CopyrightC 1977 American Society for Microbiology PrintedinU.SA.
Inhibition of Host Cell Protein Synthesis by UV-Inactivated
Poliovirus
TIM HELENTJARIS* AND ELLIE EHRENFELD
Departments of Biochemistry andMicrobiology,* University of Utah Medical Center, Salt Lake City, Utah 84132
Receivedfor publication 1 July 1976
The ability of poliovirus that was irradiated with UV light at energies up to
2,160
ergs/mm2
to subsequently inhibit host cell protein synthesis wasmea-sured.Theinactivation of the host cell shutofffunction followed one-hit kinetics.
Increasingirradiation did not affect the rate ofinhibition until the multiplicity
ofinfectionafter irradiation was reduced toapproximately 1 PFU/cell. At higher
functional multiplicities,the rate was unchanged,but an increasinglag before
the onset ofinhibition was observed with increasing irradiation. The energy
levels required to inactivate virus-induced inhibition of host cell protein
synthe-sis suggestthatdamage to virus RNA rather than to virus capsid proteins is
responsible for the loss of function. When the inactivation of host cell shutoff was
compared with the inactivation of other viral functions by UV irradiation, it
correlated exactly withtheloss of infectivity but not withotherviralfunctions
measured. Guanidine treatment, which prevents detectable viral RNA and
proteinsynthesis, completely inhibited host cell shutoff by low multiplicities of
unirradiated virusinfection but nothigher multiplicities. When a high
multi-plicity of virus wasfirst reduced to a low titer by irradiation, host cell shutoff
wasstillevident in the presence of guanidine. The results demonstrate that the
complete inhibition ofhost cell protein synthesis can be accomplished by one
infectious viral genome per cell.
The inhibition ofinfected cell RNA and
pro-teinsynthesis bypicornaviruseswasrecognized
almost15yearsago, yetthe mechanismof this
viral function is still unclear. This inhibition
might be due to either input virion protein or
RNA without the need for viral RNA
transla-tion or replication or due to a product of a
functional viral genome, or possibly due to
some combination of the two. Conflicting
evi-dence has been presentedfor and against each
possibility. Penman and Summers (16), using
either puromycin orcycloheximide to
synchro-nizepoliovirusinfection, found that a period of
protein synthesis was required after the
re-moval of the inhibitor before the inhibition of
cell protein synthesis was evident. Baltimore
andco-workers(2)also found that critical levels
ofp-fluorophenylalanine, aninhibitor ofprotein
synthesis,couldblock theabilityofmengovirus
toinhibitbothcell RNAandproteinsynthesis.
These datasuggested that the inhibition of cell
protein synthesis required the translation of
the viral genome. However, Holland (7) and
Collinsand Roberts (4) havesubsequently
pre-sentedevidence demonstrating that inhibitors
ofproteinsynthesis(puromycin,
fluorophenyla-lanine, and azetidine) could selectively
disso-ciate host cell "shutoff' from detectable viral RNA and protein synthesis. Also, infection in
the presence of 1 mM guanidine, which
pre-vents detectable viral replication and
transla-tion, doesnot seem toaffect hostcellshutoffat
highermultiplicities of infection (MOIs) (7, 16).
Usingtemperature-sensitivemutants to
exam-inethisproblem,Steiner-PryorandCooper (20)
showed that the abilityto inhibithost cell
pro-teinsynthesis seemstomap in theregionofthe
structural protein genes. However, Cole and
Baltimoredemonstrated that defective
interfer-ing polio virions are able to inhibit host cell
translationdespite the fact that their
defective-nessoriginatedbecause of deletionsinthe
cap-sidgenes and resulted inaninabilitytospecify
for structuralproteins (3).
Perhaps the most compelling evidence
indi-cating the need for a functional viral genome
has been the ability of UV irradiation (1, 16)
and treatment by proflavine (7) to inactivate
theabilityof thevirus to causehost cell shutoff.
Atthe time these experimentswerereported,it
was presumed that the majorsite ofaction of
eachagentwas todamagetheviral RNA.
How-ever, theproflavineexperimentswere not
con-clusivesince, asthe authorindicated, he could
259
on November 10, 2019 by guest
http://jvi.asm.org/
not exclude the possibility that inactivation
wasdueto the oxidation ofcoat proteins. The
conclusions drawnfrom the UV light
inactiva-tion experiments could also be quesinactiva-tioned in
view of data presented by Miller and
Plage-mann(14). They investigated the effects ofUV
irradiation on mengovirus and found that the
inactivation of viral infectivity seemedto
corre-late with damage to the viral genome, but
that alterations in viral capsid proteins did
oc-curwithgreater amountsofirradiation. Other
data demonstrating the effects ofUV
irradia-tion on proteins have also been reported (11,
17).
