1 NA NA
⌬HVR1 ⬍0.01 ⬍0.01
⌬HVR1/I347L ⬍0.01 ⬍0.01
CBH23 E2 Jc1 55,68 NA
⌬HVR1 0.352 2.081
⌬HVR1/I347L 0.078 0.561
␣-SR-BI IgG SR-BI Jc1 0.480 9.279
⌬HVR1 NA NA
⌬HVR1/I347L NA NA
5A6 CD81 Jc1 0.076 0.479
⌬HVR1 0.030 0.346
⌬HVR1/I347L 0.015 0.385
CD81 LEL Viral CD81 Jc1 6.791 NA
binding site ⌬HVR1 0.103 1.55
⌬HVR1/I347L 0.038 1.119
a
Pⱕ0.001 for HVR1 virus mutants versus Jc1 for all agents except for 5A6, for whichPⱕ0.05.
b
NA, not applicable.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.585.299.540.344.700.2]viral CD81 binding site on the virus particle. All three viruses were neutralized in a dose-dependent fashion by the CD81-specific antibody with no significant differences among the
three types of virus particles (P⬎0.2) (Fig. 8A and Table 1).
Moreover, the 90% inhibitory concentration (IC90) was
com-parable between the viruses for each antibody. Together, these results argue that unlike for SR-BI, the overall efficiency of CD81 usage was not much influenced by removal of HVR1 and the I347L mutation. Strikingly, however, both viruses lacking HVR1 were much more susceptible to entry inhibition by
hCD81-LEL, since as little as 1.6 and 1.2g/ml of hCD81-LEL
inhibited infection of ⌬HVR1 and ⌬HVR1/I347L,
respec-tively, by 90% (Fig. 8B and Table 1). In contrast, a 50-fold-higher dose of hCD81-LEL neutralized only 70% of wild-type HCV. Together, these results indicated that deletion of HVR1 may have uncovered the viral CD81 binding site and that its
increased exposure was not influenced by the I347L mutation. In line with this notion, precipitation of HCV particles by hCD81-LEL was more efficient for both viruses lacking HVR1 than for wild-type virions (Fig. 8C), as ca. 6-fold more viral
RNA was precipitated when these⌬HVR1 viruses were
incu-bated with hCD81-LEL beads than wild-type virus particles. Although deletion of HVR1 also slightly increased nonspecific adherence to GST-coupled beads (Fig. 8C, left), the specific binding to CD81-LEL was clearly much increased, as is evident from the relative binding of these viruses to GST and hCD81-LEL-coupled beads (Fig. 8C right).
[image:8.585.135.450.68.446.2]Deletion of HVR1 impairs HCV fusion, and this impediment is partially compensated for by the I347L suppressor muta-tion.Dreux et al. recently reported that HVR1 was required for enhancement of HCVpp fusion by ApoC1 (10). Moreover, using the same liposome-based fusion assay, these authors also
FIG. 8. Deletion of HVR1 exposes the viral CD81 binding site. (A) Huh7-Lunet N hCD81 cells were inoculated with indicated luciferase reporter viruses normalized to equal quantities of HCV core in the presence of increasing doses of CD81-specific monoclonal antibodies or control antibodies directed against CD13. The efficiency of infection was determined 72 h later by luciferase reporter assay and is expressed relative to infections performed in the absence of antibodies. Data are means and standard deviations of triplicate measurements and are from a representative of three independent experiments. (B) Infections (as described for panel A) were conducted in the presence of increasing doses of recombinant human CD81 large extracellular loop fused to GST (hCD81-LEL) or of GST alone. Two independent experiments were conducted. (C) Equal numbers of viruses were incubated with 10g hCD81-LEL or GST. Subsequently, complexes were precipitated using glutathione-coated beads, and associated HCV RNA was quantified by qRT-PCR. Data are means of duplicate measurements. Two independent experiments were performed.
