Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Identification of Novel MicroRNA-Like Molecules Generated from
Herpesvirus and Host tRNA Transcripts
䌤
Tiffany A. Reese,
1Jing Xia,
2L. Steven Johnson,
1Xiang Zhou,
2Weixiong Zhang,
2,3* and Herbert W. Virgin
1,4*
Department of Pathology and Immunology,1Department of Computer Science and Engineering,2Department of
Genetics,3and Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Disease Research,4
Washington University in St. Louis, St. Louis, Missouri
Received 1 April 2010/Accepted 8 July 2010
We applied deep sequencing technology to small RNA fractions from cells lytically infected with murine
gammaherpesvirus 68 (␥HV68) in order to define in detail small RNAs generated from a cluster of
tRNA-related polycistronic structures located at the left end of the viral genome. We detected 10 new candidate microRNAs (miRNAs), six of which were confirmed by Northern blot analysis, leaving four as provisional. In addition, we determined that previously identified and annotated viral miRNA molecules were not necessarily represented as the most abundant sequence originating from a transcript. Based on these new small RNAs and
previously reported␥HV68 miRNAs, we were able to further describe and annotate the distinctive ␥HV68
tRNA-miRNA structures. We used this deep sequencing data and computational analysis to identify similar structures in the mouse genome and validated that these host structures also give rise to small RNAs. This reveals a possible convergent usage of tRNA/polymerase III (pol III) transcripts to generate small RNAs from both mammalian and viral genomes.
Data from deep sequencing technologies suggest that the coding complexity of many genomes, from virus and plant to mouse and human genomes, has been underestimated. Re-searchers have identified a vast array of different types of noncoding RNAs (ncRNAs) emanating from these different genomes. Importantly, some viruses, with their compact ge-nomes and tractable genetics, provide a valuable tool for un-derstanding general principles of eukaryotic genomes and a functional model for defining novel types of ncRNAs as well as their mechanisms of action.
The transcriptional complexity of eukaryotic genomes is ex-emplified by the increase in the number and types of functions of small ncRNAs. These RNAs can be subdivided to include not only microRNAs (miRNAs) (21 to 25 nucleotides [nt]), small interfering RNAs (siRNAs) (21 to 25 nt), and Piwi-interacting RNAs (piRNAs) (24 to 31 nt) (11, 14) but also the recently identified tRNA-derived RNA fragments (tRFs) (18 to 25 nt) (9). In addition to tRFs, there are a few other exam-ples of small RNAs that are associated with tRNAs and poly-merase III (Pol III). These include 19-nt Dicer-dependent RNA fragments derived from tRNAs (4), human small RNAs interspersed among Alu repeats and transcribed by Pol III (2), and tRNA-miRNA polycistrons from the murine gammaher-pesvirus 68 (␥HV68) (20).
Many DNA viruses, especially herpesviruses, encode miRNAs
and other ncRNAs that regulate viral replication and host re-sponses. For example, the adenovirus-associated (VA) RNA is an
⬃165-nt ncRNA that inhibits the antiviral effects of PKR-me-diated translation inhibition (12), the Epstein-Barr virus (EBV)-encoded RNAs (EBERs) disrupt interferon-induced apoptosis of host cells (22), and herpesvirus saimiri encodes seven small nuclear RNAs of the Sm class (HSURs) which are believed to play a role in T-cell activation (5). Additionally, functionally conserved miRNAs from different herpesviruses, including human cytomegalovirus (HCMV), EBV, and Kapo-si’s sarcoma herpesvirus (KSHV), help these viruses evade immune recognition by targeting a host transcript, MICB, a ligand of natural killer (NK) cells which controls herpesvirus replication (16, 23). These different ncRNAs may participate in posttranscriptional gene silencing (by mRNA degradation or translational regulation) or transcriptional gene silencing by acting on chromatin function and accessibility. Together, these ncRNAs constitute new classes of viral genes that may play important roles in viral infection.
␥HV68 is a murine gammaherpesvirus genetically related to EBV and KSHV. Similar to these other herpesviruses,
␥HV68 is known to encode at least 10 miRNAs with as-yet-undefined function (20). These were identified by cDNA cloning, with three of 10 confirmed by Northern blot analysis in infected B-cell lines (20) and nine of 10 detected by reverse transcription (RT)-PCR from infected fibroblasts (6). However, distinct from EBV and KSHV,␥HV68 gen-erates its primary miRNA transcripts (pri-miRNA) via Pol III, instead of Pol II, as part of larger transcripts that in-clude tRNA-like elements directly upstream of the predicted miR-NAs (3, 6, 20). Moreover, these miRmiR-NAs are generated via a noncanonical processing pathway that utilizes tRNase Z instead of Drosha to liberate the pre-miRNA hairpin from the tRNA. The subsequent pre-miRNA is thought to then
* Corresponding author. Mailing address for H. W. Virgin: Depart-ment of Pathology and Immunology, Washington University School of Medicine, 660 S. Euclid Ave., Box 8118, St. Louis, MO 63110. Phone: (314) 362-9223. Fax: (314) 362-4096. E-mail: [email protected]. Mailing address for W. Zhang: Department of Computer Science and Engineering, Washington University, One Brookings Drive, Campus Box 1045, St. Louis, MO 63130. Phone: (314) 935-8788. Fax: (314) 935-7302. E-mail: [email protected].
䌤Published ahead of print on 21 July 2010.
10344
on November 8, 2019 by guest
http://jvi.asm.org/
be processed by Dicer, similar to other miRNAs, to generate the mature miRNA (1).
To further expand our understanding of ␥HV68-expressed small RNAs, we analyzed small RNA libraries by Illumina sequencing and mapped the small RNAs to the first 7.5 kb of the genome. In the initial identification of the ␥HV68 miRNAs, tRNA-miRNA polycistrons from this region were predicted with each of the eight tRNAs followed by two stem-loop hairpins (20). The 10 previously annotated miRNAs mapped to these predicted hairpin structures from tRNA-like transcripts, but many hairpins were without an identified miRNA. From our sequencing data, we demonstrate that most of these hairpins do indeed have candidate miRNAs emanat-ing from them. Also, a tRNA with no previously identified miRNA, tRNA7, has a candidate miRNA following it. More-over, in some cases the most frequent read from a region of a previously described viral miRNA was shifted by a few nucle-otides from the published miRNA sequence. We detected an approximately 22-nt small RNA for six of the 10 candidate miRNAs by Northern blot analysis. Further mining of the sequencing data also revealed mouse transcripts with tRNAs followed by novel small RNAs that could correspond to either miRNAs or putative tRFs recently characterized from prostate cancer cell lines (9), thus suggesting a possible coevolution of virus and host resulting in similar mechanisms of expression of small ncRNAs.
