0022-538X/11/$12.00 doi:10.1128/JVI.05886-11
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Kaposi’s Sarcoma-Associated Herpesvirus Noncoding Polyadenylated
Nuclear RNA Interacts with Virus- and Host Cell-Encoded
Proteins and Suppresses Expression of Genes Involved in
Immune Modulation
䌤
Cyprian C. Rossetto and Gregory S. Pari*
The University of Nevada, Reno, School of Medicine, Department of Microbiology & Immunology, Reno, Nevada 89557
Received 3 August 2011/Accepted 22 September 2011
During lytic infection, Kaposi’s sarcoma-associated herpesvirus (KSHV) expresses a polyadenylated nuclear RNA (PAN RNA). This noncoding RNA (ncRNA) is localized to the nucleus and is the most abundant viral RNA during lytic infection; however, to date, the role of PAN RNA in the virus life cycle is unknown. Many examples exist where ncRNAs have a defined key regulatory function controlling gene expression by various mechanisms. Our goal for this study was to identify putative binding partners for PAN RNA in an effort to elucidate a possible function for the transcript in KSHV infection. We employed anin vitroaffinity protocol where PAN RNA was used as bait for factors present in BCBL-1 cell nuclear extract to show that PAN RNA interacts with several virus- and host cell-encoded factors, including histones H1 and H2A, mitochondrial and cellular single-stranded binding proteins (SSBPs), and interferon regulatory factor 4 (IRF4). RNA chromatin immunoprecipitation (ChIP) assays confirmed that PAN RNA interacted with these factors in the infected cell environment. A luciferase reporter assay showed that PAN RNA expression interfered with the ability of IRF4/PU.1 to activate the interleukin-4 (IL-4) promoter, strongly suggesting a role for PAN RNA in immune modulation. Since the proteomic screen and functional data suggested a role in immune responses, we investigated if constitutive PAN RNA expression could affect other genes involved in immune responses. PAN RNA expression decreased expression of gamma interferon, interleukin-18, alpha interferon 16, and RNase L. These data strongly suggest that PAN RNA interacts with viral and cellular proteins and can function as an immune modulator.
Kaposi’s sarcoma-associated herpesvirus or human herpes-virus 8 (KSHV/HHV-8) is the causative agent of Kaposi’s sarcoma, body cavity lymphoma, and multicentric Castleman’s disease (25, 33). In cell culture, KSHV exists primarily in a latent phase, where no virus is produced and limited viral genome copies are maintained in infected cells. Lytic reactiva-tion, resulting in the production of infectious virions, can be induced by various chemical agents as well as by the epigenetic regulation of the virus lytic switch protein encoded by the ORF50 gene, namely, K-Rta (3, 10, 19, 36, 46).
During lytic infection, similar to the case for all herpesvi-ruses, there is a cascade of viral gene expression consisting of the production of immediate-early, early, and late proteins. In the case of KSHV, lytic infection is also marked by the expres-sion of a 1.1-kb noncoding transcript known as polyadenylated nuclear RNA (PAN RNA) (32, 34). The PAN RNA transcript accumulates to very high levels in lytically infected cells, esti-mated at approximately 80% of the poly(A)⫹RNA fraction (34).
Although many factors influence or control KSHV growth, replication, and pathogenesis, the role of PAN RNA in any of these processes has been elusive. PAN RNA expression is
activated by K-Rta through a K-Rta response element (RRE) present within the PAN RNA promoter (6, 32, 35, 36). A small subset of PAN RNA is associated with high-molecular-weight ribonucleoprotein complexes (4). To date, the emphasis of published studies, outside the initial reports characterizing the regulation of gene expression and mapping of the PAN RNA locus, has been on PAN RNA sequence elements that increase the nuclear abundance of intronless mRNA and on structural analysis of PAN RNA (7, 26, 30). However, these studies failed to address the function of PAN RNA, and to date, previous studies have not definitively shown that PAN RNA contributes an essential role to KSHV growth. Also, the singular role of PAN RNA functioning as a factor that increases the abun-dance of cellular transcripts has not been established. The lack of identification of specific transcripts (cellular or viral) that are affected by PAN RNA expression suggests that PAN RNA may have other roles in KSHV lytic replication.