Because other viruses have been shown to
inhibit hostcell processes afterUVlight
inacti-vation (8, 15, 19) and because of the recent
observations of Racevskisetal. (18) suggesting
that poliovirions can inhibit translation in
re-ticulocyte lysates, wereexamined the effect of
UV irradiation on the ability of poliovirus to
inhibit cellular protein synthesis. The principal
goal of this investigation was to determine
whether a functional viral genome was
neces-sary for host cell shutoff or whether shutoff
could be ascribed solely to input virus capsid
proteins. Our results indicate that the ability of
poliovirus to inhibit cell protein synthesis is
sensitive to UV irradiation atlevels ofenergy
which are consistentwith damageto the viral
genome but nottovirionproteins. The
inactiva-tionof the shutoffabilityfollowsone-hit
kinet-ics and demonstrates that approximately 1
PFU/cell is sufficient to cause host cell shutoff.
Increasing the MOI above 1 decreases the lag
time before the virus replication cycle begins.
Moreover, when compared with the
inactiva-tion of other viral funcinactiva-tions, loss of the shutoff
ability correlates exactly with loss of virion
infectivity.
MATERIALS AND METHODS
Cells and virus. Suspension cultures of HeLa S3 cells were grown at a density of 3 x 105to 8 x 105 cells/mlinEagleminimal essential medium(MEM) supplementedwith 5%fetal calf serum.
TheMahoney strain of poliovirus type 1 was col-lected bycentrifugation from infected cell extracts treated with 1%sodiumdodecylsulfate (SDS), and thevirus was further purified on CsCldensity gra-dients. Optical absorption of 1.00 at 260 nm was assumedequal to 1.3 x 1013virusparticles, and the infectivity of recovered virus was assayed on HeLa cellmonolayers under 0.7% agarose.
Irradiation of virus. Virus was diluted to 1.5 x
1010PFU/ml in MEM without serum and irradiated inmicrotiterplates at a distance of 5 cm by a broad-spectrum UVlamp. The energyatthe distance of 5 cmwas measured to be 36ergs/mm2*s.
Evaluationofproteinsynthesis in infected cells.
Cells at 5 x 106/ml in MEM in Spinner culture were infected with virus at an MOI ofapproximately 10 PFU/cell. After 30 min, fetal calf serumw'asadded to 5%. Guanidine,whenadded,wasusedat afinal concentrationof 1 mM. Every 30 min from 0.5 to 5.0 hpostinfection, 0.5 ml of infectedcells wasremoved andexposedto0.25,ICiof[35S]methionine(specific activity, 503Ci/mmol;NewEngland Nuclear Corp.) for 10 min at37°CinSpinnerculture. The incorpora-tion wasstopped by the additionof cold Earle salt solution, and the cells werewashed by centrifuga-tion.Cellprotein wasprecipitated by 5% trichloroa-ceticacidontoWhatman GF/C filters, and the incor-porationof[35S]methionine wasevaluated ina liq-uidscintillationcounter.
Viral protein production was also examined by SDS-polyacrylamide gel electrophoresis (PAGE) of infected cell lysates. HeLa cells were infected as above with virus that had been irradiated for var-iousperiods oftime.The MOI of each samplebefore irradiationwas 10. At 30 minpostinfectioncalf se-rumwasaddedto5%. Atspecifiedtimes, 50 ,tCiof 14C-labeled amino acids (proteinhydrolysate, New England Nuclear Corp.) was added and the samples wereallowedtoincorporatethelabeledaminoacids for a period of 2 h. At this point the cells were
washed in Earle solution and resuspended inRSB (10 mMNaCl-10 mMTris [pH7.4]-1.5mMMgCl2). Nonidet P-40 wasadded to 1%andthe nuclei were discarded after centrifugation. The cytoplasm was made 1% in SDS and analyzed on polyacrylamide gels bythe method of Maizel (12).
Evaluation of RNA synthesis in infected cells. Cells wereinfected asbefore,but at 30 min postin-fection 5% serum, actinomycin D (gift of Merck, Sharp, and Dohme) at 10
jig/ml,
and ['4C]uridine(specific activity, 50 mCi/mmol; NewEngland Nu-clearCorp.) at 1.3 ,Ci/mlwere also added. At 30-minintervals from 0.5 to 5.0 hpostinfection, 0.2 ml ofinfectedcells was removed and the incorporation wasstopped bythe addition of cold Earle solution. The cells were washed by centrifugation, and the RNA was precipitated by 5% trichloroacetic acid ontoWhatmanGF/C filters.The amount of incorpo-rated [14C]uridine was then determined in a liquid scintillation counter.