on November 8, 2019 by guest
http://jvi.asm.org/
demonstrated that fusion of authentic HCV particles was stim-ulated by ApoC1. Therefore, we specstim-ulated that impaired en-try of HCV lacking HVR1 may be linked to suboptimal mem-brane fusion and compared the fusion properties of authentic HCV with or without HVR1 and the I347L mutation. Relying on the liposome-based assay previously described for the fu-sion of HCVcc (22), we compared the fufu-sion properties dis-played by our various viral constructs. As is depicted in Fig. 9, the rate of fusion was decreased, and an increase of fusion could be observed only later for HCV lacking HVR1. Inter-estingly, insertion of the I347L mutation restored fusion
com-petence of the⌬HVR1 mutant almost to the level of wild-type
HCV particles (Fig. 9). Consequently, these results suggest that deletion of HVR1 attenuates HCV entry via impaired membrane fusion and that this impediment is partially over-come by the I347L compensatory mutation in E1.
HVR1 protects conserved viral cross-neutralizing epitopes.
It is well established that HVR1 is a neutralizing epitope, since antibodies from infected patients as well as antibodies raised against HVR1 peptides bind to HVR1 and inhibit HCV infec-tion (14, 29). However, relatively little is known about the importance of HVR1 for exposure and recognition of alterna-tive, more conserved viral epitopes which may be crucial for eliminating infectious HCV through neutralizing antibodies. Interestingly, it was reported recently that HCVpp lacking HVR1 were neutralized more efficiently by HCV patient sera than the parental HCVpp (4). However, the mechanism of the increased susceptibility and the relevance of this observation for authentic HCV particles were unclear. Therefore, we ana-lyzed the susceptibility of HCVcc with or without HVR1 and the I347L mutation to a broad set of HCV E2-specific mono-clonal antibodies directed against various conserved cross-neu-tralizing epitopes. More specifically, we employed AP33, a mouse antibody that recognizes a defined linear epitope im-mediately downstream of HVR1 (52) and that cross-neutral-izes HCV genotype 1 to genotype 6 infections in the context of
HCVpp (38). In addition, we utilized a well-defined set of human monoclonal antibodies originally derived from the pe-ripheral B cells of an individual infected with genotype 1b HCV. These antibodies are known to recognize conforma-tional epitopes of the E1/E2 glycoprotein complex, and exten-sive cross-competition experiments had identified three clus-ters of overlapping epitopes with distinct properties (domains A, B, and C [30, 32]). While antibodies interacting with domain A were nonneutralizing (e.g., CBH4G), those specific to do-mains B and C (e.g., CBH5 and CBH7, respectively) inhibit E2 binding to CD81 and neutralize infection of HCVpp and HCVcc from genotypes 1 and 2 (31, 33). Finally, we quantified neutralization of these viruses by a spectrum of sera derived from patients chronically infected with genotype 1 to 4 viruses.
Interestingly, with the applied doses ranging up to 50g/ml,
none of the monoclonal antibodies markedly disturbed infec-tion of wild-type HCV, suggesting that neutralizainfec-tion of the Jc1 chimera may be very difficult and possibly less efficient than that of HCVpp (Fig. 10A and Table 1; also, see Fig. S1 in the supplemental material). In sharp contrast, all monoclonal an-tibodies strongly neutralized viruses that lacked HVR1 except for CBH4G, which binds a viral epitope that is not neutralizing (33). Neutralization was particularly strong for CBH5 and
AP33 with an IC90below 20 ng/ml (Table 1). Notably, there
was no significant difference between Jc1/⌬HVR1 and Jc1/
⌬HVR1/I347L, indicating that the suppressor mutation in E1
did not strongly influence neutralization. Together, these data suggested that deletion of HVR1 permits efficient virus neu-tralization through different conserved cross-neutralizing epi-topes. In agreement with this assumption, serum-contained antibodies from patients chronically infected with HCV geno-types 1 to 4 invariably neutralized viruses lacking HVR1 with clearly increased efficiency compared to wild-type particles (Fig. 10B). In fact, none of these sera reduced wild-type infec-tivity by more than 90% even at the highest concentration of serum employed (1:20 dilution), while in some cases the
⌬HVR1 viruses were already fully neutralized by a
250-fold-lower serum dilution (1:5,000). Although⌬HVR1 viruses are
slightly more prone to nonspecific inhibition by high concen-trations of human sera (Fig. 10B), these results nevertheless provide strong evidence that these viruses are also much more susceptible to inhibition by patient-derived antibodies. More-over, since all HCV-neutralizing monoclonal antibodies that
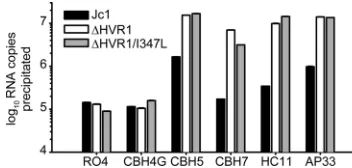
we tested also precipitated⌬HVR1 viruses from suspension
much more efficiently than wild-type HCV (Fig. 11), we con-clude that the deletion of HVR1 results in increased exposure of crucial conserved epitopes, thus facilitating effective neu-tralization.