MATERIALS AND METHODS
Animals.C57BL/6 mice were obtained from Jackson Laboratories and bred at Washington University, St. Louis, MO. Stat1KO mice were obtained from Rob-ert Schreiber (13). All mice were housed and bred in a specific-pathogen-free barrier facility in accordance with federal and institutional guidelines. Mice were infected intraperitoneally between 8 and 10 weeks of age with 1⫻106PFU of ␥HV68 and euthanized 2 days later. Peritoneal cells were collected for RNA isolation.
Cells.NIH 3T12 cells (ATCC CRL-164) and Vero cells (ATCC CCL-81) were maintained in DMEM5 (Dulbecco’s modified Eagle’s medium with 5% fetal calf serum [FCS], 2 mML-glutamine). Bone marrow-derived macrophages were
generated as follows. Bone marrow was flushed from femurs of C57BL/6 mice and incubated in BM20 (10% FCS, 20% L-cell-conditioned medium derived from a macrophage colony-stimulating factor [M-CSF]-producing fibroblast cell line, 5% horse serum, 2 mML-glutamine, and 1 mM sodium pyruvate in DMEM) for the first 7 days ofin vitroculture. On day 7, adherent cells were scraped and seeded in BM10 medium (10% FCS, 10% L-cell-conditioned medium, 5% horse serum in DMEM). On day 10, cells were infected and maintained in BM10 medium.
Virus infections.␥HV68 WUMS (ATCC VR1465) was passaged and titered on NIH 3T12 cells (19). NIH 3T12 cells and bone marrow-derived macrophages were infected at a multiplicity of infection (MOI) of 10 PFU for 1 h at 37°C and 5% CO2, washed three times with medium, and replated in media. Subsequent RNA extractions on mock and infected cells were done 18 h later.
RNA preparations. Total RNA was prepared using the mirVana miRNA isolation kit (Ambion) according to the manufacturer’s instructions for isolation of total RNA. RNA was treated with DNase I (Ambion) and run on a 15% denaturing acrylamide gel (Invitrogen) followed by staining with ethidium bro-mide to check for RNA integrity.
Small RNA preparations for sequencing.Total RNA libraries for sequencing from mock- or␥HV68-infected cells were prepared using the small RNA sample prep kit (Illumina) by following theSmall RNA v.1.5 Sample Preparation Guide. Twenty micrograms of total RNA was used as input, and small RNA was size selected by running total RNA on a 15% denaturing polyacrylamide gel and cutting out the RNA corresponding to 18 to 30 nt.
Initial processing of sequencing libraries.The raw sequence reads were first processed to remove reads with no 3⬘sequencing adaptor, of low quality, or shorter than 18 nt. The sequence adaptor trimming was done by an in-house program that recursively searches for the longest subsequence of the adaptor appearing within a sequence read. If a raw sequence read did not have a subsequence of the adaptor that was longer than 4 nt, it was considered to have no adaptor. The adaptor-trimmed sequence reads that map to the␥HV68 and mouse genomes with zero mismatches were retained for further analysis.
[image:2.585.47.540.82.290.2]Identification of candidate miRNAs.We revised and extended an miRNA identification method we developed previously (24) to find novel miRNAs in ␥HV68 and mouse genomes, which may have more mismatches and bulges in miRNA precursor foldback structures than regular miRNAs. We first mapped the adaptor-trimmed sequence reads to the genome using Bowtie (8), merged neighboring loci if they shared overlapping reads, and then examined the folding structures of the (merged) loci. Since the average length of an miRNA precursor is⬃80 nt, we took 100 nt as the length of putative pre-miRNAs in our analysis. At each genomic locus to be analyzed, a series of DNA sequence segments covering the sequence reads were extracted for secondary structure analysis. The starting sequence segment extended 80 nt upstream of the sequence reads, and subsequent segments were extracted by a sliding window of 100 nt, with an TABLE 1. ␥HV68 miRNAsa
miRNA Sequence No. of
reads Associated tRNA Confirmed by:
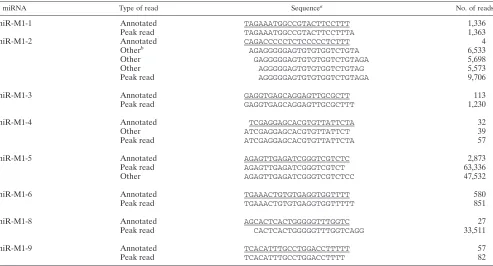
miR-M1-1 UAGAAAUGGCCGUACUUCCUUU 1,336 tRNA1 Pfeffer et al., Diebel et al.
miR-M1-2 CAGACCCCCUCUCCCCCUCUUU 4 tRNA2 Pfeffer et al., Diebel et al.
miR-M1-3 GAGGUGAGCAGGAGUUGCGCUU 113 tRNA2 Pfeffer et al., Diebel et al.
miR-M1-4 UCGAGGAGCACGUGUUAUUCUA 32 tRNA3 Pfeffer et al., Diebel et al.
miR-M1-5 AGAGUUGAGAUCGGGUCGUCUC 2,873 tRNA4 Pfeffer et al., Diebel et al.
miR-M1-6 UGAAACUGUGUGAGGUGGUUUU 580 tRNA4 Pfeffer et al., Diebel et al.
miR-M1-7-5p AAAGGUGGAGGUGCGGUAACCU 51 tRNA5 Pfeffer et al., Diebel et al.
miR-M1-7-3p GAUAUCGCGCCCACCUUUAUU 236 tRNA5 Pfeffer et al., Diebel et al.
miR-M1-8 AGCACUCACUGGGGGUUUGGUC 27 tRNA6 Pfeffer et al., Diebel et al.
miR-M1-9 UCACAUUUGCCUGGACCUUUUU 57 tRNA8 Pfeffer et al., Diebel et al.
