Dependent and Leads to Efficient but Delayed Degradation in
Endosomal Compartments
Lise-Andrée Gobeil,aRobert Lodge,a,bMichel J. Tremblaya,b
Centre de Recherche en Infectiologie, Centre Hospitalier Universitaire de Québec-CHUL,a
and Département de Microbiologie-Infectiologie et Immunologie, Faculté de médecine, Université Laval,b
Québec, Canada
HIV-1 endocytosis by a macropinocytosis-like mechanism has been shown to lead to productive infection in macrophages. How-ever, little is known of this pathway. In this study, we examined HIV-1 endocytosis using biochemical approaches and imaging techniques in order to better understand the mechanisms that allow for productive infection of these cells via the endosomal pathway. We show here that this macropinocytosis-like mechanism is not the sole pathway involved in HIV-1 endocytosis in macrophages. However, this pathway specifically requires CCR5 engagement at the cell surface, which in turn suggests that the virus and its coreceptor are present in the endosomal environment simultaneously. Furthermore, although we observed efficient viral degradation following endocytosis, analyses of HIV-1 transport through the endolysosomal pathway revealed that viral degradation is delayed following endosomal internalization, possibly allowing the virus to complete its fusion.
P
roductive entry of enveloped virus into susceptible target cellsrequires envelope mixing with the cellular membrane (1) and
can occur either directly at the plasma membrane (plasma mem-brane fusion [PMF]) or from inside endosomes after cellular en-gulfment of the viral particle (fusion after endocytosis [FAE]). These two distinct entry mechanisms are often defined as pH in-dependent or pH in-dependent. In PMF, receptor binding is neces-sary and sufficient to trigger conformational changes associated with fusion events, whereas in FAE, the low-pH environment in-side the endolysosomal machinery is also required for these changes to occur. FAE, however, can also be triggered by proteol-ysis, a mechanism involving low-pH-activated proteases but that
is nonetheless considered to be pH independent (2,3).
Viruses can exploit a variety of different endosomal pathways for their cellular entry. The most studied and the best described are clathrin-mediated endocytosis (CME), caveola-dependent
en-docytosis, and macropinocytosis (4). In CME, viruses and their
transmembrane receptors are packaged into clathrin-coated vesi-cles. Receptor nucleation at the site of endocytosis is coordinated
by adaptor proteins such as AP-2 (5). Caveolae are lipid
raft-en-riched flask-shaped plasma membrane invaginations that contain
caveolin, which is a cholesterol binding protein (6). Involvement
of caveola structures in viral internalization was described for
sim-ian virus 40 (SV40) (7) and echovirus 1 (8). In macropinocytosis,
large vesicles called macropinosomes are used by the cell to inter-nalize a large amount of solute and membranes. Their formation usually occurs in highly ruffled regions of the plasma membrane and results from its fusion with lamellipodia that fold back onto
themselves (9). Following internalization via one of the
above-mentioned mechanisms, viruses are usually transported to
lyso-somes through endosomal vesicles of increasing acidity (10),
although they can also be transported, specifically for caveola-mediated endocytosis, through neutral pH routes and organelles
such as caveosomes (11). Endosomal trafficking offers many
ad-vantages for viruses that are able to circumvent degradation in the unfriendly environment of the endolysosomal pathway. It al-lows, for example, viruses to escape immune surveillance and
to bypass restriction factors or physical obstacles such as the actin cortex. It also provides viral particles with rapid transport
to the cell nucleus (4).
Although PMF and FAE were previously thought to be mutu-ally exclusive, several studies suggest that some viruses are able to exploit both pathways. Indeed, Newcastle disease, vaccinia, and herpes simplex viruses are capable of pH-independent PMF. However, they may also infect target cells by pH-dependent FAE
(12–14). Another example is Epstein-Barr virus, for which fusion
occurs by both PMF and FAE, depending on the cell type,
al-though in that case, FAE is not triggered by pH acidification (15).
Human immunodeficiency virus type 1 (HIV-1) is another example of a virus that can use both PMF and FAE for cell entry. It has been suggested that FAE may be the result of a rapid virus engulfment after its specific primary receptor (i.e., CD4) and co-receptors (the most commonly used being CXCR4 and CCR5) are engaged at the cell surface (and are thus pH independent). The occurrence of this event would therefore depend on the balance between cellular internalization/degradation rates and the virus
fusion kinetics (16). In agreement with this model, the process of
HIV-1 entry seems to be highly cell type dependent. We and others showed that endocytosis has a major role in HIV-1 infection of
transformed cell lines such as trophoblasts (17) and epithelial cells
of intestinal (18) or cervical (19) origin. However, studies using
primary human CD4⫹T cells, the principal cellular targets of
HIV-1 in peripheral blood, showed that both PMF and FAE occur
in these cells (20), whereas only PMF seems to be important for
infection of dendritic cells (21).
Received12 July 2012 Accepted19 October 2012 Published ahead of print31 October 2012
Address correspondence to Michel J. Tremblay, michel.j.tremblay @crchul.ulaval.ca.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01802-12
on November 7, 2019 by guest
http://jvi.asm.org/
Macrophages are important targets of HIV-1 and contribute to disease progression by persisting after infection, allowing a con-stant and stable dissemination of viruses to other tissues. Their functions as phagocytes and antigen-presenting cells (APCs) are reflected by a constitutive high rate of fluid-phase pinocytosis and
vesicle fusion with lysosomes (22). Furthermore, they have
evolved to express a high level of lysosomal proteases and are therefore able to degrade antigens more efficiently than other
APCs such as dendritic cells (23). Although these characteristics
might impact the ability of HIV-1 to use the endosomal pathway to productively infect these cells, two previous reports point to a role for macropinocytosis or a macropinocytosis-like mechanism (the pathway of HIV endocytic entry in macrophages [PHEEM])
in HIV-1 infection of macrophages (24,25). However, the
mech-anisms allowing for FAE of HIV-1 in these cells are not completely understood. In the present study, we combined biochemical ap-proaches and imaging techniques to analyze HIV-1 endocytosis and trafficking in macrophages in order to better understand these two processes. Our results suggest that PHEEM is not the sole pathway involved in HIV-1 endocytosis in macrophages. How-ever, CCR5 engagement is required to induce HIV-1 internaliza-tion by PHEEM, suggesting that both HIV-1 and CCR5 are present in endosomal compartments after this specific type of en-docytosis. Furthermore, although HIV-1 endocytosis led to effi-cient viral degradation in endolysosomal compartments, virus transport analyses showed that its degradation was delayed, pos-sibly allowing a relatively small number of virions to complete fusion.