DISCUSSION
In this study, we characterized the role of the hepatitis C virus HVR1 for virus replication in cell culture using authentic HCV particles. In particular, we analyzed the relevance of this determinant for individual steps of the viral replication cycle and for receptor interactions, membrane fusion, and neutral-ization by virus-specific antibodies. Our data indicate that this domain is dispensable for RNA replication, virus assembly, and infection; however, HCV particles produced from ge-nomes lacking HVR1 differ from wild-type particles in a
num-FIG. 9. Impaired fusion of viruses lacking HVR1 is partially com-pensated for by the I347L suppressor mutation in E1. Equal quantities of cell culture-produced viruses (normalized for core protein content) were used for fusion assays with fluorescently labeled liposomes. De-quenching of R18 was used as indicator of fusion between liposomes and viral membrane. Maximal dequenching of R18 was determined after 0.1% Triton X-100 was added to the liposomes. Data are from a representative of five independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:9.585.84.239.67.226.2]ber of important ways. First, these viruses have a moderately decreased specific infectivity compared to particles carrying HVR1. Second, the virus population produced is skewed and comprises fewer particles with very low density. Third, unlike wild-type HCV, which is highly infectious in the
very-low-density range between 1.02 and 1.08 g/ml, ⌬HVR1 particles
with equivalent density are not infectious. In contrast,
infec-tivity of wild-type and⌬HVR1 viruses with intermediate
den-sity (1.12 g/ml) is indistinguishable. Therefore,⌬HVR1
parti-cles are influenced by this deletion to variable degrees depending on the particle density. In turn, these data suggest that the dependence of HCV particles on HVR1 varies be-tween very low and intermediate density and that the require-ments for infection may differ between HCV particle types.
It is well established that HCV interacts with lipoproteins. In
fact, the variable density of HCV particles in vivo has been
[image:10.585.82.504.65.538.2]attributed to differential association with lipoproteins and an-tibodies (23, 54, 55), and careful purification of HCV from
FIG. 10. Deletion of HVR1 enhances neutralization by monoclonal antibodies and patient sera and causes increased exposure of conserved epitopes. Huh7-Lunet N hCD81 cells were inoculated with viruses normalized to equal quantities of HCV core in the presence of increasing doses of human or murine HCV E2-specific monoclonal antibodies (A) or sera from HCV infected patients or a healthy donor (B). The RO4 monoclonal antibody, recognizing a cytomegalovirus protein, served as a control. Infection efficiency was determined by luciferase assays 72 h postinoculation and is expressed relative to that of infections performed in the absence of antibodies and sera. Data are means and standard deviations of triplicate measurements and are from one representative of at least two independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
low-density fractions of human sera revealed that these HCV RNA-carrying structures, sometimes designated “lipoviro par-ticles,” also contained at least core and apolipoprotein B (ApoB) (2). Moreover, HCV produced in cell culture, and therefore in the absence of antibodies, also exhibits heteroge-neous density (35), and it was recognized that production of HCV by human hepatocytes depends on apolipoprotein E (ApoE), ApoB, and microsomal triglyceride transfer protein (MTP), host factors that are essential for secretion of very low density lipoproteins (VLDL) (8, 19, 26). Finally, coprecipitation of cell culture-derived HCV with ApoE- or ApoC1-specific anti-bodies as well as neutralization of infection by these antianti-bodies suggests a direct interaction between HCVcc and ApoE- and ApoC1-comprising lipoprotein complexes (8, 10, 36).