The discovery of significant functional roles for long non-coding RNAs (ncRNAs) in human cells with respect to their association with chromatin-modifying complexes (13, 15, 16) suggested to us that KSHV PAN RNA may function to control gene expression in a targeted but global manner. These previ-ously described ncRNAs play key regulatory roles, such as regulating the activity or localization of proteins and acting as scaffolds to regulate gene expression (11, 28).
The overall goal of this study was to identify cellular and viral factors that interact with PAN RNA and to use these data to predict one possible function for the transcript. In an effort * Corresponding author. Mailing address: University of Nevada,
Reno, School of Medicine, Department of Microbiology and Immu-nology, 1664 N. Virginia St., Center for Molecular Medicine Building/ Mailstop 320, Reno, NV 89557. Phone: (775) 784-4824. Fax: (775) 327-2332. E-mail: [email protected].
䌤Published ahead of print on 28 September 2011.
13290
on November 7, 2019 by guest
http://jvi.asm.org/
to identify cellular and viral binding partners for PAN RNA, we employed anin vitro affinity column protocol. Using this protocol, PAN RNA covalently linked to cyanogen bromide (CNBr) agarose beads was used as bait for factors present in induced, infected BCBL-1 cell nuclear extract. Eluted proteins were resolved by two-dimensional (2D) gel electrophoresis, followed by protein identification by liquid chromatography-mass spectrometry (LC-MS) analysis. PAN RNA was shown to interact with virus-encoded proteins ORF59 and ORF26 and with cellular histones H1 and H2A and mitochondrial and cellular single-stranded DNA binding proteins (SSBPs). Addi-tionally, the cellular protein interferon regulatory factor 4 (IRF4) was identified as a putative binding partner for PAN RNA. Transient expression assays demonstrated that expres-sion of PAN RNA efficiently downregulated activation of an IRF4-responsive promoter, possibly implicating PAN RNA as a factor that especially affects the function of IRF4, and they suggested a role in immune modulation. To follow up on the possible broader role of PAN RNA in immune responses, a BJAB cell line that constitutively expressed PAN RNA was generated and used to evaluate the expression profiles of sev-eral interferon response and signaling genes by quantitative PCR (qPCR). The results show a decrease in the expression of several genes that regulate the immune response and strongly suggest that PAN RNA plays a role in immune modulation.
MATERIALS AND METHODS
Cells.HEK293 and Cos 7 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% bovine growth serum (HyClone, Logan, UT). BCBL-1 and BJAB cells were maintained in RPMI medium sup-plemented with 10% fetal bovine serum (FBS). The BJAB-PAN cell line was created by electroporating 10⫻106
BJAB cells in 0.5 ml DMEM with 100 mg sheared salmon sperm DNA (Ambion) and 1 mg pcDNA-PAN (provided by N. Conrad, University of Texas). BJAB cell mixtures were put into a 4-mm-gap mammalian electroporation cuvette and pulsed at 200 V, with the capacitance set to 1,600F. Cells were allowed to recover for 5 min on ice and then plated on RPMI medium with 10% FBS. Twenty-four hours after electroporation, cells were pelleted and plated on RPMI medium-10% FBS with 2 mg/ml G418 to select for BJAB cells that contained the pcDNA-PAN plasmid.
RNA purification.PAN RNA was generated from a linearized plasmid con-taining the PAN locus or antisense PAN by using Megascript T7 (Ambion). PAN RNA was subjected to DNase treatment to remove any residual plasmid DNA, and RNA was then extracted with phenol-chloroform, precipitated with isopro-panol, and resuspended in Tris-EDTA (TE) with RNaseOut (Invitrogen). A sample of PAN RNA was electrophoresed in a 1% agarose gel containing 6% formaldehyde in 1⫻MOPS (morpholinepropanesulfonic acid) to visually check RNA integrity.
BCBL-1 and BJAB cell nuclear extracts.To isolate proteins binding with PAN RNA, nuclear extracts were prepared from BCBL-1 cells induced with 12-tetradecanoyl phorbol 13-acetate (TPA) and sodium butyrate. BCBL-1 cells (1⫻ 107) were induced with 25 ng/ml TPA and 0.3 mM sodium butyrate for 72 h
before being harvested for nuclear extract. BJAB cells (1⫻107
) were used to harvest control nuclear extract. Nuclear extract was prepared using a CelLytic NuCLEAR extraction kit (Sigma). Western blots for K-Rta and K-bZIP were performed on purified nuclear extract to verify that the BCBL-1 cells had been induced to enter the lytic cycle. Nuclear extract was diluted 1:4 in buffer A (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 5 mM dithiothreitol [DTT], 0.5 mM
phenylmethylsulfonyl fluoride [PMSF], protease inhibitors) before being loaded onto the prepared PAN RNA column.