MATERIALS AND METHODS
Reagents and antibodies.Chlorpromazine (CPZ) was obtained from Calbiochem (EMD Millipore, Toronto, ON, Canada) and kept as a stock solution of 10 mg/ml in ethanol (EtOH). Dimethyl amiloride (DMA), 5-(N-ethyl-N-isopropyl)amiloride (EIPA), and bafilomycin A1 were all obtained from Sigma-Aldrich (St. Louis, MO) and kept in dimethyl sul-foxide (DMSO) at concentrations of 100 mM, 25 mM, and 100M, respectively. Maraviroc (MVC) was obtained from the NIH AIDS Re-search and Reference Reagent Program (Germantown, MD) and was kept in DMSO at the concentration of 10 mM. The anti-p24 hybridomas 31-90-25 and 183-H12-5C were respectively obtained from the American Type and Culture Collection (Manassas, VA) and the NIH AIDS Research and Reference Reagent Program and were used in our in-house sandwich enzyme-linked immunosorbent assay (ELISA) (26). Both antibodies were purified using MabTrap protein affinity columns (Pharmacia Biotech, Piscataway, NJ) according to the manufacturer’s instructions. For immu-nofluorescence microscopy, mouse anti-EEA1 (BD Biosciences, Franklin Lakes, NJ), mouse anti-CD63 (H5C6; Developmental studies Hybrid-oma Bank, University of Iowa, Iowa City, IA), and mouse anti-Lamp1 (H4A3, DSHB, University of Iowa) were used in combination with Alexa 555-coupled goat anti-mouse IgG (Molecular Probes, Invitro-gen, Life Technologies, Burlington, ON, Canada). DRAQ5 was ob-tained from Biostatus (Leicestershire, United Kingdom) and Lyso-tracker Red from Lonza (Allendale, NJ).
Preparation of macrophages.Peripheral blood mononuclear cells from healthy donors were isolated by Ficoll-Hypaque gradient centrifu-gation. Monocytes were separated from other cells, mostly lymphocytes, by adhesion for 2 h at 37°C in 150- by 20-mm tissue culture dishes fol-lowed by washing with endotoxin-free phosphate-buffered saline (PBS) (Sigma-Aldrich). Monocytes were then allowed to differentiate for 4 days in RPMI 1640 medium supplemented with 5% autologous plasma, 25 ng/ml macrophage colony-stimulating factor (M-CSF), and antibiotics and then washed and allowed to differentiate for 3 more days. Monocyte-derived macrophages (here called macrophages) were recovered by
scrap-ing followscrap-ing incubation with Accutase (eBioscience, San Diego, CA) and further cultured in RPMI 1640 medium supplemented with 5% autolo-gous plasma and antibiotics. Experiments were performed in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and antibiotics.
Molecular constructs.The X4-tropic pNL4-3 (27) and the R5-utiliz-ing pYU-2 (28,29) full-length molecular HIV-1 clones were obtained from the NIH AIDS Research and Reference Reagent program. The pNL4-3BalenvR5-tropic molecular clone was a kind gift of R. Pomerantz (Thomas Jefferson University, Philadelphia, PA) (30). The pNL4-3env⫺ molecular clone was obtained by introducing a⫺1 frameshift in the en-velope (Env) precursor and was a gift from D. E. Ott (National Cancer Institute, Frederick, MD). pJRFLenvand pHCMV-G were from N. R. Landau (NYU School of Medicine, New York, NY) and J. C. Burns (UCSD, San Diego, CA), respectively. The pNL4-3gag-iCFPBalenv con-struct was obtained by replacing the X4-tropic NL4-3envgene with that of the Bal virus in the pNL4-3gag-iCFP construct, which was kindly pro-vided by B. K. Chen (Mount Sinai School of Medicine, New York, NY) (31), using the SalI and BamHI restriction sites. Both pNLC4.3Balenvand pNLC4.3gag-eGFPBalenv were obtained by replacing the X4-tropic NL4-3env gene with that of the Bal virus in pNLC4.3gag-eGFP and pNLC4.3 constructs, which were kindly provided by H. G. Kräusslich (Heidelberg University, Heidelberg, Germany) (32), using the same re-striction sites. The integrity of all constructs was verified by rere-striction analysis and by sequencing.
Virus production.Viruses were produced by calcium phosphate trans-fection of 293T cells as described previously (26). JRFLenv(R5-tropic) and vesicular stomatitis virus (VSV)-G-pseudotyped viruses were obtained by cotransfection of pNL4-3env⫺with pJRFLenvor pHCMV-G, respectively. To obtain NL4-3gag-eGFPBalenvparticles, 293T cells were cotransfected using equimolar amounts of pNLC4.3Balenvand pNLC4.3gag-eGFPBalenv.For live-cell experiments, viruses were inactivated with 2-aldrithiol (AT-2). This mild oxidizing reagent induces covalent modification of the free sulfhydryl groups of the cysteines of internal virion proteins, in particular, the nucleo-capsid proteins. This inactivation method preserves the structural and func-tional integrity of Env glycoproteins on the virus surface and therefore has no impact on its interaction with cell surface receptors (33). Briefly, viral suspen-sions were incubated overnight at 4°C in the presence of 1 mM AT-2 and then purified by ultracentrifugation. A sandwich ELISA previously developed in our laboratory (26) was used to quantify the amount of p24 in each of these viral preparations. Virus infectivity was determined using the TZM-bl re-porter cell line (34,35).
Viral intracellular p24 assay.Macrophages (105cells/well in 24-well
plates) were either left untreated or preincubated with CPZ, DMA, EIPA, MVC, MVC⫹EIPA, or their respective drug vehicle for 30 min and then put in contact with different virus stocks (20 ng of p24/105cells) for 2.5 h
at 37°C to allow for virus internalization (i.e., fusion and endocytosis). Final drug concentrations were 10g/ml, 100M, 25M, and 200 nM for CPZ, DMA, EIPA, and MVC, respectively. Cells were then washed and treated with trypsin for 5 min to remove uninternalized viruses from their surface. This treatment efficiently removed 75 to 80% of the viruses on the cell surface (20 ng of p24/105cells) after a 2-h binding experiment at 4°C
(data not shown). After additional washes, cells were disrupted in lysis buffer (PBS, 0.05% Tween 20, 2.5% Triton X-100, and 0.02% thimerosal), and intracellular p24 was evaluated by ELISA (26), with a limit of detec-tion of 31 pg/ml. The experiment was performed in triplicate, and the amount of intracellular p24 was normalized on cellular viability measured on drug-treated cells. In data illustrated below (seeFig. 3), ultracentri-fuged viruses were used to ensure that no free p24 was present in viral preparations. Typical p24 concentrations in virus preparations used in this work ranged from 0.2 to 3.6 ng/ml, depending on the blood donors, type of viruses, and drug treatments.
Viral degradation assay. Macrophages (105 cells/well in 24-well
plates) were put in contact with different virus stocks (20 ng of p24/105
cells) at 37°C to allow for virus internalization (i.e., fusion and endocyto-sis). Following 2 h of incubation, cells were washed and treated with
on November 7, 2019 by guest
http://jvi.asm.org/
sin for 5 min to remove uninternalized viruses from their surface (time zero) and then incubated at 37°C to allow for virus degradation. Cell lysis was performed at time zero and at 1.5 and 3 h, and the intracellular p24 content was measured at every time point using our in-house ELISA (26). As controls, macrophages were preincubated with bafilomycin A1 (100 nM) for 30 min before addition of viruses and were left in contact with the drug for the whole duration of the experiment. The experiment was per-formed in triplicate.