miR-M1-10-5p UAAGAACCCUCAGUGCAAUCACU 79 tRNA1 N, RT-PCR
miR-M1-10-3p UGAUUACACGGAAGGUUCUUUU 122 tRNA1 N, RT-PCR
miR-M1-11 UUUGGUGUGGGAGUCCUACCCCUU 85 tRNA5 N, RT-PCR
miR-M1-12 UGGGAAGAGUCUGUUGAGUGGC 3,674 tRNA6 N, RT-PCR
miR-M1-13a-5p CCCGUUCUGGAUGCUGUGGGAC 709 tRNA7 N, RT-PCR
miR-M1-13a-3p UGCUACAGCGUGCAGAACGUUU 100 tRNA7 N, RT-PCR
miR-M1-13b-5p UGUGGUGGGACUCUGCAGA 1 tRNA7 RT-PCR
miR-M1-13b-3p UUGGCAGUUCUGCAGCAGUCAGCG 1 tRNA7 RT-PCR
miR-M1-14-5p ACCCGCGUGGCCGGAGUGUUU 1 tRNA8 RT-PCR
miR-M1-14-3p CCCUCUAACCCACCUUCCGG 1 tRNA8 RT-PCR
aBold indicates novel candidate miRNA. N, Northern blot analysis.
on November 8, 2019 by guest
http://jvi.asm.org/
increment of 1 nt, until the window reached 80 nt downstream of the sequence reads. Each of these 80-nt segments was folded by the RNA-fold program (7). Segments lacking stems of at least 18 nt and segments lacking sequence reads that mapped to any of their stems were excluded. A representative segment was chosen from those that have the same or a similar folding structure. The top five folding structures with a free energy no greater than⫺18 kCal/mol were further visually inspected. We retained those segments that formed a typical hairpin foldback structure with up to 5 mismatches on one stem region. We examined multiple folding structures in order to identify miRNA candidates, since a com-putationally predicted optimal RNA folding structure may not be necessarily functionally relevant. We extracted a longer region that included a candidate miRNA and its neighboring tRNA and folded the region using the RNA-fold program.
Northern blot analysis.Northern blotting for miRNAs was done using the enhanced method for detection (18). Twenty micrograms of total RNA was run on a 15% denaturing acrylamide gel (Invitrogen) from mock-infected 3T12 cells, ␥HV68-infected 3T12 cells, and, in some cases, Vero cells. Decade markers were run on each gel to determine nucleotide length (Ambion). Blots were probed with RNA probes generated usingin vitrotranscription (mirVana miRNA probe construction kit [Ambion]). Probe templates were as follows (boldface indicates a complementary sequence to the T7 promoter primer): miR-M1-10-5p, TAAG AACCCTCAGTGCAATCACTCCTGTCTC; miR-M1-10-3p, TGATTACACG GAAGGTTCTTTTCCTGTCTC; miR-M1-11, TTTGGTGTGGGAGTCCTAC CCCTTCCTGTCTC; miR-M1-12, TGGGAAGAGTCTGTTGAGTGGCCCTG TCTC; miR-M1-13a-5p, CCCGTTCTGGATGCTGTGGGACACCTGTCTC;
13a-3p, TGCTACAGCGTGCAGAACGTTTCCTGTCTC; miR-M1-13b-5p, TGTGGTGGGACTCTGCAGACCTGTCTC; miR-M1-13b-3p, TTGG CAGTTCTGCAGCAGTCAGCGCCTGTCTC; miR-M1-14-5p, ACCCGCGTG GCCGGAGTGTTTCCTGTCTC; miR-M1-14-3p, CCCTCTAACCCACCTTC CGGCCTGTCTC; tRNA 717/209 miR, TTGGGACTGGTACTCCTTTACCTG TCTC; tRNA 84 miR, TCTGGACACATGTGGCTTTTCCTGTCTC; tRNA 717 miR hairpin, TTGGGACTGGTACTCCTTTATTAGCCGGGTGTCCT GTCT; tRNA 209 miR hairpin, TTGGGACTGGTACTCCTTTATTTGCAT CATTGGCTAATCTAGTCATCTCCCAATGTTGCTAGTTTTAAAAATG CCTGTCT; tRNA 84 miR hairpin, TCTGGACACATGTGGCTTTTTGTTT CTATTTTGGGACACAAAAGGTCAGGTCCGTCTGGCCTGTCT.
[image:3.585.48.541.81.206.2]RT-PCR for miRNAs.RT-PCR detection of miRNAs was performed using the miScript PCR system from Qiagen. Primer assays for each novel miRNA were designed using the sequences in Table 1. One microgram of total RNA was reverse transcribed using Qiagen miScript reverse transcriptase mix, which in-cludes a blend of poly(A) polymerase, reverse transcriptase, oligo(dT) primers, and random primers. Expression of miRNAs was normalized to the small nuclear RNA (snRNA) RNU6B. Products from mock- and␥HV68-infected samples were analyzed both by melting curve and by gel electrophoresis. An HCMV miRNA miR-US33-5p was used as the negative control and the mouse miRNA-16 was used as the positive control for miRNA size when products were run on a gel. Following quantitative PCR, products were run on a 3% NuSieve GTG agarose gel to determine the relative size of the amplified products com-pared with mouse miRNA-16 and the DNA marker V (Roche).