PAN RNA column and nuclear extract preparation.A CNBr-activated Sep-harose 4 Fast Flow column (GE Heathcare Life Sciences) was used to immobi-lize the PAN RNA. In order to couple the PAN RNA with CNBr-activated Sepharose, the CNBr-activated Sepharose (1 g) was reconstituted with 10 ml cold 1 mM HCl for 5 min and then washed with 2 ml cold 1 mM HCl for 5 min, and this was repeated three times. Beads were washed once with coupling solution (0.1 M NaHCO3[pH 8.3], 0.5 M NaCl), and PAN RNA was diluted in 2 ml
coupling solution with 20l RNaseOut (Invitrogen), added to CNBr beads (approximately 2 ml PAN RNA coupling solution to 4 ml packed beads), and incubated overnight with rotation at 4°C. PAN RNA-coupled beads were washed once with 5 volumes of coupling buffer to remove excess PAN RNA. Nonreactive groups on the CNBr-activated Sepharose were blocked with 1 volume of 0.1 M Tris-HCl (pH 8.0) with 125g/ml of poly(dI-dC) for 2 h at room temperature. The PAN RNA-bound beads were then washed three times with alternating low-pH buffer (0.1 M Tris acetate [pH 3.5], 0.5 M NaCl) and high-pH buffer (0.1 M Tris-HCl [pH 8.5], 0.5 M NaCl). Beads were washed with buffer A and loaded onto a column, and excess buffer was allowed to flow out by gravity. Nuclear extract from either induced BCBL-1 or BJAB cells was added to the PAN RNA column, and the eluant that passed through the PAN RNA-bound beads was stored as the unbound fraction. After the column was washed once with buffer A, the bound proteins were eluted with the same buffer containing sequentially increasing concentrations of NaCl (50 mM, 150 mM, 500 mM, and 1 M). Eluted proteins were used for 2D gel analysis.
PAN proteomics with MS analysis.BCBL-1 and control cell proteins from different salt elutions were matched and differentially labeled with Cy5 (control cells) or Cy3 (BCBL-1 cells). Proteins were separated using 2D gel electropho-resis, and protein spots were compared using DeCyder 2D 7.0 software (9). A total of 8 spots were identified that were on the PAN RNA BCBL-1 2D gels and were not present on the control gels. These protein spots were picked and identified by mass spectrometry.
Luciferase reporter assay.HEK293 cells were cotransfected with the pGL3-il4 promoter, IRF4, PU.1 (provided by M. Kaplan, Indiana University), and pcDNA-PAN. Cells were lysed, and standard luciferase detection was performed (Promega). Each assay was measured in triplicate, and each experiment was repeated three times.
Subclones carrying PAN RNA were generated and ligated into the phCMV-xi vector (Genlantis) by using a CloneEZ kit (Genscript). Primers were constructed to amplify the PAN RNA locus as two fragments, named PAN1 (consisting of 563 nucleotides [nt], corresponding to coordinates 28661 to 29223 in the HHV-8 genome [GenBank accession no. AF148805]) and PAN2 (consisting of 559 nt, corresponding to coordinates 29224 to 29782). Primers specific to each PAN half were constructed with ends homologous to the multicloning site within the phCMV-xi vector to allow for insertion of the PCR product through homologous recombination. The PCR primers were as follows (the lowercase sequence is homologous to the phCMV-xi vector sequence, and the uppercase sequence is the PAN RNA locus sequence): PAN-1Forward, 5⬘-cgacttaacagatctcgagctcaagc
ttcgaattcTTTAGCACTGGGACTGCCCAGTCACCTTGGC; PAN-1Reverse,
5⬘-cccgggcccgcggtaccgtcgactgcagaattcCAGATTGTCACATTTAGGGCAAAGT GGCC; PAN-2Forward, 5⬘-cgacttaacagatctcgagctcaagcttcgaattcGATGTGTATC TTATTGGTGCGTTGTGAAGCA; and PAN-2Reverse, 5⬘-cccgggcccgcggtaccg tcgactgcagaattcCCATCCCAATCGACGCAAGTCAAGACACAA.