Cellular viability assay.Cellular metabolism was monitored by incu-bating control drug vehicle- or drug-treated cells (105cells in 24-well
plates) with the CellTiter 96 Aqueous One Solution Cell Proliferation Assay reagent (Promega, Madison, WI) for 1 h at 37°C according to the manufacturer’s instructions. The supernatant absorbance at 490 nm was read using an ELx808 Absorbance Microplate Reader (BioTek, Winooski, VT). The assay was performed in triplicate.
Live cell imaging. Macrophages (105cells/well) were cultured in
8-well Lab-Tek II chambered coverglasses (Nalge Nunc International, Thermo Fisher Scientific, Rochester, NY) and loaded with Lysotracker Red (100 nM) for 1.5 h at 37°C. AT-2-inactivated NL4-3gag-iCFPBalenv -labeled viruses (20 ng of p24/105cells) were then allowed to bind to cells at
4°C for 2 h (in order to obtain synchronized viral entry). Cells were then transferred to warm media containing Lysotracker Red to allow for virus internalization (time zero). Cells were imaged (12 fields) on a WaveFx Spinning Disk confocal microscope (Quorum Technologies, Guelph, ON, Canada) equipped with the appropriate lasers and filters.
Immunofluorescence microscopy. Macrophages (105 cells/well)
were seeded on coverslips in 24-well plates and then allowed to interact with NL4-3gag-eGFPBalenv(20 ng of p24/105cells) at 4°C for 2 h for
synchronization of virus entry. Cells were then washed to remove un-bound viruses and transferred to 37°C to allow for virus internalization (i.e., fusion and endocytosis). After the different incubation times indicated, cells were fixed with 4% paraformaldehyde (in PBS) for 30 min and then processed for imaging as follows: fixed cells were first permeabilized and blocked with a 0.1% (vol/vol) Triton X-100 (Sigma-Aldrich), 1% (wt/vol) bovine serum albumin (Sigma-Aldrich), 10% (vol/vol) human plasma, and 20% (vol/vol) normal goat serum (Jackson ImmunoResearch, West Grove, PA) solution. Macrophages were stained for EEA1 (1/100), CD63 (1/300), or Lamp1 (1/300) for 1 h, washed in PBS, and further stained with secondary antibody (1/500) and the DNA probe DRAQ5 (1/1,000) for 30 min. Micro-scope slides were then mounted using Fluoromount G (Southern Biotech, Birmingham, AL). Cells were imaged (4 fields per marker per time point) by confocal laser scanning microscopy, using a FluoView FV300 microscope (Olympus, Center Valley, PA) equipped with the appropriate lasers and fil-ters.
Image processing and deconvolution and colocalization analyses. Image processing was made using either the Volocity 4.2.1 (PerkinElmer, Waltham, MA) or NIH ImageJ 1.42 software. Image deconvolution, co-localization analyses, and the number of intracellular particles (using the measurement protocol) were obtained using Volocity 4.2.1. In particular, for estimating the number of intracellular viruses, HIV-1 particles were identified according to pixel intensity using the same threshold as that used in colocalization analyses.
Statistical analysis.Each data set was independently analyzed for Gaussian distribution using the Kolmogorov-Smirnov (KS) normality test. Paired Studentttests (seeFig. 2C,3, and5) and one-samplettests (see
Fig. 1,2AandB, and4) were performed using GraphPad Prism 5 software.
RESULTS
Macropinocytosis is involved in HIV-1 endocytosis in
macro-phages.Two separate groups have shown that HIV-1 replication
is reduced in macrophages infected in the presence of amiloride
derivatives such as DMA and EIPA (24,25). These drugs
specifi-cally inhibit macropinocytosis by acting on Na(⫹)/H(⫹)exchange
and submembranous pH, thereby affecting F-actin remodeling
(36). The authors concluded that macropinocytosis is involved in
HIV-1 infection in such cells. However, only EIPA was efficient in reducing HIV-1 integration, as opposed to DMA, which had no
significant effect (24). Therefore, we set out to determine if this
discrepancy could reflect an unspecific effect of amiloride deriva-tives on the late steps of the viral cycle. We designed an intracel-lular HIV-1 p24 assay that allows for quantification of virus inter-nalization and measures the effects of these drugs on the early events of the virus cycle. In this case, intracellular p24 represents both endosomal and cytosolic viral particles at a given time point and does not discriminate between fusion (PMF and FAE) and unproductive endocytosis. However, the assay will detect any sig-nificant reduction in HIV-1 endocytosis, as the vesicular fraction was shown to represent at least 45% of total intracellular p24 in
macrophages in previous fractionation studies (25).
Cells were first pretreated with EIPA or DMA for 30 min before allowing for the internalization of viral particles for 2.5 h at 37°C. After a 5-min trypsin treatment, cell lysis was performed and in-tracellular p24 was quantified by ELISA. Data were normalized
according to cellular metabolic activity as measured by anin vitro
cellular proliferation assay (as described in Materials and Meth-ods). We first tested the reliability of our assay using a VSV-G-pseudotyped virus, as its entry occurs exclusively by endocytosis and as it has been shown to exploit macropinocytosis in some cells
(37–39). As expected, both EIPA and DMA reduced VSV-G
inter-nalization in macrophages, as mean intracellular p24 concentra-tions were, respectively, 1.27 ng/ml, 0.79 ng/ml, and 0.92 ng/ml
for DMSO-, EIPA-, and DMA-treated cells (Fig. 1A). However,
this effect was greater for EIPA, with a reduction of 38% compared
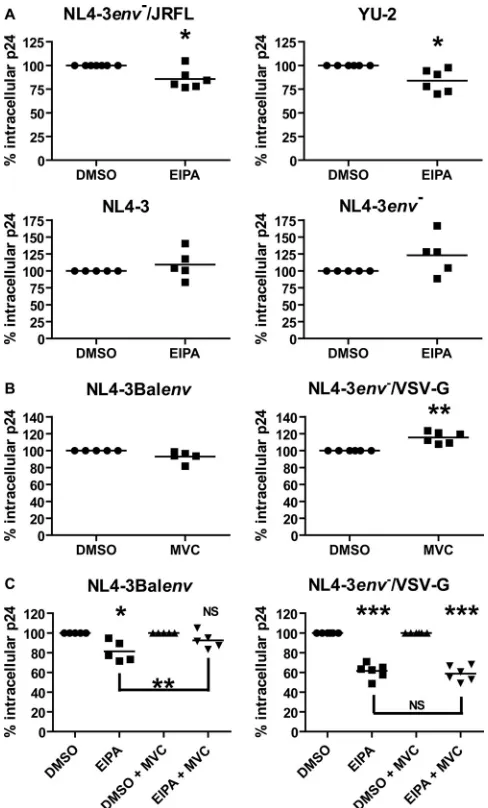
FIG 1HIV-1 endocytosis involves macropinocytosis in macrophages. Cells were pretreated for 30 min with EIPA (25M), DMA (100M), or the equiv-alent amount of DMSO (as vehicle control) before allowing for internalization of NL4-3env⫺/VSV-G (A) or NL4-3Balenv(B) viruses for 2.5 h at 37°C. Fol-lowing a 5-min treatment with trypsin to remove membrane-bound viruses, cell lysis was performed. Total intracellular p24 was quantified by ELISA and normalized for cell viability. Results represent mean values of at least 11 inde-pendent experiments performed with macrophages derived from different blood donors and are expressed as the percentage of intracellular p24 com-pared to that found in cells treated with DMSO (considered 100%). Asterisks denote statistically significant differences (**,P⬍0.01; ***,P⬍0.001) com-pared with DMSO-treated cells.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.354.487.62.278.2]to 24% with DMA. These results confirm that our assay is sensitive enough to detect variations in HIV-1 endocytosis. Therefore, we
tested a fully replicative R5-tropic virus, NL4-3Balenv, in the same
assay. EIPA successfully reduced NL4-3Balenvinternalization in
macrophages (mean measured intracellular p24 concentrations of 0.64 ng/ml for DMSO and 0.53 ng/ml for EIPA, for a reduction of 17% after normalizing for cell viability), although with a less
pro-nounced effect than with the VSV-G-pseudotyped virus (Fig. 1B).