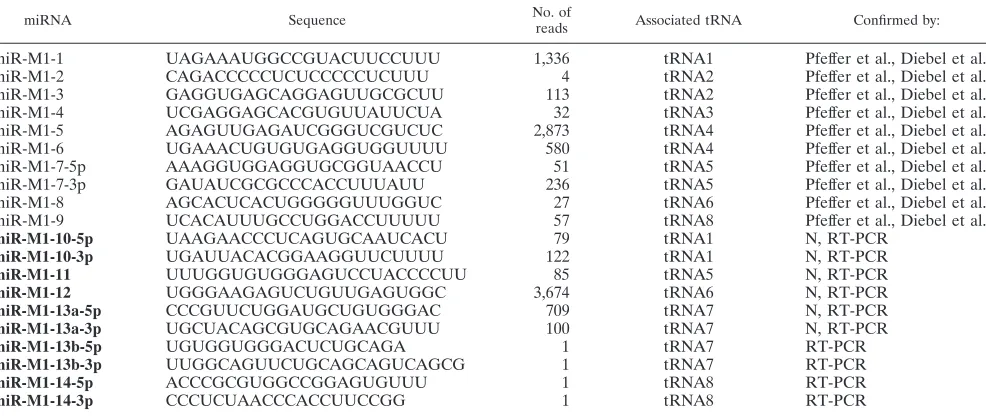
[image:3.585.93.492.485.661.2]FIG. 1. Identification of novel candidate viral miRNAs in␥HV68-infected fibroblasts. (A) Size-selected small RNA (18 to 30 nt) from 3T12 mouse fibroblasts infected with␥HV68 was Solexa sequenced. The resulting reads were mapped to the␥HV68 genome, and the first 7.5 kb of the genome is shown. The top line represents the position in the genome, and the annotated positive- and negative-strand ORFs are shown as green arrows. Yellow arrows represent annotated tRNAs on the positive strand, and blue arrows represent annotated miRNAs on the positive strand. Below that, the numbers of positive and negative strand reads per nucleotide are plotted on a log2scale. Thus, a peak represents all the reads in the region that overlap a particular nucleotide. Red arrows point to the position at which novel candidate miRNAs map. Data from three independent experiments were combined for the final analysis.
TABLE 2. Statistics of 3 small RNA sequencing libraries
Category of reads
No. of reads
Description ␥⌯V68-1 ␥⌯V68-2 ␥⌯V68-3
Total 6,411,884 10,722,962 13,792,659 Total raw sequencing reads No adaptor 3,498,979 3,436,380 4,568,762 Reads with no adaptor
Low quality 1,874 171,139 15,088 Reads with middle Ns
Short 1,040,150 1,486,925 756,171 Reads⬍18 nt after adapter trimmed Total qualified 1,870,881 5,628,518 8,452,638 Total high-quality readsⱖ18 nt
Total mapped to␥HV68 6,176 48,289 208,530 Total high-quality reads mapped to␥HV68 genome Total mapped to mouse 1,229,450 4,295,302 6,329,431 Total high-quality reads mapped to mouse genome Unique qualified 188,558 251,101 310,202 Unique high-quality readsⱖ18 nt
Unique mapped to␥HV68 997 3,388 9,461 Unique high-quality reads mapped to␥HV68 genome Unique mapped to mouse 28,327 94,931 83,660 Unique high-quality reads mapped to mouse genome
on November 8, 2019 by guest
http://jvi.asm.org/
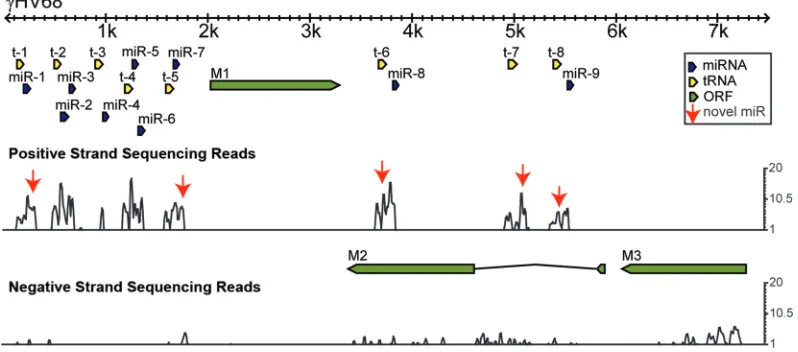
FIG. 2. Predicted structure of novel candidate␥HV68 miRNAs. Predicted secondary structures of tRNA-miRNA transcripts, with annotated miRNAs and peak reads depicted in red. Novel candidate miRNAs are boxed. Novel and previously annotated miRNAs were analyzed for peak read. #, annotated miRNAs for which the most abundant read is shifted slightly off and does not correspond to annotation. #*, an annotation that differs significantly from the peak read such that the peak read is on the opposite arm from miRNA annotation. Graphs depict read frequency across the nucleotide position of the structures for each polycistron. For tRNA7 and tRNA8, boxed regions depict areas with possible alternate folded structures.
on November 8, 2019 by guest
http://jvi.asm.org/
RESULTS
Identification of novel miRNAs from␥HV68-infected cells.
To identify novel ␥HV68 miRNAs, we performed Illumina deep sequencing on size-selected (⬍30-nt) RNA from lytically infected fibroblasts. Total RNA was harvested after 18 h of infection of 3T12 cells, and small RNA libraries were prepared for sequencing. We obtained more than 30.9 million raw se-quence reads from three replicate small RNA libraries, among which 262,995 mapped to the␥HV68 genome and 11,854,183 mapped to the mouse genome. The reads that mapped to the genomes with no mismatches were used in the analysis pre-sented here. A summary of the sequencing data from three independent experiments is given in Table 2. The majority of the reads that mapped to the␥HV68 genome were 21 nt or 23 nt long, while the ones that mapped to the mouse genome were 22 nt or 23 nt long (data not shown). Figure 1 shows the reads that mapped to the first 7.5 kb of the ␥HV68 genome, with novel miRNA peaks indicated by red arrows.
After mapping reads to the␥HV68 genome, we found five novel sequencing peaks directly following the␥HV68 tRNAs, similar but distinct from the 10 known␥HV68 miRNAs (Fig. 1). These novel sequences from the positive strand all mapped to the already-predicted tRNA-miRNA secondary structures (Fig. 2) (20). The few reads from the negative strand are of unknown significance and were not further evaluated. Graph-ical displays of read frequencies across the tRNA-miRNA transcripts are shown in Fig. 2, and sequences of the novel candidate miRNAs and their read counts are listed in Table 1. Candidate miRNAs shown in Fig. 2 were chosen based on the following criteria: (i) occurrence of sequence reads on the arms
of predicted hairpin structures, (ii) frequencies of peak reads on predicted hairpins, (iii) presence of possible miRNA* se-quence reads, and (iv) presence of possible 2- to 3-nt 3⬘ over-hangs on miRNA/miRNA* duplexes. miRNA* is the sequence from the opposite arm of an miRNA/miRNA* duplex formed by the pre-miRNA hairpin. We found two putative folding structures for tRNA7 and tRNA8 with compatible low folding energies, which yielded alternate possible pre-miRNA struc-tures (Fig. 2). When we examined the reads across the region of tRNA7, we found reads that mapped to both possible stem-loops, although the frequencies for any given read for the miR-M1-13b stem-loop were very low (Table 1). tRNA8 was similar; there were reads across the alternate stem-loop en-compassing miR-M1-14, but their frequency was low. There was one read that occurred 46 times and included the sequence for miR-M1-14-5p; however, that read was 29 nucleotides long (data not shown). We included miR-M1-13b and miR-M1-14 in our subsequent analyses to attempt to confirm them by other methods.