Virion production by cellsinfected with irradi-ated virions. Cells wereinfectedasbefore and,at 30 minpostinfection, 5% serum, actinomycin D at 10 ,ug/ml, and ["4C]uridinewere addedto 1.3 ,Ci/ml. At 6 hpostinfection, the cell cytoplasm was collected by treatment of the cells with 1% Nonidet P-40 and subsequent centrifugation to remove nuclei. SDS (1%) was added to the cytoplasmic extract along with purified poliovirus labeled with [3H]uridine, which served as the marker. The extract was lay-ered onto a 7 to 47% sucrose gradientinRSB and centrifuged at 40,000 rpm in an SW41 rotor for 2.5 h. The gradient was fractionated and each fraction was assayedfor 3H and 14Cacid-precipitable radioactiv-ity in a liquidscintillation counter. The 14C in the peak corresponding to irradiated virus was
com-paredwith thepatterndemonstrated by 3H-labeled virus that had notbeenirradiated. Since no obvious changeinthe sedimentationprofile was seenwith
on November 10, 2019 by guest
http://jvi.asm.org/
increasing irradiation,all of the 14C radioactivity in the virus peakwastotaled and used as a relative
indicator of virionproduction.
In other experiments unlabeled progeny virus
from irradiatedvirus-infectedextractswasprepared and titered on HeLa cell monolayers under 0.7% agarose.
RESULTS
Effect of irradiation on host cell shutoff.
Polioviruswas irradiatedasdetailed above for
periods of 0, 10, 20, 30, 40, and 50 s. These
irradiated preparationswereusedtoinfect cells
at an MOI that corresponded to 10 PFU/cell
before irradiation. Protein synthesiswas
evalu-ated inthese cells and the resultsareshownin
Fig. 1. The rate of [35S]methionine
incorpora-tion in the uninfected cells increases linearly
from 0.5 to about 3.5 h, where it decreases
slightly until 5.0 h. Incorporation by the cells
infected with unirradiated virus appears
nor-mal until 1.0hpostinfection. At this time, the
incorporationrate declines linearly until 2.0 h
postinfection, where it again beginstoincrease.
6-
5-
4-r;T)
I0
z
s
3-
C-"I
r
2-
1-\50SEC
\40 SEC
\30 SEC
20 SEC
MO SEC 'O SEC
2 3 4 5
TIME POST-INFECTION IHRS.)
FIG. 1. Host cellshutoff by irradiatedvirus. Vi-rus was irradiatedas indicated above and used to
infect HeLa cells. Protein synthesis in the infected
cells was evaluated by 10-min pulses with
[35S]methionineevery0.5 hfrom0.5to5.0 h postin-fection. Samples were trichloroacetic
acid-precipi-tated andevaluated inaliquid scintillationcounter. Control line represents protein synthesis in
unin-fectedcells.
This increase continues for a short time and
then it, too, decreases. From examination ofthe
proteinsmadeatthislater timeby SDS-PAGE,
ithas beendemonstrated that nearly all of the
proteinsmade by the cell after 2.0 h
postinfec-tion are virus specific (data notshown).
Irradiation of poliovirus prior to infection has
two effects on the host cell shutoff pattern.
First, there is a significant delay before the
inhibition of cell protein synthesis is seen, and
this delay increases withincreased irradiation
up to 30 s. For instance, whereas unirradiated
virusbegins to inhibit cell protein synthesisat
about 1.15 hpostinfection, 10 s of irradiation
de-lays the onsetofinhibition to about 1.50h and
protein synthesis in cells infected byvirus
ir-radiatedfor 20 s appears normal until 1.85 h
postinfection. A total of 30 s of irradiation de-lays the shutoff until about 2.20 h postinfection,
butincreased irradiation of theinfecting virus
failed to further delaythe shutoff past 2.25 h
postinfection. It is evident from the effect on
viral proteinsynthesis as well asfromdata to
be described below that the viral replication
cycle as awholeisdelayed by irradiation in a
manneranalogous tothedelayonshutoff. The
second effect of irradiation on shutoffcan be
discerned fromtheslopeof the decrease in the
rateof cell proteinsynthesis. After10and20s
ofirradiation, although theonsetof inhibition
was delayed, the rate of inhibition of cell
pro-teinsynthesis was notsignificantly altered.
Be-ginning at 30 sofirradiation,however, therate
ofinhibitionbegins to decrease with increasing
irradiation. This change in slope continues
un-til 50 sof irradiation, which was the greatest
amount of irradiation used in these
experi-ments.
Figure 2 summarizestheeffect of increasing
UVirradiation ofinfecting virions on the rate
ofhost cell shutoff. The slopes ofthe shutoff
curvesfrom the onsetof the inhibition,
ignor-ingthelag,until the beginning of viral protein
synthesis havebeentaken as a measure ofthe
rateofshutoff. The slope of the line generated
by unirradiated virus was expressed as 1.00,
and theslopesoftheother lines were calculated
as fractions of 1.00. As shown in Fig. 1, low
levelsofirradiation(less than 25 s) resulted ir
nochangeintherateofinhibition. Thus,Fig.2
shows a noticeable shoulderonthe curve until
about 25 sofirradiation, wherethe inhibition
decreased logarithmically. This shoulder does
not appear to be due to multiple-hit
inactiva-tionkinetics,sincethe cells were infected at an
MOI greater than one before irradiation.