However, DMA failed to reduce HIV-1 endocytosis (mean intra-cellular p24 concentration of 0.74 ng/ml), confirming the
previ-ously reported lower efficiency of this drug (24). Considering our
observations using EIPA, we conclude that a macropinocytosis-like mechanism is involved in HIV-1 endocytosis in macrophages. Binding of gp120 to CCR5 is required for HIV-1
macropi-nocytosis in macrophages.In order to investigate the
require-ments for HIV-1 macropinocytosis in macrophages, we repeated
the experiment illustrated inFig. 1without DMA, using viruses
that bind different receptors, i.e., NL4-3env⫺/JRFLenvand YU-2
(R5-tropic), which can bind both CD4 and CCR5, and NL4-3 (X4-tropic), which can bind both CD4 and CXCR4. Their inter-nalization in the presence of EIPA was compared with that of an
Env-deficient virus (i.e., NL4-3env⫺) that can bind none of the
above and was used as a control. As shown inFig. 2A, the presence
of EIPA reduced the mean intracellular p24 concentration
follow-ing a 2.5-h virus internalization of either NL4-3env⫺/JRFLenv
(1.18 ng/ml for DMSO and 0.92 ng/ml for EIPA) or YU-2 (0.79 ng/ml for DMSO and 0.68 ng/ml for EIPA), confirming our
re-sults obtained with the NL4-3Balenvvirus. As for 3 and
NL4-3env⫺viruses, we could not significantly reduce their endocytosis
using EIPA (mean measured intracellular p24 concentrations of 0.43 ng/ml and 0.51 ng/ml and of 0.38 ng/ml and 0.53 ng/ml for
DMSO- and EIPA-treated cells for NL4-3 and NL4-3env⫺,
respec-tively). These results suggest that gp120 binding to CD4 is not sufficient and that CCR5 interaction is required to promote an HIV-1 macropinocytosis-like mechanism in macrophages.
In order to confirm the requirement of CCR5 in such a mech-anism, we tested HIV-1 endocytosis in the presence of MVC, a
CCR5 antagonist. As shown inFig. 2B, NL4-3Balenv
internaliza-tion was only slightly reduced (0.98 ng/ml, compared to 1.03 ng/ml for DMSO) in the presence of MVC, whereas that of
NL4-3env⫺/VSV-G was increased (1.09 ng/ml, compared to 0.94 ng/ml
for DMSO). However, the effect of EIPA on NL4-3Balenv
inter-nalization was significantly reduced by MVC (Fig. 2C), while that
on NL4-3env⫺/VSV-G was not. Indeed, the mean intracellular
p24 concentrations measured for NL4-3Balenv in cells treated
with DMSO or EIPA were 1.03 ng/ml or 0.77 ng/ml in the absence of MVC, respectively, and 0.98 ng/ml or 0.82 ng/ml, respectively, with MVC. These results correspond to reductions of viral inter-nalization in the presence of EIPA of 25% and 16%, respectively,
following normalization for cell viability. As for NL4-3env⫺/
VSV-G, we obtained 0.94 ng/ml and 0.53 ng/ml of intracellular p24 in DMSO- and EIPA-treated cells, as opposed to 1.09 ng/ml
and 0.57 ng/ml in DMSO⫹MVC- and EIPA⫹MVC-treated
mac-rophages, respectively, corresponding to reductions in intracellu-lar p24 of 56% and 53%. These data confirm the role of CCR5 in the macropinocytosis-like HIV-1 endocytosis.
CCR5 binding is involved but not required for efficient HIV-1
endocytosis in monocyte-derived macrophages (MDMs). As
CCR5 binding by gp120 is required to induce macropinocytosis of HIV-1 in macrophages, we investigated if this interaction was
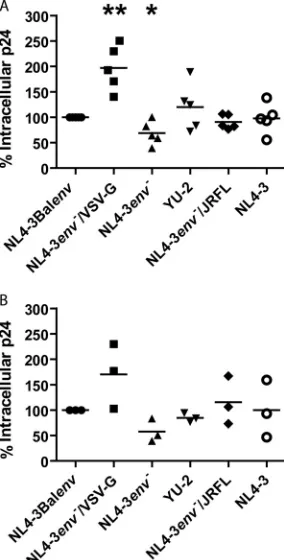
re-quired for its efficient endocytosis. We therefore compared the amounts of intracellular p24 of the different viruses following a 2.5-h internalization at 37°C. In these experiments, we used ultra-centrifuged viruses, eliminating the possibility that free HIV-1 p24 present in our viral preparations could be internalized and affect results. As expected, R5-tropic viruses were all internalized with
the same efficiency (Fig. 3A), as similar mechanisms are involved
in their internalization. However, endocytosis efficiency was
FIG 2CCR5 binding by gp120 is required for macropinocytosis of HIV-1 in macrophages. Cells were pretreated for 30 min with EIPA (25M) (A), mara-viroc (MVC, 200 nM) (B), EIPA⫾MVC (C), or the equivalent amount of DMSO (as vehicle control), before allowing for NL4-3env⫺/JRFLenv(top left), YU-2 (top right), NL4-3 (bottom left), or NL4-3env⫺(bottom right) (A), or NL4-3Balenv(left) or NL4-3env⫺/VSV-G (right) (B and C) viruses to be in-ternalized for 2.5 h at 37°C. After a 5-min trypsin treatment to remove mem-brane-bound viruses, cell lysis was performed. Total intracellular p24 was quantified by ELISA and normalized for cell viability. Results represent mean values of 5 or more independent experiments performed with macrophages derived from different blood donors and are expressed as the percentage of intracellular p24 compared to that found in cells treated with DMSO (consid-ered 100%). Asterisks denote statistically significant differences (*,P⬍0.05; **,P⬍0.01; ***,P⬍0.001; NS, not significant) compared with DMSO-treated cells.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.299.543.66.470.2]highly variable for macrophages derived from one blood donor to another (0.5 to 2.4 ng of p24/ml). The amount of intracellular p24 measured following endocytosis of the VSV-G-pseudotyped virus was greater than that of other viruses (1.0 to 3.2 ng of p24/ml). This was also expected, as the endocytosis of this virus is known to be very efficient, and its degradation rate is expected to be lower than that of R5-tropic viruses. Intracellular p24 detected following
endocytosis of NL4-3env⫺virus was only slightly lower than that
of its R5-tropic counterparts (0.4 to 1.7 ng of p24/ml), and similar amounts of intracellular p24 were obtained for NL4-3 (0.5 to 2.4 ng of p24/ml) and R5-tropic viruses, even though NL4-3 and
NL4-3env⫺do not bind CCR5. As these experiments were all
per-formed using the same set of viral productions, and to ensure that results were reproducible, we repeated the same experiment with 3 different viral productions in macrophages derived from the same
blood donor and obtained similar results (Fig. 3B), although we
observed different endocytosis efficiency levels from one viral pro-duction to another. Taken together, these results indicate that efficient HIV-1 endocytosis can occur in the absence of CCR5 binding.