Sequence abundance of previously identified miRNAs.
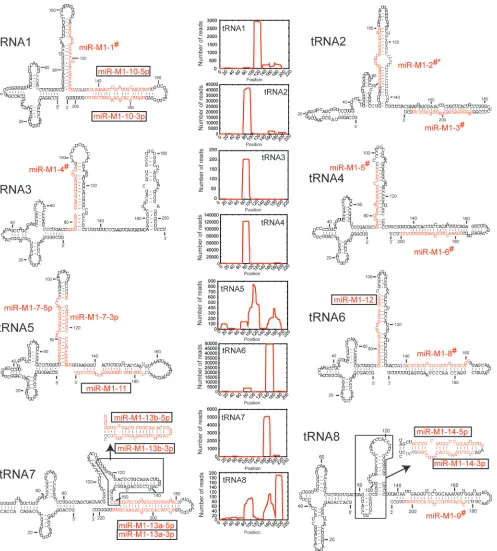
[image:5.585.47.540.80.346.2]In-terestingly, when we examined our sequencing results for the previously identified␥HV68 miRNAs, we found that the most abundant sequence did not always correspond to the annotated miRNA sequence (20). This is indicated in Fig. 2 by the pound symbol (#) next to the name of the miRNA. In all but one case, the peak read for a given stem-loop is shifted off from the annotated miRNA by only 1 to 3 nt (Table 3). For miR-M1-1, -3, -5, -6, and -9, the differences are all at the 3⬘end and may represent differences in processing or could be an artifact of adapter ligation for sequencing. However, miR-M1-2, -4, and
TABLE 3. Discrepancy between viral miRNA annotation and peak read
miRNA Type of read Sequencea No. of reads
miR-M1-1 Annotated TAGAAATGGCCGTACTTCCTTT 1,336
Peak read TAGAAATGGCCGTACTTCCTTTA 1,363
miR-M1-2 Annotated CAGACCCCCTCTCCCCCTCTTT 4
Otherb AGAGGGGGAGTGTGTGGTCTGTA 6,533
Other GAGGGGGAGTGTGTGGTCTGTAGA 5,698
Other AGGGGGAGTGTGTGGTCTGTAG 5,573
Peak read AGGGGGAGTGTGTGGTCTGTAGA 9,706
miR-M1-3 Annotated GAGGTGAGCAGGAGTTGCGCTT 113
Peak read GAGGTGAGCAGGAGTTGCGCTTT 1,230
miR-M1-4 Annotated TCGAGGAGCACGTGTTATTCTA 32
Other ATCGAGGAGCACGTGTTATTCT 39
Peak read ATCGAGGAGCACGTGTTATTCTA 57
miR-M1-5 Annotated AGAGTTGAGATCGGGTCGTCTC 2,873
Peak read AGAGTTGAGATCGGGTCGTCT 63,336
Other AGAGTTGAGATCGGGTCGTCTCC 47,532
miR-M1-6 Annotated TGAAACTGTGTGAGGTGGTTTT 580
Peak read TGAAACTGTGTGAGGTGGTTTTT 851
miR-M1-8 Annotated AGCACTCACTGGGGGTTTGGTC 27
Peak read CACTCACTGGGGGTTTGGTCAGG 33,511
miR-M1-9 Annotated TCACATTTGCCTGGACCTTTTT 57
Peak read TCACATTTGCCTGGACCTTTT 82
a
Underlining indicates annotated, mature miRNA. b
Abundant read close to the miRNA.
on November 8, 2019 by guest
http://jvi.asm.org/
-8 were more substantially different from the previous annota-tion. For miR-M1-4 and miR-M1-8, both showed differences between the peak read and the previous annotation at the 5⬘ end of the miRNA. This could significantly alter the predicted seed sequences or target mRNAs and change conclusions re-garding the function of these two miRNAs. In the case of
miR-M1-2, the peak read illustrated in Fig. 2 is actually on the 5⬘ arm of the stem-loop, while the annotated miRNA is sup-posed to be on the 3⬘ arm of the stem-loop. The miRNA sequence for miR-M1-2 is detected only four times from the sequencing, while the reads from the other arm of the stem-loop are detected at a much higher frequency and are
[image:6.585.43.545.420.666.2]repre-FIG. 3. Detection of candidate viral miRNAs. Northern blot analysis of novel candidate miRNAs from 3T12 cells mock infected (M) or infected with␥HV68 (V). Ethidium bromide staining is shown to indicate RNA loading. Blots are representative of 2 or 3 independent experiments. In many cases, blots were probed for one miRNA and later stripped and reprobed for a different miRNA.
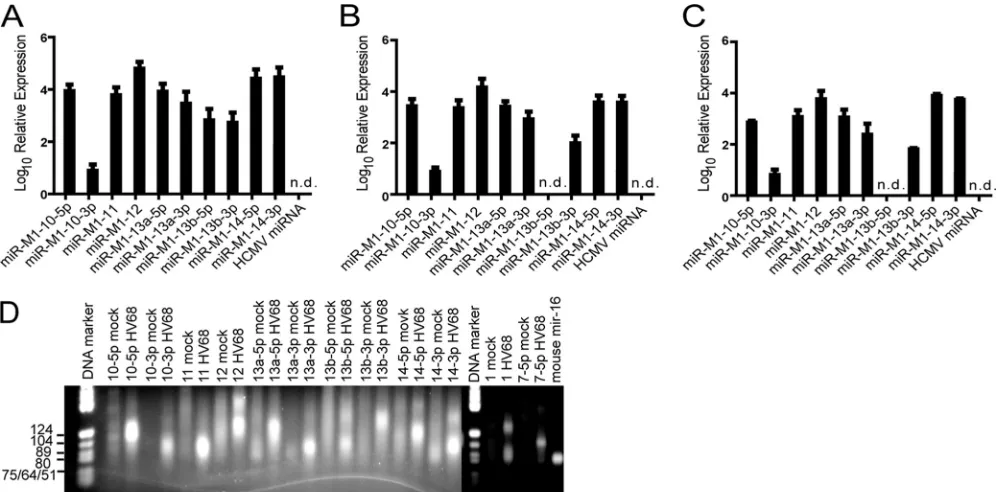
FIG. 4. Detection of candidate viral miRNAsin vitroandin vivo. (A) RT-PCR detection of candidate miRNAs from lytically infected 3T12 cells. (B) RT-PCR detection of candidate miRNAs from bone marrow-derived macrophages infected with␥HV68 for 20 h. (C) RT-PCR detection of candidate miRNAs from peritoneal cells isolated from Stat1KO mice infected with ␥HV68. Data represent results from two or three independent experiments. Error bars are standard errors of the mean. (D) Quantitative PCR products from mock or infected 3T12 cells described for panel A were run on a 3% NuSieve GTG agarose gel. Sizes of markers are indicated on the left. miRNA-amplified products run with the 80-to 89-nt markers.