In spite of these findings, it remains unclear how HCV actually associates with lipoproteins and which viral determi-nants modulate this interaction. Our observation that deletion of HVR1 reduces the number of very low density HCV parti-cles suggests that this domain influences the interplay between HCV and lipids or lipoproteins. However, beyond lipids or lipoproteins, other factors may influence the density of HCV particles, including for instance variable abundance of particle-associated viral proteins (e.g., E1/E2 complexes). Alterna-tively, a single virus particle type with constant composition may interact with a variable lipoprotein partner differing in lipid or apolipoprotein content. Further work is needed to distinguish between these possibilities and to determine how HVR1 modulates the density of HCV particles.
Surprisingly,⌬HVR1 particles with low density (1.02 to 1.08)
are not infectious or are poorly infectious, whereas⌬HVR1
par-ticles with an intermediate density of 1.12 g/ml were not impaired with regard to infectivity. Importantly, both particle types have a similar ratio between HCV RNA and core protein, arguing that the selective loss of infectivity is not related to aberrant packaging of core or RNA.
Considering that HCV membrane fusion is likely a cooper-ative process involving a number of fusion proteins, low abun-dance of particle-associated glycoprotein complexes may, in principle, preclude infection and could therefore selectively impede infection of particles with low numbers of glycopro-teins. In line with this hypothesis, a decreased fusion compe-tence of E1/E2 complexes lacking HVR1 may selectively ablate infectivity of particles with low numbers of E1/E2 complexes but may still be sufficient to support fusion and infection of particles with a high abundance of E1/E2 complexes.
Corre-spondingly, a compensatory mutation that rescues fusion ac-tivity, such as the I347L mutation identified in this study, may restore infectivity of these low-density particles by re-establish-ing fusion competence to a level sufficient to permit infection even with a low number of E1/E2 complexes. Alternatively, a lack of interaction of HCV particles devoid of HVR1 with a crucial entry factor may be specific to particles with very low density and may consequently also limit infectivity of these virions. Future experiments dissecting the composition of HCV particles with different densities should help to distin-guish between these hypotheses.
Besides the findings discussed above, our comparison of receptor utilization by HCV with and without HVR1 revealed two important features of HVR1. First, this domain influences utilization of SR-BI by HCV. These results extend previous reports that assessed the influence of HVR1 on the interplay with SR-BI using soluble E2 or HCVpp (5, 45). Previous stud-ies suggested that deletion of HVR1 may decrease or even ablate interaction with SR-BI, since soluble E2 lacking HVR1 no longer interacted with SR-B1 and since HCVpp devoid of this domain were no longer susceptible to neutralization by an SR-B1-specific serum (5, 45). Our data obtained with HCVcc lacking HVR1 support this notion, since entry of these parti-cles was also no longer inhibited by polyclonal anti SR-BI immunoglobulins.
Second, our data indicate that HVR1 masks the viral CD81 binding site. This conclusion is based on the observation that
soluble CD81 (hCD81-LEL) neutralizes and precipitates⌬HVR1
particles much more readily than wild-type particles. Possibly, the conserved CD81 binding site in wild-type particles is mostly hid-den until interaction with a host factor elicits a conformational change, fully exposing the CD81 binding site and thus preparing the virus for contact with this crucial receptor. Such a strategy is reminiscent of the interplay of human immunodeficiency virus type (HIV-1) with CD4 and the chemokine receptors, where binding to CD4 induces a conformational change that facilitates contact with the chemokine receptors of the CXCR4 or CCR5 type (summarized in reference 61). This may facilitate successful evasion from neutralizing immune responses through protection of conserved viral epitopes necessary for essential receptor inter-actions.