CME is not significantly involved in HIV-1 endocytosis in
macrophages. As CCR5 binding is not required for efficient
HIV-1 engulfment in macrophages, macropinocytosis is thus not the sole mechanism involved in virus endocytosis. We therefore investigated which other endocytosis mechanism(s) could be in-volved in HIV-1 internalization in macrophages. We first dis-carded caveolin-mediated endocytosis, given that Carter and col-leagues previously showed that both caveolin-1 RNA transcripts
and protein were not detectable in macrophages (24). Although
we did detect caveolin-1 RNA transcripts in our macrophage preparations, we were unable to detect the protein by either West-ern blotting or immunofluorescence microscopy using antibodies from different sources (data not shown). We then investigated whether CME could be involved in HIV-1 endocytosis. CME is unlikely to lead to productive HIV-1 infection in macrophages, given that previous reports have shown that inhibition of this
pathway by CPZ has no effect on HIV-1 integration (24).
How-ever, electron microscopy studies of macrophages that had inter-nalized HIV-1 particles determined that the virus can be found in
clathrin-coated vesicles (25), suggesting that CME may be
in-volved in CCR5-independent HIV-1 internalization. Thus, we took advantage of our HIV-1 internalization assay, which detects intracellular p24 following either productive or nonproductive infection, in order to investigate the role of CME in endocytosis of HIV-1 in macrophages. Cells were pretreated with CPZ or its ve-hicle for 30 min prior to the internalization of different virus par-ticles for 2.5 h at 37°C. Macrophages were then treated with tryp-sin for 5 min and extensively washed prior to lysis and quantification of intracellular p24 by ELISA, and data were nor-malized according to cellular metabolic activity. We found that CPZ had either no effect or slightly increased HIV-1 internaliza-tion in macrophages derived from most of the blood donors tested (Fig. 4), therefore suggesting that CME is not a major internaliza-tion pathway in macrophages.
HIV-1 particles are targeted for degradation following their
endocytosis in macrophages.In an attempt to investigate the fate
of virions following endocytosis and vesicular transport in
mac-rophages, we adapted ourin vitrointracellular p24 assay to follow
intracellular p24 in a time course study. In this experiment,
treat-ment with bafilomycin A1, an inhibitor of vesicular H⫹-ATPase
(40), blocks vesicular acidification and hence should inhibit viral
degradation. Briefly, macrophages were treated with bafilomycin A1 for 30 min or left untreated prior to their contact with
NL4-3Balenv, NL4-3env⫺, or NL4-3env⫺/VSV-G viruses and then
fur-ther incubated for 2 h at 37°C to allow for virus internalization. Cells were then washed and treated with trypsin for 5 min to remove bound viruses (time zero). Macrophages were then re-turned to 37°C, in the presence or absence of the drug, and intra-cellular p24 amounts were measured over time using our home-made p24 ELISA, following cell lysis at different time points. We observed a loss of intracellular p24 over time, in untreated
mac-rophages, for all the viruses tested (Fig. 5). However, the reduction
in intracellular p24 was much slower, over time, for the VSV-G-pseudotyped virus, which is consistent with efficient viral escape from endosomes following their acidification. Surprisingly, we did not detect a significant difference between the rates of loss in
intracellular p24 for the NL4-3Balenv and NL4-3env⫺viruses,
suggesting that the great majority of HIV-1 particles are degraded in macrophages following virus internalization. Furthermore, ba-filomycin A1 almost completely inhibited intracellular p24 de-crease over time for all the viruses, confirming that the dede-crease of
FIG 3CCR5 binding is involved but not required for efficient HIV-1 endo-cytosis in macrophages. Cells were allowed to internalize equal amounts (stan-dardized in term of p24 content) of the listed virus stocks (i.e., ultracentrifuged viral productions) for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. Total intracellular p24 was quantified by ELISA. Results are expressed as percentages of NL4-3Balenv intracellular p24 and represent mean values from 5 independent experiments performed with the same viral production on macrophages derived from 5 different blood donors (A) or mean values from a single experiment per-formed with 3 different viral productions on macrophages derived from a single blood donor (B). Asterisks denote statistically significant differences (*, P⬍0.05; **,P⬍0.01) compared with macrophages exposed to NL4-3Balenv viruses (considered 100%).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.92.234.68.348.2]intracellular p24 over time was due to endosomal degradation. It can be concluded that the majority of internalized HIV-1 particles are degraded after their internalization in macrophages.
HIV-1 is transported into compartments of increasing
acid-ity following endocytosis in macrophages. We next studied
HIV-1 transport into vesicular compartments in live cell experi-ments. Macrophages were first labeled using Lysotracker Red, a cell-permeant dye that gradually becomes fluorescent when
ex-posed to the low pH of acidified vesicles. Next, NL4-3gag
-iCFP-Balenvfluorescent HIV-1 particles were allowed to bind cells at
4°C for 2 h. The temperature was then shifted to 37°C (time zero) to permit viral internalization. This technique allowed us to syn-chronize viral endocytosis and to increase viral detection, there-fore facilitating colocalization studies. Cells were thus imaged
ev-ery 20 to 30 min. As depicted in Fig. 6, the average virus
colocalization with Lysotracker Red-labeled vesicles increased over time, reaching a peak level at 60 min following the
tempera-ture shift, and then decreased slowly after 90 min. This suggests that HIV-1 particles are transported to acidified vesicles following their endocytosis in macrophages. Peak colocalization coefficients varied from 0.50 to 0.75 in cell samples derived from the three blood donors tested. However, overall HIV-1 colocalization with highly acidified (highly positive for Lysotracker Red) endosomes was not significant (data not shown), thus suggesting that viruses were efficiently degraded before reaching these intracellular com-partments. These results indicate that HIV-1 is transported and degraded in acidified vesicles following its endocytosis in macro-phages.