on November 8, 2019 by guest
http://jvi.asm.org/
sented by over 9,700 reads (Table 3). This suggests that miR-M1-2 may be misannotated and the actual miRNA is on the 5⬘ arm, and the 3⬘sequence, with lower read abundance, is actu-ally miRNA*.
Confirmation of candidate␥HV68 miRNAs.Putative novel
miRNAs were confirmed using both Northern blot analysis and RT-PCR. Six of the 10 novel candidate␥HV68 miRNAs could be detected by Northern blot analysis from 3T12 cells (Fig. 3), and those that could not, probably due to low relative abun-dance, were also tested by RT-PCR (Fig. 4A). RT-PCR was performed on␥HV68-infected 3T12 cells (Fig. 4A), bone mar-row-derived macrophages (Fig. 4B), as well as directlyex vivo
peritoneal cells of infected Stat1-deficient mice (Fig. 4C), dem-onstrating that the␥HV68 candidate miRNAs were expressed during the course ofin vivoinfection. PCR products from 3T12 cDNA were run on a gel to confirm specific detection of miRNA products (Fig. 4D). This RT-PCR assay works by polyadenylating all RNA and adding a universal tag during the reverse transcription step. The subsequent PCR is performed using a universal primer and an miRNA-specific primer. Thus, both the mature miRNA sequence and the pre-miRNA can be detected using this method. In multiple cases for our candidate miRNAs, the predominant PCR product was larger than ex-pected for an miRNA (as determined by detection of miR-M1-1 from␥HV68 and miR-16 from mouse), suggesting that the PCR may be detecting pre-miRNA products. Even for miR-M1-1, the PCR assay amplified a small band correspond-ing to the mature miRNA, as well as a large band that could result from amplification of the pre-miRNA (Fig. 4D). It is possible that our predicted miRNA sequences are not exactly correct, and a different primer that is shifted by even 1 to 2 nt could amplify an miRNA-sized product. In the cases of miR-M1-10-5p, miR-M1-12, miR-M1-13a-5p, miR-M1-13a-3p, M1-13b-5p, M1-13b-3p, M1-14-5p, and miR-M1-14-3p, we found the PCR detection method to be incon-clusive. However, because of positive detection by Northern blot analysis, we can conclude that an miRNA-sized product exists for M1-10-5p, M1-10-3p, M1-11, miR-M1-12, miR-M1-13a-5p, and miR-M1-13a-3p. miR-M1-13b and miR-M1-14 remain provisional, because we could not con-firm their existence by Northern blot analysis or RT-PCR.
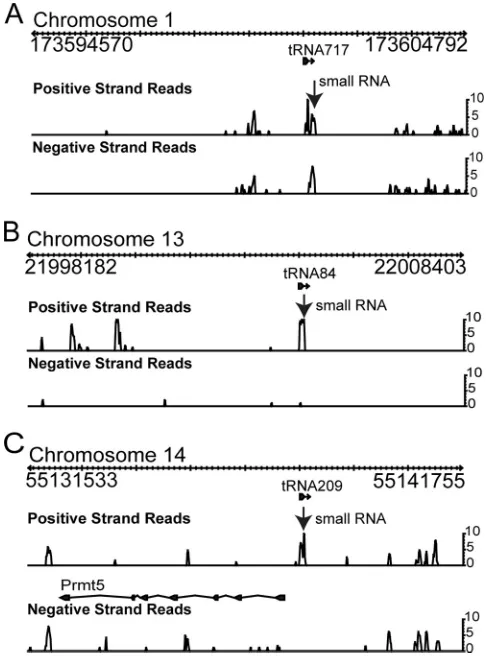
Mouse tRNA-small RNA polycistrons.Given the interesting
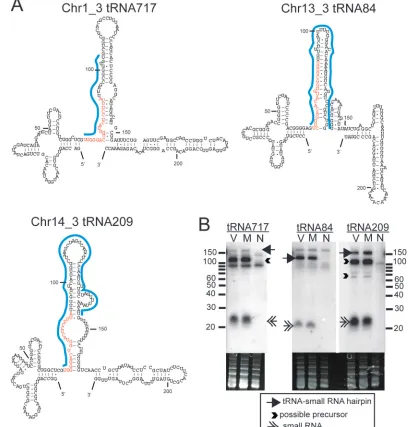
structures of the tRNA-miRNA polycistrons in␥HV68 and the assumption that viruses and their hosts utilize similar genetic mechanisms to regulate gene expression, we interrogated the sequencing data for possible mouse tRNA-small RNA polycis-trons. Three tRNA-small RNA polycistrons were identified from the sequencing data (Fig. 5). Novel small RNAs mapped to regions just 3⬘of known tRNAs (Fig. 5) and could be folded into putative tRNA-miRNA polycistrons with structures simi-lar to those in ␥HV68 (Fig. 6A). Interestingly, small RNA 717/209 mapped to two different chromosomal locations fol-lowing two different tRNAs, tRNA717 and tRNA209 on chro-mosomes 1 and 14, respectively (Table 4; Fig. 6A). Small RNA 84 mapped uniquely to the region downstream of tRNA84. Notably, the tRNA717-small RNA is directly preceding a short interspersed element (SINE). Northern blot analysis using the probes shown in Fig. 6A confirmed the existence of a 20- to 22-nt RNA species, as well as a possible precursor of⬎100 nt for tRNA717 and tRNA209 and a putative tRNA-small RNA
band of⬎130 nt for all three small RNAs, thus indicating the existence of larger transcripts that could include a pre-miRNA hairpin (Fig. 6B). Probes just for the miRNA sequence gave similar results (data not shown). The longer probes for tRNA84 and tRNA209 should bind uniquely to the structures in Fig. 6A, but we were unable to design a unique probe for tRNA717. The small RNA sequence for the putative miRNA is the same as that in tRNA209, and the sequence directly after the small RNA is part of a SINE; therefore, the long probe for the tRNA717 hairpin may have cross-hybridized to tRNA209-small RNA or to a SINE. Finally, while the tRNA209-small RNAs were identified from our sequencing data on infected cells, Northern blot analysis showed that these RNAs are present in both uninfected and infected 3T12 cells but not in the negative-control monkey cell line Vero.