Extrapolation of the slope of the straight-line
segmentinFig.2tothe ordinateinterceptsat a
point that would suggestapproximately 10-hit
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.505.76.211.320.570.2]262
cell. Thus, inhibition of host cellprotein
syn-thesis occurs at the same rate, albeit after a
variablelag, aslongasthere isapproximately
one infectious particle per cell; thereafter the
rateof inhibition isreduced.
Effect of irradiation on viral RNA and pro-tein synthesis. Another function of the viral
genome,virus-specific RNA synthesis, was
ex-amined after irradiation. Cells were infected
with irradiated virus at an MOI of 10PFU/cell
before irradiation and the viral RNA synthesis
in these cells was measured. The results are
shown inFig.4.Therewas nosignificant
incor-porationby uninfected cells over the period of1
to 5 h after the addition ofactinomycin D and
[14C]uridine (data not shown). In the cells in-fected with unirradiated virus, no viral RNA
synthesis is evident until 2.25 hpostinfection.
There is then a linearincorporation until about
4.00hpostinfection when synthesisceases. As
with host cellshutoff, the irradiation of virus
before infection demonstrates two effects on
viral RNA synthesis. First, there is a
signifi-cant delay before the onset of viral RNA
syn-thesis until about 3.50 h with 30 s ofirradiation,
10 20 30 40 50
TIME OF IRRADIATION (SECS.)
FIG. 2. Host cell shutoffby irradiated virus. The slopes of thestraight-line segments during host cell shutoff by irradiated virus inFig.1 wereplottedas fractionsoftheshutoffdemonstrated byunirradiated virus(solid line).Thedashed line represents extrap-olation ofthestraight-linesegmentfrom30to 50 s.
inactivation kinetics of the input. Since the
cellswereinfectedat anMOIofapproximately
10, this corresponds to one-hit kinetics in the
inactivation of the host cell shutoff ability.
Thatthis is the case was verified bydoing the
sameexperiment at anMOIbefore irradiation
of about 1,000. Under these conditions, the
shoulderwasextended from 25 sof irradiation
toapproximately threetimesthat value (70 s),
as expected ifthe shoulderwere MOI
depend-ent.
Effect of irradiation on viral infectivity.
The same virus used in the previous
experi-mentwas assayedforinfectivity by plaque
as-say before and after irradiation for 10 to 60 s
andthe resultsare showninFig. 3. No
shoul-der is evident and one-hit inactivationkinetics
areobserved. The37%survivaldose(D37)is306
ergs/mm2, and about3 logs of viral infectivity
arelost within 60 s of irradiation.Inagreement
withthe results showninFig. 2,25 sof
irradia-tionreduced theinfectivity of the virus inputin
these experiments to approximately 1 PFU/
11
U-Cl.
0D
0
z
z 0
C-0
CL
.1
-.01
-
.001-10 20 30 40 50
TIME OF IRRADIATION (SECS.)
FIG. 3. Infectivity loss after irradiation. Virus samples wereirradiatedfortheindicated times and
theinfectivitywasassayedonHeLacellmonolayers.
Theresults (0)wereplottedasfractionsof the
unir-radiated samples.
7-0 z
5
C,)
z
0
CL.
* \
60 J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.505.100.233.70.357.2] [image:4.505.279.456.338.608.2]
4-3- O SEC
_ 20SEC
z
O0SEC
30 SEC
c-I,
C-)~~ ~ ~~~~~0SEC
50 SEC 60 SEC
2 3 4 5 6
TIME POST-INFECTION (HRS)
FIG. 4. Viral RNA synthesis by irradiated virus. Virus was irradiated as indicated above and used to infect HeLa cells. At 0.5 h after infection actinomycin D (to 3.3
mg/ml)
and[14C]uridine (to 125 Xilml) wereadded. Incorporation was allowed to continue and0.2-ml samplesweretaken every0.5hfrom0.5 to 6.0 h postinfection. Incorporation was stopped with cold Earle solution, and the trichloroacetic acid-precipitable radioactivity was assayed by aliquid scintillation counter.
and furtherincreases inirradiationareunable
to further delay RNA synthesis. Second, the
rateof RNAsynthesis, asdemonstratedby the
slope of theincrease inuridineincorporation, is
decreasedby increasingamountsofUV
irradia-tion.
As before we have ignored the delay factor
and havetakenthe
slope
of the linegenerated
byunirradiated virustobe1.00 andexpressed
theothers as fractions of it. The loss of RNA
synthetic activity with increasing irradiation
was plotted in Fig. 5, as are the other results
presented inthis report. As can be seen, viral
RNAsynthesis isinactivatedby one-hit
kinet-ics, with noshoulder evident. The D37 for this
activity isapproximately 1,150
ergs/mm2.