In line with this view, our results highlight the importance of HVR1 for protection of neutralizing epitopes. Remarkably, cross-neutralization of wild-type HCV (Jc1) by patient-derived antibodies was limited and much more efficient when HVR1 was lacking. These results suggest that all these patients had
successfully induced strong cross-neutralizing antibodies
[image:11.585.70.246.69.153.2]against epitopes different from HVR1. However, these anti-bodies are relatively ineffective against viruses possessing HVR1, possibly because key epitopes are only poorly accessi-ble. Congruent with this interpretation, neutralization and vi-rus precipitation through a series of monoclonal antibodies recognizing defined viral epitopes of the E1/E2 complex were much enhanced for viruses lacking HVR1. Notably, all of these antibodies interfere with E2 binding to CD81 (CBH5, CBH7, CBH23, AP33, HC1, HC11 [32, 37]), thus reinforcing the idea that removal of HVR1 has indeed exposed the CD81 binding site and highlighting the notion that interference with this receptor interaction is a crucial mechanism for virus neutral-ization. It is worth mentioning here that the adaptive mutation
FIG. 11. Deletion of HVR1 causes increased exposure of con-served epitopes. Equal numbers of viruses (normalized to HCV RNA) were incubated with 1g of the specified antibodies. Subsequently, immune complexes were precipitated using protein G-coated beads, and associated HCV RNA was quantified by qRT-PCR. Data are representative of the results of two independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
in assembly and release of virus particles with optimal compo-sition, thus conferring high infectivity on viruses with low den-sity. Second, it influences the HCV membrane fusion process. Third, this domain physically protects a conserved neutralizing epitope, the viral CD81 binding site. Together, these charac-teristics of HVR1 are likely crucial for establishment and maintenance of a chronic infection. On the other hand, this knowledge should be useful for further dissection of the mech-anisms of HCV cell entry and for devising vaccine formulations that induce potent neutralizing antibody responses.
ACKNOWLEDGMENTS
We are grateful to Takaji Wakita and Jens Bukh for JFH1 and J6CF isolates, respectively, to Charles Rice for Huh-7.5 cells and the E9E10 antibodies, to Shoshana Levy for providing the hCD81-LEL-GST ex-pression construct, and to Arvind Patel for the gift of the E1-specific polyclonal antiserum. We thank Genentech for providing AP33 anti-bodies. We also thank all members of the Department of Experimental Virology for helpful suggestions and discussions and Thomas von Hahn for critical reading of the manuscript.
This work was supported by an Emmy Noether Fellowship (PI 734/ 1-1) from the Deutsche Forschungsgemeinschaft (DFG), by grants from the Initiative and Networking Fund of the Helmholtz Association within the Helmholtz Alliance on Immunotherapy of Cancer SO-024 and HA-202 to T.P., by PHS grant HL079381 to S.K.H.F., and a grant from the Agence Nationale de Recherche contre le SIDA et les he ´pa-tites virales (to E.-I.P.). D.B. was funded by a stipend from the inter-national research training group 1273 (IRTG 1273) provided by the DFG.
REFERENCES
1.Alter, M. J.2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol.13:2436–2441.
2.Andre, P., F. Komurian-Pradel, S. Deforges, M. Perret, J. L. Berland, M. Sodoyer, S. Pol, C. Brechot, G. Paranhos-Baccala, and V. Lotteau.2002. Characterization of low- and very-low-density hepatitis C virus RNA-con-taining particles. J. Virol.76:6919–6928.
3.Bartosch, B., J. Dubuisson, and F. L. Cosset.2003. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein com-plexes. J. Exp. Med.197:633–642.
4.Bartosch, B., G. Verney, M. Dreux, P. Donot, Y. Morice, F. Penin, J. M. Pawlotsky, D. Lavillette, and F. L. Cosset. 2005. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol.79:8217– 8229.
5.Bartosch, B., A. Vitelli, C. Granier, C. Goujon, J. Dubuisson, S. Pascale, E. Scarselli, R. Cortese, A. Nicosia, and F. L. Cosset.2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tet-raspanin and the SR-B1 scavenger receptor. J. Biol. Chem.278:41624–41630. 6.Blight, K. J., J. A. McKeating, and C. M. Rice.2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Vi-rol.76:13001–13014.