DISCUSSION
[image:7.585.299.542.69.400.2]Using Illumina deep sequencing of ␥HV68-infected 3T12 cells, we identified 10 novel candidate miRNAs, six of which were confirmed by Northern blot analysis. These novel miRNAs emanate from the same Pol III transcripts previously identified to generate the 10 known␥HV68 miRNAs. These
FIG. 5. Identification of novel tRNA-small RNA polycistrons in mouse. Sequencing data from Table 2 were also analyzed for mouse small RNAs associated with tRNAs. Three novel small RNAs (arrow) immediately downstream of tRNA717 on chromosome 1 (A), tRNA84 on chromosome 13 (B), and tRNA209 on chromosome 14 (C) were identified.
on November 8, 2019 by guest
http://jvi.asm.org/
data increase the number of␥HV68 miRNAs substantially and further define the unusual ␥HV68 tRNA-miRNA structures that generate pre-miRNA stem-loops from larger Pol III tran-scripts. While the previous work to identify␥HV68 miRNAs was done using a cDNA cloning method (20), our data using
[image:8.585.83.491.73.500.2]deep sequencing technology provide a more extensive picture of miRNA expression from the ␥HV68 genome. Moreover, this technique allowed us to examine small RNA expression from the mouse genome in parallel and to identify similar tRNA-small RNA polycistrons in mouse.
FIG. 6. Detection of tRNA-small RNA polycistrons in mouse. (A) Predicted secondary structures of three mouse tRNA-small RNA transcripts. Nucleotides of the small RNAs are depicted in red. Blue lines indicate the sequences that were used to probe the Northern blots shown in panel B. (B) Northern blotting for tRNA-associated small RNAs was performed on RNA from mock-infected 3T12 (M),␥HV68-infected 3T12 (V), and uninfected Vero (N) cells. Arrows and arrowheads indicate small RNAs, putative precursor structures, and possible polycistrons. Blots are representative of results from two independent experiments.
TABLE 4. Novel mouse tRNA-associated small RNAs
miRNA Sequence No. of
reads Associated tRNA(s) Confirmation
tRNA717/209 small RNA UUGGGACUGGUACUCCUUUA 278 tRNA717 and -209 Northern blot analysis
tRNA84 small RNA UCUGGACACAUGUGGCUUUU 717 tRNA84 Northern blot analysis
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.43.546.677.725.2]One interesting conundrum revealed by this sequencing data is the incongruity between previous miRNA annotation using different methods and miRNA identification using deep se-quencing. As discussed above, one of the main criteria for prediction of an miRNA, after position on a predicted stem-loop, was relative sequence abundance. We and others (21, 24) have used the peak read within the arm of a stem-loop to identify an miRNA. However, when we applied that criterion to already-annotated miRNAs from the ␥HV68 genome, we found that only two (miR-M1-7-5p and miR-M1-7-3p) actually met that standard. All of the rest were either slightly shifted off the most abundant sequence or, in the case of miR-M1-2, on the complete opposite arm of the stem-loop. Similar results were observed for Illumina sequencing of embryonic stem cells (15), and these sequence variations from the “reference” se-quence were termed isomiR sese-quences. It is possible that isomiR sequences can vary across tissue or cell type (the orig-inal clones were from a B-cell line, and our results are from fibroblasts) and that 3⬘modifications and RNA editing repre-sent differences in cell type or time point of infection. Along the same lines, miR-M1-2 could be biologically active in la-tently infected B cells but otherwise represents the inactive miR-M1-2* in lytically infected cells (17). isomiR sequences may also derive from variability in a Dicer cleavage site. This raises the question of whether the current␥HV68 annotations are actually representative of the most prevalent isomiR se-quence. In the cases of miR-M1-10 and miR-M1-13a, we were able to detect both the 5⬘and 3⬘arms by Northern blot anal-ysis, which were similar to results from extensive sequencing of KSHV that revealed abundant miRNA* sequences that were detectable by Northern blot analysis, along with their “non-star” miRNAs (10). In the case of KSHV, some miRNA* sequences were found to be biologically active. Further work will be required to determine if the same is true for all the derivatives of␥HV68 miRNAs.
Also important to note is the relationship between read count and relative abundance. Our data demonstrate that read abundance does not always correlate with actual abundance of a transcript or with ease of detection by other methods (e.g., Northern blotting). For example, miR-M1-11 and mir-M1-13a-3p were present at a relatively low read frequency but were readily detected by Northern blot analysis. On the other hand, other candidate miRNAs, such as miR-M1-12 and mir-M1-13a-5p, have greater numbers of reads associated with them but are barely detectable by Northern blot analysis (Table 2 and Fig. 3). While there could be a difference in probe affinity and labeling, this was reproducible with different preparations of probes. This indicates that read number alone is not neces-sarily a quantitative measure of miRNA abundance.