The RNA synthesized in cells infected with
irradiatedvirus was examinedby sucrose
gra-dient velocity sedimentation and displayed a
normal sedimentationprofileand RNase
sensi-tivity(datanotshown). Noevidence for smaller
RNAfragmentswas detectablebythisanalysis.
Viralprotein synthesis was also evaluated by
SDS-PAGE andanautoradiographof thegel is
showninFig. 6.Cells infectedwith virus
inac-tivated for 0, 20, 40, and 60 s were labeled with 14C-amino acids for 2-h periods to coincide with
thepeak of viralprotein synthesisineach case
(Os, 2.25 to 4.25 hpostinfection;20s,2.50to 4.50
h postinfection; 40 s, 3.50 to 5.50 hpostinfection;
60 s, 4.00 to 6.00 hpostinfection). The gel
pat-tern of samples infected with virus after
in-creasing amounts of irradiation is not
signifi-cantly affected, except foraslightly increased
incorporation in several detectable host cell
proteins, possiblydue to less effective shutoff.
Somewhatunexpectedinview of thelarge loss
ofinfectivityinthe input virus duetothe
irra-diation, there is a significant amount of viral
protein being made even in the sampleinfected
with virusirradiated for 60 s.
Ability of irradiated virions to produce
progeny virions. The synthesis of relatively
larger amounts of virus-specific proteins and
RNA by cells infected with virus, which was
demonstrated by plaque assay to be markedly reduced in infectivity, prompted us to measure
the production ofassembled viral particles in
these cells. Cells infectedwith irradiated virus
in the presence of actinomycin D and
[14C]uridinewereharvestedasdescribedabove,
andcytoplasmic extracts were analyzedon
su-z
c-cn
z
0
cr-LL.
10 20 30 40 50 60 TIMEOF IRRADIATION (SECS)
FIG. 5. Loss ofviralfunctions after irradiation. Lossofinfectivityandshutoffareplottedas before. Lossofviral RNAsynthesiswasdeterminedby plot-tingtheslopesofthe increase in viral RNAsynthesis (fromFig. 4) for irradiatedvirus asfractionsof the slope demonstrated by unirradiated virus. Virion productionwas determinedasdescribed inthe text, and the total counts from irradiated preparations
wereplotted asfractions ofthecountsfrom unirra-diated viruspreparations. PFUproductionwas de-terminedasdescribedinthe text, and the virusyield forirradiatedsampleswasplottedasfractionsof the virusyield fromanunirradiatedvirussample.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.505.47.234.53.315.2] [image:5.505.256.441.370.535.2]264 HELENTJARIS AND EHRENFELD
VPI
VP2
VP3
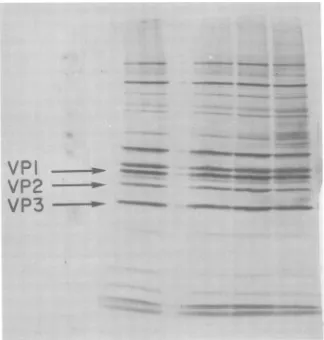
FIG. 6. Viralproteinsynthesisafter irradiation. Virus wasirradiatedfor 20, 40,and 60sand used to
infect cells, as detailed in the text. Atspecified times 50 XCi of 14C-labeled aminoacids was added, and incorporation wasallowedto continue for 2 h. Thecytoplasms were made 1%SDS andanalyzed on10%
polyacrylamidegels.ArrowsindicateVP1, 2,and 3asmarkers. Thefourcolumnsrepresent cellcytoplasms
infectedwith virusirradiatedfor 0, 20, 40, and 60 s,respectively.
crose velocity gradients for the production of
virion particles. There were no differences in
the sedimentation of unirradiated virus and
progenyviruswhoseparentalvirionshad been
irradiatedforupto60s.Theamountof
radioac-tivity in thepeakof theprogenyfrom
nonirra-diated virus was expressed as 1.00, and all otherpeakswerecalculatedasfractions of 1.00.
The loss of the ability of irradiated virus to
specifyforprogenyviruswithequivalent
sedi-mentation properties is plotted in Fig. 5. The
curveislinear withintercept 1, indicating
sin-gle-hit inactivationkinetics. Interestingly, the
slope oftheinactivationcurvedoesnotcoincide with lossofinfectivity but with the slope of the
inactivationofviralRNA synthesis.
Unlabeled progeny virus wasprepared in a
likemannertoassayfortheloss of the ability of
irradiated virus to specifyfor infectious
prog-eny. The progenyvirus was
assayed
forinfec-tivityonHeLacellmonolayers (Fig. 5). There
isanoticeableshoulderinthecurveuntil30sof
irradiation, where a decrease in infectious
progenyproductionisseen.There appearstobe
noclearexplanationofwhy infectious progeny
production should have a higher D37 than
150s particle production or why the former
inactivationcurvehasashoulderonitwhereas
thelatter appears linearinitsinactivation.