The endolysosomal pathway is involved in HIV-1 transport
and degradation following endocytosis in macrophages.The
en-dolysosomal route is the main vesicular pathway involved in cargo transport to lysosomes for degradation, and it has also been shown
to be involved in transport of macropinocytosed cargos (41). We
therefore sought to determine its involvement in HIV-1 transport
FIG 4CME is not significantly involved in HIV-1 endocytosis in macrophages. Cells were pretreated for 30 min with CPZ (10g/ml) or the equivalent amount of ethanol (EtOH, as vehicle control) before being allowed to internalize the different virus preparations for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. Total intracellular p24 was quantified by ELISA and normalized for cell viability. Results represent mean values from 6 independent experiments performed on macrophages derived from different blood donors and are expressed as the percentage of intracel-lular p24 compared to that found in cells treated with ethanol (considered 100%).
FIG 5HIV-1 particles are targeted for degradation following their endocytosis in MDMs. Cells were either left untreated (left panel) or pretreated with 100 nM bafilomycin A1 (right panel) for 30 min at 37°C before allowing for NL4-3Balenv, NL4-3env⫺/VSV-G, or NL4-3env⫺viruses to be internalized for 2 h at 37°C. Cells were then washed, trypsinized to remove bound viruses (time zero), and incubated at 37°C (with or without the drug) to allow for virus degradation. Intracellular p24 was quantified over time by ELISA after cell lysis. Results are expressed as the percentage of the initial amount of intracellular p24 at time zero and represent mean⫾standard error of the mean (SEM) values from 3 independent experiments performed on macrophages derived from different blood donors. Asterisks denote statistically significant differences (**,P⬍0.01; ***,P⬍0.001) compared with macrophages exposed to NL4-3env⫺/VSV-G viruses.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.136.450.65.227.2] [image:6.585.135.452.509.667.2]in macrophages. We first allowed NL4-3gag-eGFPBalenvviruses to bind to macrophages grown on coverslips for 2 h at 4°C and then shifted the temperature to 37°C to synchronize viral internal-ization. Macrophages were then fixed at different time points, stained for EEA1, CD63, and Lamp1, which are specific markers for early endosomes, multivesicular bodies/late endosomes, and lysosomes, respectively, and mounted for microscopy. As ex-pected, we observed colocalization between HIV-1 and all the
en-dolysosomal markers tested (Fig. 7). In all cases, colocalization
was first low or absent and then increased to reach a peak level as viruses entered specific intracellular compartments and finally creased as viruses progressed in the pathway or were being de-graded. Peak colocalization for EEA1, CD63, and Lamp1 was se-quential in time, suggesting viral transfer from early to late endosomes and then to lysosomes. However, colocalization re-mained high for EEA1 throughout the experiment, indicating that some viruses remained in this compartment for a longer time period before being transferred. The loss of green fluorescent pro-tein (GFP)-labeled particles occurred 30 min after the tempera-ture shift, corresponding to peak Lamp1 colocalization. This
sug-gests that viral degradation begins when viruses reach Lamp1-positive compartments, corresponding to lysosomes, and that transport to these compartments is important for HIV-1 degrada-tion. Overall, these results indicate that the endolysosomal path-way is involved in HIV-1 transport and degradation following endocytosis in macrophages.
DISCUSSION
Previous studies have shown that HIV-1 internalization by mac-ropinocytosis, specifically PHEEM, could lead to productive
in-fection in macrophages (24, 25), even though these cells were
shown to degrade antigens more efficiently than other APCs such
as dendritic cells (23), which are restrictive to FAE and only allow
PMF (21). In the current study, we confirmed the contribution of
a macropinocytosis-like pathway in the process of HIV-1 infec-tion in macrophages and identified factors that may contribute to the occurrence of FAE.
Our intracellular p24 assay confirmed the role of macropi-nocytosis in HIV-1 internalization in macrophages, although the effect of EIPA was only moderate compared to previously
re-FIG 6HIV-1 transits into increasingly acidic compartments following its endocytosis in macrophages. Cells were loaded with 100 nM Lysotracker Red for 1.5 h and then allowed to bind NL4-3gag-iCFPBalenvparticles at 4°C for 2 h for viral entry synchronization prior to internalization at 37°C (time zero). Cells were imaged for 180 min, and colocalization analysis was performed. Confocal images (z-projection of 8 to 12 1-m slices) shown in the upper panel are representative of HIV-1 colocalization with Lysotracker Red at different time points, whereas the graph in the lower panel shows HIV-1 colocalization coefficients over time. The experiment was performed on macrophages derived from 3 different blood donors, and data presented are from one representative donor. Bar⫽5m.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.136.453.63.435.2]ported observations (24). However, this is attributable to the na-ture of our assay, which measures all viruses in endosomal com-partments, whether these pathways are productive or not. Furthermore, this assay does not discriminate between endosomal
and cytosolic p24. We did not specifically fully characterize this macropinocytosis as PHEEM. Nevertheless, we obtained results similar to those of Carter and colleagues concerning the effects of DMA and EIPA on the early stages of the HIV-1 viral cycle.
In-FIG 7HIV-1 transits through the endolysosomal pathway in macrophages. Cells were allowed to bind NL4-3gag-eGFPBalenvparticles at 4°C for 2 h for viral entry synchronization prior to internalization at 37°C (time zero). Cells were then fixed at different time points, permeabilized, stained for EEA1, CD63, or Lamp1, and mounted prior to fluorescent confocal microscopy. Image deconvolution, colocalization, and virus count analyses were then performed as described in Materials and Methods. (A) Confocal images (z-projection of 8 to 12 0.5-m slices) are representative of HIV-1 colocalization with EEA1 (top panels), CD63 (middle panels), or Lamp1 (bottom panels) at the peak of colocalization. (B) Three graphs represent colocalization coefficients at different time points for all three endolysosomal markers. (C) Comparison of colocalization coefficients for all markers. (D) Total number of fluorescent viral particles detected over time in 12 random fields. The experiment was performed on macrophages derived from 4 different blood donors, and data presented are from one representative donor. Bar⫽5m.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.111.475.62.592.2]deed, we observed a significant effect of EIPA on HIV-1 endocy-tosis, whereas DMA had no significant effect. Considering that DMA reduced VSV-G-pseudotyped virus internalization by only 24% compared to 38% for EIPA, it is clear that DMA is less effi-cient than EIPA in reducing macropinocytosis, as suggested by Carter et al. This is much more likely to explain the discrepancy between the effects of DMA on HIV-1 infection and on viral inte-gration presented in their paper, rather than a nonspecific effect of amiloride derivatives on the late steps of the viral cycle.
In addition to confirming previously reported
macropinocy-tosis (24,25), our assay further allowed us to show that
macropi-nocytosis is not the only pathway involved in HIV-1 endocytosis in macrophages. Indeed, treating the cells with EIPA did not sig-nificantly impact the internalization of X4-tropic and Env-defi-cient viruses, although virus internalization efficiency was the same for R5- and X4-tropic viruses. Furthermore, using the same p24 intracellular assay, we were able to identify CCR5 binding as a prerequisite for macropinocytosis of HIV-1. When comparing different viruses, we observed that macropinocytosis was involved only in R5-virus internalization. Furthermore, using MVC, we
significantly inhibited macropinocytosis of NL4-3Balenv.