7.Brown, R. S.2005. Hepatitis C and liver transplantation. Nature436:973– 978.
8.Chang, K. S., J. Jiang, Z. Cai, and G. Luo.2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol.81:13783–13793.
9.Clayton, R. F., A. Owsianka, J. Aitken, S. Graham, D. Bhella, and A. H. Patel.2002. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol.76:7672–7682.
the viral quasispecies. Science288:339–344.
14.Farci, P., A. Shimoda, D. Wong, T. Cabezon, D. DeGioannis, A. Strazzera, Y. Shimizu, M. Shapiro, H. J. Alter, and R. H. Purcell.1996. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. U. S. A.93:15394–15399.
15.Farci, P., R. Strazzera, H. J. Alter, S. Farci, D. DeGioannis, A. Coiana, G. Peddis, F. Usai, G. Serra, L. Chessa, G. Diaz, A. Balestrieri, and R. H. Purcell.2002. Early changes in hepatitis C viral quasispecies during inter-feron therapy predict the therapeutic outcome. Proc. Natl. Acad. Sci. U. S. A.99:3081–3086.
16.Flint, M., T. von Hahn, J. Zhang, M. Farquhar, C. T. Jones, P. Balfe, C. M. Rice, and J. A. McKeating.2006. Diverse CD81 proteins support hepatitis C virus infection. J. Virol.80:11331–11342.
17.Forns, X., R. Thimme, S. Govindarajan, S. U. Emerson, R. H. Purcell, F. V. Chisari, and J. Bukh.2000. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl. Acad. Sci. U. S. A.97:13318–13323.
18.Friebe, P., J. Boudet, J. P. Simorre, and R. Bartenschlager.2005. Kissing-loop interaction in the 3⬘end of the hepatitis C virus genome essential for RNA replication. J. Virol.79:380–392.
19.Gastaminza, P., G. Cheng, S. Wieland, J. Zhong, W. Liao, and F. V. Chisari. 2008. Cellular determinants of hepatitis C virus assembly, maturation, deg-radation, and secretion. J. Virol.82:2120–2129.
20.Gottwein, J. M., T. K. Scheel, T. B. Jensen, J. B. Lademann, J. C. Prentoe, M. L. Knudsen, A. M. Hoegh, and J. Bukh.2009. Development and char-acterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology49:364–377.
21.Hadlock, K. G., R. E. Lanford, S. Perkins, J. Rowe, Q. Yang, S. Levy, P. Pileri, S. Abrignani, and S. K. Foung.2000. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol.74:10407–10416.
22.Haid, S., T. Pietschmann, and E. I. Pecheur. 2009. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J. Biol. Chem.284:17657–17667. 23.Hijikata, M., Y. K. Shimizu, H. Kato, A. Iwamoto, J. W. Shih, H. J. Alter, R. H. Purcell, and H. Yoshikura.1993. Equilibrium centrifugation studies of hepatitis C virus: evidence for circulating immune complexes. J. Virol.67: 1953–1958.
24.Hoofnagle, J. H.2002. Course and outcome of hepatitis C. Hepatology 36:S21–S29.
25.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating.2003. Hepatitis C virus glycoproteins mediate pH-depen-dent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A.100:7271–7276.
26.Huang, H., F. Sun, D. M. Owen, W. Li, Y. Chen, M. Gale, Jr., and J. Ye.2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848–5853.
27.Ka¨rber, G.1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Archiv Exp. Pathol. Pharmakol.162:480–487.
28.Kato, N., Y. Ootsuyama, H. Sekiya, S. Ohkoshi, T. Nakazawa, M. Hijikata, and K. Shimotohno.1994. Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J. Virol.68:4776–4784. 29.Kato, N., H. Sekiya, Y. Ootsuyama, T. Nakazawa, M. Hijikata, S. Ohkoshi,
and K. Shimotohno.1993. Humoral immune response to hypervariable re-gion 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. J. Virol.67:3923–3930.