Effect of guanidine on inhibition of cell
protein synthesisby irradiated virions.
Infec-tionofcellsinthe presence of1mMguanidine
resultsinnodetectablepoliovirus-directed
pro-teinand RNA synthesis (7, 16). Athigh MOIs
in the presence ofguanidine, host cell shutoff
occurs at near-normal rates, whereas at low
MOIs
the inhibition issignificantly
decreased.This suggests that infection at alow MOI
re---so
um
.-.I IAPMM WSW
-- VP ..%
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.505.108.432.76.416.2]quires some amplification of a viral function
necessary for shutoff. To examine what effect
guanidine treatmentmight have onshutoff by
irradiated virus, the inhibition of cell protein
synthesis was evaluated in the presence and
absence of guanidine in cells infected by
un-treated virus at MOIsof 10 and 1 and virus at
anMOI of10 that had been irradiated for 30 s
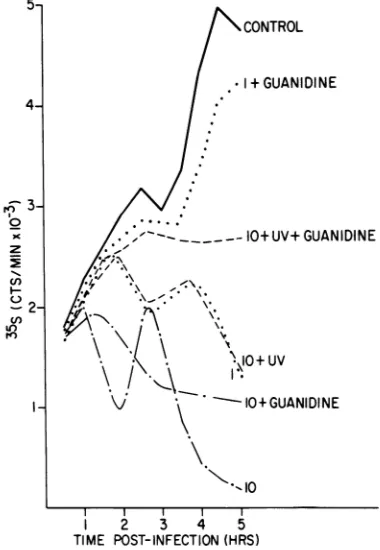
(Fig. 7).The inhibition by unirradiated virus at
anMOI of10 isreduced by the guanidine
treat-ment, but shutoff is still evident. Inhibition
begins at the same time in both
guanidine-treated and untreated, infected cells, but both
the slope and the degree of inhibition are de-creased by treatment. In the absence of guani-dine, cells infected with untreated virus at an
MOI of 1 show a significant delay in both
shu-toff and viral protein synthesis, as compared
with infection at an MOI of 10. This suggests
that at lower MOIs complete host cell shutoff
andviralreplicationmight requirethe
produc-tionofsome component,with the kineticsofits
I-| I+ GUANIDINE
____ _-10+ UV+ GUANIDINE
*\..-..\- \;.
*AK-
\.* \ \. +u
(
~~\I10+
UVv \
-- 10+GUANIDINE
\ 10
1 2 3 4 5
TIME POST-INFECTION (HRS)
FIG. 7. Effect of guaindine treatment on shutoff by irradiated virus. Untreated virus was used to
infect cells at an MOI of10 (- - ) and 1 (....) both with and without1 mMguanidine. Virus was
also irradiatedfor30sand usedtoinfectcellsatan
MOIof10 before irradiation (---) both with and without 1mMguanidine.Proteinsynthesiswas
eval-uatedandplottedasdescribed in thelegendtoFig.1.
production being dependent upon the MOI.
Guanidinetreatment atthis MOI not only
re-ducesshutoff but also completely aborts it. The
curvegenerated bythe combinationof infection
at an MOI of 1 withguanidine treatment does
not significantly differ from uninfected,
un-treated cells.
When thecells areinfected inthe absence of
guanidine with virus at an MOI of 10 that has
been irradiated for 30 s, the shutoff and viral
protein synthesis curve coincide exactly with
untreated virus infection at an MOI of 1. After
30 s of irradiation, there remains
approxi-mately 0.4 PFU/cell, as determinedby plaque
titration. That this sample generates a curve
similar to the one by untreated virus at a
higher MOI suggests that thedamaged genomes
aresomehowabletoparticipate inshutoff.This
possibility is also suggested by examiningthe
effect ofguanidineon the inhibitionby
irradi-atedvirus.Although guanidinetreatment
com-pletelyaborts theinhibitionbyuntreatedvirus
at anMOIof1, thereis stillsignificant
inhibi-tionbythe irradiated virus at aneffectiveMOI
of 0.4 PFU/cell. Again, the sample manifests
moreofaneffectthan wouldbeexpectedonthe
basis of the remaining plaque-forming ability
alone.
DISCUSSION
The principal goalof this investigation was
toexaminethe effect ofUV irradiationon the
abilityofpoliovirus to causeinhibition of host
cellproteinsynthesis.Effective shutoffrequires
one infectious virus particle per cell, and the
kinetics of inhibition are independent of the
MOI, except for an increasing delay before the
onsetof inhibition at decreasing MOIs. If the
delayfactor is ignored, theUV inactivation of
theability of the virus to induceshutoffcanbe
shown to follow one-hit inactivation kinetics,
with aD37of 306ergs/mm2, avalueidenticalto
that forthe inactivation ofinfectivity.