Al-though it may be surprising that MVC does not significantly
re-duce NL4-3Balenvinternalization, this can be easily explained by
the drug’s action. Indeed, while EIPA allows for the binding of HIV-1 to CCR5 without internalization, MVC blocks this bind-ing. In such an event, the virus will use an alternative endocytosis receptor.
The involvement of CCR5 in macropinocytosis is significant, as the FAE route can occur only when both HIV-1 and its core-ceptor are present simultaneously in the same endosomal com-partment, as a result of coreceptor engagement and engulfment at
the plasma membrane (16). This unique characteristic of
mac-ropinocytosis explains its specific disposition for HIV-1 infection. Indeed, this pathway is the only one that has been associated with
productive HIV-1 infection in macrophages (24,25).
We were unable to specifically identify another major route of HIV-1 endocytosis in this specific cell type. It is possible that a variety of receptors are involved in nonproductive internalization of HIV-1 in these cells. It is known that gp120 can bind to a variety of cell surface receptors present at the cell surface, specifically via interactions of its glycosylated residues with sugar-binding pro-teins or via electrostatic interactions between its positive charges
and the negative charges of surface molecules (42). Furthermore,
different cellular constituents can be incorporated onto the viral envelope upon viral budding and may mediate HIV-1 internaliza-tion via interacinternaliza-tion with their natural counterreceptor at the target
cell surface (43). These molecules are likely to be responsible for
endocytosis of Env-deficient HIV-1 particles used in our experi-ments. It should be further taken into consideration that these yet-to-be-characterized pathways may vary in macrophages derived from different blood donors and that the membrane expression of differ-ent receptors is dependdiffer-ent on the cellular activation status of
macro-phages (44). In this regard, we previously demonstrated that HIV-1 is
efficiently internalized by CME in M1- and M2a-activated MDMs in a CCR5-independent manner and that macropinocytosis is not a
ma-jor internalization pathway in these cells (45).
Following endocytosis, virus degradation was monitored by measuring intracellular p24 in a time course experiment. Surpris-ingly, we did not detect any difference in degradation between
NL4-3Balenvand NL4-3env⫺viruses, although NL4-3Balenvis
fusion competent and some of these viruses should undergo PMF or efficiently escape degradation in endosomal compartments by
FAE. This unexpected result is not due to a loss in NL4-3Balenv
cytosolic p24 following fusion (PMF⫹FAE). Indeed, intracellular
p24 of VSV-G-pseudotyped HIV-1, which is mostly cytosolic given this virus’s efficient pH-dependent FAE, did not decrease significantly over time in untreated cells. Furthermore, any
poten-tial loss of intracellular p24 following fusion (PMF⫹FAE), in the
case of NL4-3Balenvvirus, should translate into a time-dependent
decrease in capsid protein even in bafilomycin A1-treated cells; we did not observe such a decrease. Another possible explanation for
the observed NL4-3Balenvdegradation rate would be that the
ma-jority of internalized viruses are present in endosomal compart-ments and therefore that cytosolic p24 contribution to total intra-cellular p24 is negligible. If such is the case, HIV-1 fusion, either at the plasma or at the endosomal membranes, is a relatively rare and inefficient event in macrophages. This hypothesis is in contradic-tion with fraccontradic-tionacontradic-tion studies that showed that cytosolic p24 rep-resents about 55% of total intracellular p24 in macrophages after a 2-h viral internalization of fusion-competent R5-tropic viruses
(25). However, cytosolic p24 is likely to be overestimated in this
study, as the author showed that 30% of an Env-deficient virus was also detected in the cytosolic fraction.
The gradual gain and following loss in colocalization of HIV-1 with Lysotracker Red in live cell confocal microscopy suggest that viral particles are efficiently transported to compartments of grad-ually lowering pH after their endocytosis-mediated internaliza-tion in macrophages. Although colocalizainternaliza-tion with Lysotracker Red was highly significant, colocalization with endosomes that were highly positive for Lysotracker Red remained low through-out the experiment. This suggests that HIV-1 does not reach highly acidic compartments and thereby confirms the efficient degradation of viral particles during their transport. Our further observations showed that viral particles are efficiently transferred from early to late endosomes and then to lysosomal compart-ments, following their internalization. However, a decrease in GFP-positive particle detection, which corresponds to viral deg-radation, was observed only after 30 min of internalization, cor-relating with HIV-1 entry into Lamp1-positive compartments. Therefore, during macropinocytosis, as the HIV-1 coreceptor is engaged before internalization, few viral particles may undergo fusion before their subsequent degradation. Indeed, blocking HIV-1 fusion at a temperature-sensitive step that allows for virus binding to its coreceptor (temperature-arrested stage) and
shift-ing to 37°C result in rapid fusion, occurrshift-ing in as fast as 5 min (46).
Furthermore, although we observed that the majority of HIV-1 particles progressed efficiently into the endolysosomal pathway following internalization, some particles remained in EEA1-posi-tive endosomes for a longer period prior to sorting. This is signif-icant given that FAE efficiency is dependent on viral degradation rates and that these compartments represent a friendly
environ-ment for the virus (32). A previous study by Lakadamyali and
colleagues has shown that there are two types of early endosomal
compartments, which differ in their maturation speed (47).
Ac-cording to this study, the slow-maturing early-endosome popula-tion is involved in the sorting of cargos to the recycling endo-somes, whereas the fast-maturing population is involved in the
progression of cargos into the endolysosomal pathway (47). It would
be interesting to distinguish these two types of early endosomal com-partments in further studies to establish their respective involvement
on November 7, 2019 by guest
http://jvi.asm.org/
in HIV-1 transport, as well as their possible implication in FAE, par-ticularly given that HIV-1 was previously observed in
transferrin-positive sorting compartments in macrophages (24).
Taken together, our data suggest that although endocytosis represents a dead end for the majority of HIV-1 particles in mac-rophages, macropinocytosis specifically offers favorable condi-tions for FAE, as this pathway depends on the previous interaction of HIV-1 gp120 with CCR5. Furthermore, a small number of virus particles may benefit from a slow progression into the endolyso-somal pathway, resulting in their delayed degradation.
ACKNOWLEDGMENTS
We thank Julie-Christine Lévesque, Ann Rancourt, and Sachiko Sato (IDRC BioImagery Platform) for helpful discussions and assistance. We also thank Odette Simard, Caroline Côté, and Marc-André Roy for technical help.
This study was supported by an Emerging Team Grant in HIV Pathogenesis to M.J.T. from the Canadian Institutes of Health Re-search (CIHR) (grant number HET-85519). L.-A.G. is the recipient of CIHR and Fonds de la Recherche en Santé du Québec Doctoral Awards. M.J.T. holds the Canada Research Chair in Human Immuno-Retrovirology (Tier 1 level).