In addition to highlighting the abundance of miRNAs ema-nating from the␥HV68 genome, these sequencing data also reveal the possible relationship between the ␥HV68 tRNA-miRNA structures and similar structures in mouse. While we have not shown whether the mouse small RNAs identified in Fig. 5 to 7 are miRNAs or some other small RNA, such as a tRF (9), the structures predicted for these tRNA-small RNAs are strikingly similar to those for the␥HV68 miRNAs. More-over, some tRFs are thought to be processed by tRNase Z (9) from a Pol III transcript consisting of a tRNA-tRF, similar to the␥HV68 tRNA-miRNAs (1). The mouse tRNA-small RNAs
we found all have Pol III termination signals (stretches of multi-ple U’s) at the end of the small RNAs. However, they also have subsequent termination signals, suggesting that a larger transcript could exist. Our Northern blot analysis confirms that both small and large transcripts are produced (Fig. 6B). Further work will be required to determine the function of these small RNAs and their relationship to the viral transcripts. Moreover, additional investi-gation into the possible presence of these types of transcripts in other viruses and hosts is under way.
ACKNOWLEDGMENTS
This work was supported by the ARRA administrative supplement MRCE 2 U54 AI057160 (H.W.V. and W.Z.), NIH grant AR058681 (W.Z.), and NSF grant DBI-0743797 (W.Z.). T.A.R. is supported by the Damon Runyon Cancer Research Foundation.
REFERENCES
1.Bogerd, H. P., H. W. Karnowski, X. Cai, J. Shin, M. Pohlers, and B. R. Cullen.2010. A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral microRNAs. Mol. Cell37:135–142. 2.Borchert, G. M., W. Lanier, and B. L. Davidson.2006. RNA polymerase III
transcribes human microRNAs. Nat. Struct. Mol. Biol.13:1097–1101. 3.Bowden, R. J., J. P. Simas, A. J. Davis, and S. Efstathiou.1997. Murine
gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J. Gen. Virol.78:1675–1687.
4.Cole, C., A. Sobala, C. Lu, S. R. Thatcher, A. Bowman, J. W. Brown, P. J. Green, G. J. Barton, and G. Hutvagner.2009. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA15:2147–2160.
5.Cook, H. L., J. R. Lytle, H. E. Mischo, M. J. Li, J. J. Rossi, D. P. Silva, R. C. Desrosiers, and J. A. Steitz.2005. Small nuclear RNAs encoded by herpes-virus saimiri upregulate the expression of genes linked to T cell activation in virally transformed T cells. Curr. Biol.15:974–979.
6.Diebel, K. W., A. L. Smith, and L. F. Van Dyk.2010. Mature and functional viral miRNAs transcribed from novel RNA polymerase III promoters. RNA 16:170–185.
7.Hofacker, I. L.2009. RNA secondary structure analysis using the Vienna RNA package. Curr. Protoc. Bioinformatics12:unit 12.2.
8.Langmead, B., C. Trapnell, M. Pop, and S. L. Salzberg.2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol.10:R25.
9.Lee, Y. S., Y. Shibata, A. Malhotra, and A. Dutta.2009. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev.23:2639–2649. 10.Lin, Y. T., R. P. Kincaid, D. Arasappan, S. E. Dowd, S. P. Hunicke-Smith,
and C. S. Sullivan.2010. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA16:1540–1558. 11.Malone, C. D., and G. J. Hannon.2009. Small RNAs as guardians of the
genome. Cell136:656–668.
12.Mathews, M. B., and T. Shenk.1991. Adenovirus virus-associated RNA and translation control. J. Virol.65:5657–5662.
13.Meraz, M. A., J. M. White, K. C. F. Sheehan, E. A. Bach, S. J. Rodig, A. S. Dighe, D. H. Kaplan, J. K. Riley, A. C. Greenlund, D. Campbell, K. Carver-Moore, R. N. DuBois, R. Clark, M. Aguet, and R. D. Schreiber.1996. Targeted disruption of the Stat 1 gene in mice reveals unexpected physio-logic specificity of the JAK-STAT signaling pathway. Cell84:431–442. 14.Moazed, D.2009. Small RNAs in transcriptional gene silencing and genome
defence. Nature457:413–420.
15.Morin, R. D., M. D. O’Connor, M. Griffith, F. Kuchenbauer, A. Delaney, A. L. Prabhu, Y. Zhao, H. McDonald, T. Zeng, M. Hirst, C. J. Eaves, and M. A. Marra. 2008. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res.18:610–621.
16.Nachmani, D., N. Stern-Ginossar, R. Sarid, and O. Mandelboim.2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe5:376–385. 17.Okamura, K., M. D. Phillips, D. M. Tyler, H. Duan, Y. T. Chou, and E. C. Lai.
2008. The regulatory activity of microRNA* species has substantial influence on microRNA and 3⬘UTR evolution. Nat. Struct. Mol. Biol.15:354–363. 18.Pall, G. S., and A. J. Hamilton.2008. Improved Northern blot method for
enhanced detection of small RNA. Nat. Protoc.3:1077–1084.
19.Pavlova, L. V., H. W. Virgin, and S. H. Speck.2003. Disruption of gamma-herpesvirus 68 gene 50 demonstrates that Rta is essential for virus replica-tion. J. Virol.77:5731–5739.
20.Pfeffer, S., A. Sewer, M. Lagos-Quintana, R. Sheridan, C. Sander, F. A. Grasser, L. F. Van Dyk, C. K. Ho, S. Shuman, M. Chien, J. J. Russo, J. Ju, G. Randall, B. D. Lindenbach, C. M. Rice, V. Simon, D. D. Ho, M. Zavolan,
on November 8, 2019 by guest
http://jvi.asm.org/
and T. Tuschl.2005. Identification of microRNAs of the herpesvirus family. Nat. Methods2:269–276.
21.Rajagopalan, R., H. Vaucheret, J. Trejo, and D. P. Bartel.2006. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev.20:3407–3425.
22.Ruf, I. K., K. A. Lackey, S. Warudkar, and J. T. Sample.2005. Protection from interferon-induced apoptosis by Epstein-Barr virus small RNAs is not mediated by inhibition of PKR. J. Virol.79:14562–14569.
23.Stern-Ginossar, N., N. Elefant, A. Zimmermann, D. G. Wolf, N. Saleh, M. Biton, E. Horwitz, Z. Prokocimer, M. Prichard, G. Hahn, D. Goldman-Wohl, C. Greenfield, S. Yagel, H. Hengel, Y. Altuvia, H. Margalit, and O. Mandel-boim.2007. Host immune system gene targeting by a viral miRNA. Science 317:376–381.
24.Sunkar, R., X. Zhou, Y. Zheng, W. Zhang, and J.-K. Zhu.2008. Identifica-tion of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol.8:25.