The energy required to inactivate the host
cell shutofffunction ofpoliovirushasbeen
com-paredwith valuesreportedinthe literature for
theinactivation of a variety of biological
func-tions and activities (Table 1). It is apparent
thattheD37for host cellshutoffis quitesimilar
to the D37 for the destruction ofmengovirus,
poliovirus, vesicular stomatitisvirus, and
mu-rineleukemia virusinfectivity, butitis
signifi-cantly smaller than theD37for the inactivation
ofcarboxypeptidase A, reverse transcriptase,or
the hemagglutination activity of mengovirus. The former is thought to be due to damage to the viral genomes, whereas the latter involves
damagetotheproteins themselves. The
dispar-ityin energies required forthe inactivation of
5-
4-3
3-0
x
z
cn
-
2-U)
r-)
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.505.49.239.307.582.2]AND EHRENFELD
TABLE 1. UV irradiation survivalof RNA and protein functions
Target (reference) D37(ergs/mm2)
Mengovirus (14) 700
Poliovirus (this investigation) 306 Vesicular stomatitis virus (15) 52.3 Rauscherleukemia virus (11) 2,000
Carboxypeptidase A (17) 300,000
Reversetranscriptase (11) 23,000-31,000
Mengovirus hemagglutina- 80,000
tionactivity (15)
the two groupsof functions is wide enough to
determine withsomecertainty that the
inacti-vation of thepoliovirus host cell shutoff activity
is due to damage to the viral RNA. Also, as
poliovirions are thought to contain 60 copies
each of four different capsid proteins, itseems
unlikely thatonehitpervirionwouldreflectan
effect of irradiationon aparticular capsid
pro-tein. In addition, we can detect no gross
changes in poliovirus capsid proteins by
SDS-PAGE afteras muchas 10minof irradiation.
The requirement, therefore, ofa functional
viralgenomefor the inhibition of host cell
pro-teinsynthesisseemsimperative, but the
partic-ipation of capsid proteins by a combinational
mechanism cannotbedisproved. To exclude a
role forcapsid proteins inhost cell shutoff, we
have attemptedtoexamineprotein synthesisin
cells infected withpurified viralRNA, but we
have been unabletoachieve asynchronous
in-fection of a large enough fraction of the cell
populationtoreliablyevaluate the rates of
pro-teinsynthesis.
By comparing the D37 of the poliovirus host
cellshutoffactivitywiththeD37 forother
polio-virusfunctions, it should be possible to
deter-mine the target size of the responsible RNA segment.Figure5showsthat theD37 forshutoff
is identical to that for infectivity, indicating
thatahit anywhereinthegenomeis sufficient
to inactivate that genome's capacity to effect
shutoff. However,ifone assumesthat the
syn-thesis of poliovirus proteinsoccursby the
cleav-age ofa single precursor polypeptide, then it
might be possible thatdamage anywhereinthe
RNAwouldpreventthepropercleavageofany
translation product as well as prevent the
translation ofsequences distal tothe UV hit.
In this case, all viral functions would display
identical inactivation curves. The slope
ob-served for the inactivation of viral RNA
syn-thesis, however, is approximately four times
that observed for the inactivation of host cell
shutofforinfectivity (Fig. 5),whichapparently
dissociates host cell shutoff from viral RNA
synthesis. One interpretation of this data is
that viral RNA synthesis is inactivated only
after a hit within the gene for the
virus-specified component(s) of the RNA
polymerase
and that this information occupies
approxi-matelyone-fourth of the genome. However,we
have not been able to rule out the possibility
of "product complementation" between
dam-aged genomes. Possibly the D37 for RNA
synthesis is actually much smaller, but,
be-cause the experiments were done at an MOI
of10, complementation between damaged
ge-nomes may slow the observed inactivation.
This possibility is suggested by two other
re-sults.Althoughinfectivity decreases withaD37
of 306 ergs/mm2, the ability to produce
infec-tious virions after infection at an MOI of 10
decreasesat amuchslowerratethanexpected
(Fig. 5). That no complementation is seen in
theinactivation ofinfectivityisnotsurprising
sinceaplaque titration by design is doneat an
MOI of much lessthan 1. The slower rate for
infectious virus production may actually
repre-sent complementation between damaged
ge-nomes or it may represent a shoulder due to
infection at an MOI higher than 1 and still be
consistent with one-hit inactivation kinetics
(analogousto host cell shutoff). Finally, when
cells wereinfected with irradiated virus andin
the presence ofguanidine under conditions in
which the effective MOI was reduced to less
than 1by irradiation, shutoff was still
demon-strable, whereas unirradiated virus at an MOI
of 1 in the presence ofguanidine resulted inno
detectable shutoff. Again, the irradiated sam-ple manifests more of an effect than would be
expectedonthebasis ofinfectivity alone. Both
oftheseare consistent withsome sortof
comple-mentation between or participation by
dam-aged genomes.