30.Keck, Z. Y., T. K. Li, J. Xia, B. Bartosch, F. L. Cosset, J. Dubuisson, and S. K. Foung.2005. Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. J. Virol.79:13199–13208.
31.Keck, Z. Y., T. K. Li, J. Xia, M. Gal-Tanamy, O. Olson, S. H. Li, A. H. Patel, J. K. Ball, S. M. Lemon, and S. K. Foung.2008. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutral-izing human monoclonal antibodies. J. Virol.82:6061–6066.
32.Keck, Z. Y., D. B. Op, K. G. Hadlock, J. Xia, T. K. Li, J. Dubuisson, and S. K.
on November 8, 2019 by guest
http://jvi.asm.org/
Foung.2004. Hepatitis C virus E2 has three immunogenic domains contain-ing conformational epitopes with distinct properties and biological functions. J. Virol.78:9224–9232.
33.Keck, Z. Y., J. Xia, Z. Cai, T. K. Li, A. M. Owsianka, A. H. Patel, G. Luo, and S. K. Foung.2007. Immunogenic and functional organization of hepatitis C virus (HCV) glycoprotein E2 on infectious HCV virions. J. Virol.81:1043– 1047.
34.Koutsoudakis, G., A. Kaul, E. Steinmann, S. Kallis, V. Lohmann, T. Pi-etschmann, and R. Bartenschlager.2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 80:5308–5320.
35.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice.2005. Complete replication of hepatitis C virus in cell culture. Science309:623–626.
36.Meunier, J. C., R. S. Russell, R. E. Engle, K. N. Faulk, R. H. Purcell, and S. U. Emerson.2008. Apolipoprotein c1 association with hepatitis C virus. J. Virol.82:9647–9656.
37.Owsianka, A., R. F. Clayton, L. D. Loomis-Price, J. A. McKeating, and A. H. Patel.2001. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol.82:1877–1883.
38.Owsianka, A., A. W. Tarr, V. S. Juttla, D. Lavillette, B. Bartosch, F. L. Cosset, J. K. Ball, and A. H. Patel.2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glyco-protein. J. Virol.79:11095–11104.
39.Penin, F., C. Combet, G. Germanidis, P. O. Frainais, G. Deleage, and J. M. Pawlotsky.2001. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol.75:5703–5710.
40.Pietschmann, T., A. Kaul, G. Koutsoudakis, A. Shavinskaya, S. Kallis, E. Steinmann, K. Abid, F. Negro, M. Dreux, F. L. Cosset, and R. Barten-schlager.2006. Construction and characterization of infectious intrageno-typic and intergenointrageno-typic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A.103:7408–7413.
41.Pileri, P., Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, A. J. Weiner, M. Houghton, D. Rosa, G. Grandi, and S. Abrignani.1998. Binding of hepatitis C virus to CD81. Science282:938–941.
42.Ploss, A., M. J. Evans, V. A. Gaysinskaya, M. Panis, H. You, Y. P. de Jong, and C. M. Rice.2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature457:882–886.
43.Ray, S. C., Y. M. Wang, O. Laeyendecker, J. R. Ticehurst, S. A. Villano, and D. L. Thomas.1999. Acute hepatitis C virus structural gene sequences as predictors of persistent viremia: hypervariable region 1 as a decoy. J. Virol. 73:2938–2946.
44.Roccasecca, R., H. Ansuini, A. Vitelli, A. Meola, E. Scarselli, S. Acali, M. Pezzanera, B. B. Ercole, J. McKeating, A. Yagnik, A. Lahm, A. Tramontano, R. Cortese, and A. Nicosia.2003. Binding of the hepatitis C virus E2 glyco-protein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol.77:1856–1867.
45.Scarselli, E., H. Ansuini, R. Cerino, R. M. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese, and A. Vitelli.2002. The human scaven-ger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J.21:5017–5025.
46.Seeff, L. B.2002. Natural history of chronic hepatitis C. Hepatology36:S35– S46.
47.Semmo, N., M. Lucas, G. Krashias, G. Lauer, H. Chapel, and P. Klenerman. 2006. Maintenance of HCV-specific T-cell responses in antibody-deficient patients a decade after early therapy. Blood107:4570–4571.