REFERENCES
1.Smith AE, Helenius A.2004. How viruses enter animal cells. Science
304:237–242.
2.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM.2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science308:1643–1645.
3.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P.2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A.102:11876 – 11881.
4.Mercer J, Schelhaas M, Helenius A.2010. Virus entry by endocytosis. Annu. Rev. Biochem.79:803– 833.
5.Doherty GJ, McMahon HT.2009. Mechanisms of endocytosis. Annu. Rev. Biochem.78:857–902.
6.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG.1992. Caveolin, a protein component of caveolae membrane coats. Cell68:673– 682.
7.Anderson HA, Chen Y, Norkin LC.1996. Bound simian virus 40 trans-locates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell7:1825–1834. 8.Marjomaki V, Pietiainen V, Matilainen H, Upla P, Ivaska J, Nissinen L,
Reunanen H, Huttunen P, Hyypia T, Heino J.2002. Internalization of echovirus 1 in caveolae. J. Virol.76:1856 –1865.
9.Lim JP, Gleeson PA.2011. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol. Cell Biol.89:836 – 843.
10. Weisz OA.2003. Acidification and protein traffic. Int. Rev. Cytol.226: 259 –319.
11. Pelkmans L, Kartenbeck J, Helenius A.2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol.3:473– 483.
12. Cantin C, Holguera J, Ferreira L, Villar E, Munoz-Barroso I. 2007. Newcastle disease virus may enter cells by caveolae-mediated endocytosis. J. Gen. Virol.88:559 –569.
13. Nicola AV, McEvoy AM, Straus SE.2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol.77:5324 –5332.
14. San Roman K, Villar E, Munoz-Barroso I.1999. Acidic pH enhancement of the fusion of Newcastle disease virus with cultured cells. Virology260: 329 –341.
15. Miller N, Hutt-Fletcher LM.1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol.66:3409 –3414.
16. Permanyer M, Ballana E, Este JA.2010. Endocytosis of HIV: anything goes. Trends Microbiol.18:543–551.
17. Vidricaire G, Imbeault M, Tremblay MJ. 2004. Endocytic host cell machinery plays a dominant role in intracellular trafficking of incoming
human immunodeficiency virus type 1 in human placental trophoblasts. J. Virol.78:11904 –11915.
18. Bourinbaiar AS, Phillips DM.1991. Transmission of human immuno-deficiency virus from monocytes to epithelia. J. Acquir. Immune Defic. Syndr.4:56 – 63.
19. Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB.2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endo-somes. Cell137:433– 444.
20. Schaeffer E, Soros VB, Greene WC.2004. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in hu-man CD4 T lymphocytes. J. Virol.78:1375–1383.
21. Janas AM, Dong C, Wang JH, Wu L.2008. Productive infection of human immunodeficiency virus type 1 in dendritic cells requires fusion-mediated viral entry. Virology375:442– 451.
22. Swanson JA.1989. Phorbol esters stimulate macropinocytosis and solute flow through macrophages. J. Cell Sci.94(Pt 1):135–142.
23. Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. 2005. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science307:1630 –1634.
24. Carter GC, Bernstone L, Baskaran D, James W.2011. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology409:234 –250.
25. Maréchal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O.
2001. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol.75:11166 –11177.
26. Bounou S, Leclerc JE, Tremblay MJ.2002. Presence of host ICAM-1 in laboratory and clinical strains of human immunodeficiency virus type 1 increases virus infectivity and CD4(⫹)-T-cell depletion in hu-man lymphoid tissue, a major site of replicationin vivo.J. Virol.76: 1004 –1014.
27. Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA.1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol.59:284 –291.
28. Li Y, Hui H, Burgess CJ, Price RW, Sharp PM, Hahn BH, Shaw GM.
1992. Complete nucleotide sequence, genome organization, and bio-logical properties of human immunodeficiency virus type 1in vivo: evidence for limited defectiveness and complementation. J. Virol.66: 6587– 6600.
29. Li Y, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH.1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol.65:3973– 3985.
30. Dornadula G, Zhang H, Shetty S, Pomerantz RJ.1999. HIV-1 virions produced from replicating peripheral blood lymphocytes are more infec-tious than those from nonproliferating macrophages due to higher levels of intravirion reverse transcripts: implications for pathogenesis and trans-mission. Virology253:10 –16.
31. Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK.2007. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol.81:12596 –12607. 32. Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich
HG.2004. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol.78: 10803–10813.
33. Rossio JL, Esser MT, Suryanarayana K, Schneider DK, Bess JW, Jr, Vasquez GM, Wiltrout TA, Chertova E, Grimes MK, Sattentau Q, Arthur LO, Henderson LE, Lifson JD.1998. Inactivation of human immunodefi-ciency virus type 1 infectivity with preservation of conformational and func-tional integrity of virion surface proteins. J. Virol.72:7992– 8001.
34. Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D.1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophage-tropic isolates of human immunodeficiency virus type 1. J. Virol.72:2855– 2864.
35. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC.2002. Emergence of resistant human immuno-deficiency virus type 1 in patients receiving fusion inhibitor (T-20) mono-therapy. Antimicrob. Agents Chemother.46:1896 –1905.
on November 7, 2019 by guest
http://jvi.asm.org/
36. Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S.2010. Amiloride inhibits macropi-nocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol.188:547–563.
37. Cernescu C, Constantinescu SN, Popescu LM.1990. Electron micro-scopic observations of vesicular stomatitis virus particles penetration in human fibroblasts. Rev. Roum. Virol.41:93–96.
38. Quinn K, Brindley MA, Weller ML, Kaludov N, Kondratowicz A, Hunt CL, Sinn PL, McCray PB, Jr, Stein CS, Davidson BL, Flick R, Mandell R, Staplin W, Maury W, Chiorini JA.2009. Rho GTPases modulate entry of Ebola virus and vesicular stomatitis virus pseudotyped vectors. J. Virol.
83:10176 –10186.
39. Sun X, Yau VK, Briggs BJ, Whittaker GR. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology338:53– 60.
40. Bowman EJ, Siebers A, Altendorf K.1988. Bafilomycins: a class of in-hibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U. S. A.85:7972–7976.
41. Racoosin EL, Swanson JA.1993. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J. Cell Biol.121:1011–1020. 42. Clapham PR, McKnight A.2002. Cell surface receptors, virus entry and
tropism of primate lentiviruses. J. Gen. Virol.83:1809 –1829.
43. Tremblay MJ, Fortin J-F, Cantin R. 1998. The acquisition of host-encoded proteins by nascent HIV-1. Immunol. Today19:346 –351. 44. Benoit M, Desnues B, Mege JL.2008. Macrophage polarization in
bac-terial infections. J. Immunol.181:3733–3739.
45. Gobeil L-A, Lodge R, Tremblay MJ.2012. Differential HIV-1 endocyto-sis and susceptibility to virus infection in human macrophages correlate with cell activation status. J. Virol.86:10399 –10407.
46. Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS.2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol.151:413– 423.
47. Lakadamyali M, Rust MJ, Zhuang X. 2006. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell124:997–1009.