ACKNOWLEDGMENTS
This work was supported by grant GB18026 from the National Science Foundation and Public HealthService grant AI12387 from theNational Institute ofAllergy and Infectious Diseases.E.E. isthe recipient of Public Health Service careerdevelopmentaward AI 00096from the Na-tionalInstitute ofAllergy and Infectious Diseases.
LITERATURE CITED
1. Bablanian,R. 1972.Depression of macromolecular syn-thesis in cells infected with guanidine-dependent poliovirus under restrictive conditions. Virology 47:255-259.
2. Baltimore, P., R. Franklin, and J. Callendar. 1963. Mengovirus-induced inhibition of host ribonucleic acid and protein synthesis. Biochim. Biophys. Acta 76:425-430.
3. Cole, C., and D. Baltimore.1973.Defectiveinterfering particles ofpoliovirus. II. Nature of the defect. J. Mol.Biol. 76:325-343.
4. Collins,F.D.,and W. K. Roberts.1972.Mechanism of mengovirus-inducedcellinjuryinL-cells:useof in-hibitorsof proteinsynthesis to dissociate
virus-spe-J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
cific events. J. Virol. 10:969-978.
5. Franlcin, R., and D. Baltimore. 1962. Patterns of mac-romolecular synthesis in normaland virus-infected mammalian cells. Cold Spring Harbor Symp.Quant. Biol. 27:175-198.
6. Haase,A. T.,S. Baron, H. Levy, andJ. A.Kasel.1969. Mengovirus-inducedcytopathic effectinL-cells: pro-tectiveeffect ofinterferon.J. Virol. 4:490-495. 7. Holland,J. 1964.Inhibition of host cellmacromolecular
synthesis by high multiplicitiesofpoliovirus under conditions preventingvirussynthesis. J. Mol. Biol. 8:574-581.
8. Huang,A.,and R. Wagner.1965.Inhibition ofcellular RNAsynthesis by non-replicating vesicular stomati-tisvirus. Virology32:337-343.
9. Katagiri, S., Y. Hinuma, and N. Ishida. 1968. Rela-tionship between theadsorptiontocells andantigenic properties in poliovirus particles. Virology 34:797-799.
10. Lawrence, C.,and R.E.Thach.1974. Encephalomyo-carditis virus infection ofmouseplasmacytomacells. I. Inhibition of cellular protein synthesis. J. Virol. 14:598-610.
11. Lovinger, G. G., H. Ping Ling, R.V.Gilden, and M. Hatanka. 1975. Effect ofUVlightonRNA-directed DNApolymeraseactivityofmurineoncornaviruses. J. Virol.15:1273-1275.
12. Maizel, J. 1971. Acrylamide gelelectrophoresis of pro-teinsand nucleicacids,p. 180-244. In K.Mamurosch
and H. Koprowski (ed.), Methods in virology. Aca-demic Press Inc., New York.
13. Marcus, R., and M. Sekellick. 1975. Cell killing by viruses.I. Cell killing by vesicular stomatitis virus: arequirementfortranscription. Virology 63:176-190. 14. Miller, R. L., and G. W. Plagemann. 1974. Effect of
ultraviolet light on mengovirus: formation ofuracil dimers, instability and degradation of capsid, and covalentlinkage of protein to viral RNA. J. Virol. 13:729-739.
15. Moss, B. 1968. Inhibition of HeLa cell protein synthesis bythe vacciniavirion.J. Virol.2:1028-1037. 16. Penman, S., and D. Summers. 1965. Effects on host cell
metabolism following synchronous infection with poliovirus. Virology27:614-620.
17. Piras, R., and B. Vallee. 1967. Carboxypeptidase A quantumyieldsonultravioletirradiation. Biochem-istry 6:2269-2272.
18. Racevskis,J., S. Kerwar, and G. Koch. 1976. Inhibition ofprotein synthesisinreticulocytelysates by poliovi-rions. J. Gen. Virol. 31:135-138.
19. Shaw,J. E.,and D.C. Cox. 1973. Early inhibition of cellularDNAsynthesis by highmultiplicities of in-fectious and UV-inactivated reovirus. J. Virol. 12:704-710.
20. Steiner-Pryor, A., andP.Cooper. 1973. Temperature-sensitivepoliovirus mutants defective in repression of hostproteinsynthesisarealso defectivein struc-turalprotein.J. Gen. Virol.21:215-225.
on November 10, 2019 by guest
http://jvi.asm.org/
![Fig. 1.tion The rate of [35S]methionine incorpora- in the uninfected cells increases linearlyfrom 0.5 to about 3.5 h, where it decreases](https://thumb-us.123doks.com/thumbv2/123dok_us/1551207.107671/3.505.76.211.320.570/fig-methionine-incorpora-uninfected-cells-increases-linearlyfrom-decreases.webp)