48.Simmonds, P., J. Bukh, C. Combet, G. Deleage, N. Enomoto, S. Feinstone, P. Halfon, G. Inchauspe, C. Kuiken, G. Maertens, M. Mizokami, D. G. Mur-phy, H. Okamoto, J. M. Pawlotsky, F. Penin, E. Sablon, I. Shin, L. J. Stuyver,
H. J. Thiel, S. Viazov, A. J. Weiner, and A. Widell.2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepa-tology42:962–973.
49.Spearman, C. 1908. The method of “right and wrong cases” (“constant stimuli”) without Gauss’s formulae. Br. J. Psychol.2:227–242.
50.Steinmann, E., F. Penin, S. Kallis, A. H. Patel, R. Bartenschlager, and T. Pietschmann.2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog.3:e103.
51.Taniguchi, S., H. Okamoto, M. Sakamoto, M. Kojima, F. Tsuda, T. Tanaka, E. Munekata, E. E. Muchmore, D. A. Peterson, and S. Mishiro.1993. A structurally flexible and antigenically variable N-terminal domain of the hepatitis C virus E2/NS1 protein: implication for an escape from antibody. Virology195:297–301.
52.Tarr, A. W., A. M. Owsianka, J. M. Timms, C. P. McClure, R. J. Brown, T. P. Hickling, T. Pietschmann, R. Bartenschlager, A. H. Patel, and J. K. Ball. 2006. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology43:592–601. 53.Thimme, R., C. Neumann-Haefelin, T. Boettler, and H. E. Blum.2008.
Adaptive immune responses to hepatitis C virus: from viral immunobiology to a vaccine. Biol. Chem.389:457–467.
54.Thomssen, R., S. Bonk, C. Propfe, K. H. Heermann, H. G. Kochel, and A. Uy. 1992. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. Berl.181:293–300.
55.Thomssen, R., S. Bonk, and A. Thiele.1993. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. Berl.182:329–334. 56.Voisset, C., N. Callens, E. Blanchard, D. B. Op, J. Dubuisson, and N.
Vu-Dac.2005. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem.280:7793–7799. 57.von Hahn, T., B. D. Lindenbach, A. Boullier, O. Quehenberger, M. Paulson, C. M. Rice, and J. A. McKeating.2006. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 43:932–942.
58.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang.2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med.11:791–796.
59.Weiner, A. J., H. M. Geysen, C. Christopherson, J. E. Hall, T. J. Mason, G. Saracco, F. Bonino, K. Crawford, C. D. Marion, K. A. Crawford, et al.1992. Evidence for immune selection of hepatitis C virus (HCV) putative envelope glycoprotein variants: potential role in chronic HCV infections. Proc. Natl. Acad. Sci. U. S. A.89:3468–3472.
60.Witteveldt, J., M. J. Evans, J. Bitzegeio, G. Koutsoudakis, A. M. Owsianka, A. G. Angus, Z. Y. Keck, S. K. Foung, T. Pietschmann, C. M. Rice, and A. H. Patel.2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmis-sion in hepatoma cells. J. Gen. Virol.90:48–58.
61.Wyatt, R., and J. Sodroski.1998. The HIV-1 envelope glycoproteins: fuso-gens, antifuso-gens, and immunogens. Science280:1884–1888.
62.Zeisel, M. B., F. L. Cosset, and T. F. Baumert.2008. Host neutralizing responses and pathogenesis of hepatitis C virus infection. Hepatology48: 299–307.
63.Zeisel, M. B., G. Koutsoudakis, E. K. Schnober, A. Haberstroh, H. E. Blum, F. L. Cosset, T. Wakita, D. Jaeck, M. Doffoel, C. Royer, E. Soulier, E. Schvoerer, C. Schuster, F. Stoll-Keller, R. Bartenschlager, T. Pietschmann, H. Barth, and T. F. Baumert.2007. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology46:1722–1731.
64.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A.102:9294– 9299.