In order to clone the PAN RNA PCR product, the phCMV-xi vector had to first be linearized by EcoRI restriction enzyme digestion. After the vector was determined to be linearized, the vector and PCR insert were incubated together with CloneEZ enzymes and buffer according to the manufacturer’s instructions. Correct clones were screened by restriction enzyme digestion patterns and sub-sequent sequencing.
Real-time PCR.Total cell RNA was harvested from 10⫻106
cells by using a PureLink RNA minikit (Invitrogen) followed by removal of genomic DNA by use of Turbo DNA-free reagent (Ambion). cDNA was synthesized from 25g of total RNA in the presence of random hexamers, deoxynucleoside triphosphates (dNTPs), and Superscript III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. cDNA was diluted in Power SYBR master mix (ABI) along with specific primers and then was distributed in triplicate into wells of a standard 96-well plate. The following standard qPCR program was used: 1 cycle of 50°C for 2 min, 1 cycle of 95°C (hot start) for 5 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Primers used for detection of specific gene expression were as follows: RNase L Forward, 5⬘-AAGACAAAGGAGGATCAAGAG CGG; RNase L Reverse, 5⬘-CATGGATCAAGGCATTTCTGCCCA; IFN␣16 Forward, 5⬘-CAGGAGTGTAAAGAAGCATCGTGT; IFN␣16 Reverse, 5⬘-AG
ATGAATAGAGACATCAGCATGGTC; IFN␥Forward, 5⬘-TGAACTGTCGC
CAGCAGCTAAA; IFN␥ Reverse, 5⬘-AGGCAGGACAACCATTACTGG
GAT; IL-18 Forward, 5⬘-GCAAGGAATTGTCTCCCAGTGCAT; and IL-18 Reverse, 5⬘-TTCCTTTCCTCTTCCCGAAGCTGT.
IFN-␥ELISA.BJAB and BJAB-PAN cells (5⫻105) were plated into 6 wells
of a 96-well plate; 3 wells served as unstimulated controls, and 3 wells of each cell type were stimulated for 48 h with 20 ng/ml phorbol myristate acetate (PMA) and 1.5M ionomycin (Sigma). Cell supernatants were harvested and analyzed for gamma interferon (IFN-␥) expression by human IFN-␥enzyme-linked
on November 7, 2019 by guest
http://jvi.asm.org/
nosorbent assay (ELISA) according to the manufacturer’s instructions (R&D Systems).
RNA chromatin immunoprecipitation (rChIP) assay.BCBL-1 cells (50⫻106
) were treated with 0.3 mM sodium butyrate. Forty-eight hours after treatment, cells were harvested, washed once with 1⫻phosphate-buffered saline (PBS), and fixed in 1% methanol-free formaldehyde for 10 min. Cells were pelleted, washed once with PBS, and quenched with 125 mM glycine for 5 min. After a final wash with PBS, cells were brought up in 2 ml RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with a protease inhibitor cocktail (Sigma), RNaseOut (Invitrogen), and 1 mM PMSF. Cells were sonicated, and the extracts were centrifuged at 800⫻gfor 5 min at 4°C to remove debris. RNA was precipitated by adding 300l lysate, 5l antibody, 50l protein G agarose beads (Santa Cruz Biotechnology), and 1l RNaseOut. This mixture was rotated overnight at 4°C. The input control was 98
l of lysate mixed with 2l of 5 M NaCl and frozen at⫺80°C until the proteinase K digestion step the following day. After the overnight incubation, the beads were washed with 1 ml of RIPA buffer, once with 1 ml of low-salt wash (0.1 SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.0], 150 mM NaCl), once with 1 ml of high-salt wash (0.1 SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.0], 500 mM NaCl), once with 1 ml of LiCl wash (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris [pH 8.0]), and twice with 1 ml of TE. The beads were resuspended in 150l elution buffer (1% SDS, 100 mM NaHCO3[pH 9.0]) for 15 min. The elution step was repeated and the fractions
combined, 60l 1 M Tris-HCl (pH 6.8) was added to the elution complexes, proteinase K was added at 0.2 mg/ml to the samples and input, and the samples were incubated at 37°C for 60 min. The cross-links were reversed at 65°C for 18 h, the beads were pelleted, and supernatant was moved into 1 ml TRIzol LS (Invitrogen) and incubated for 5 min at room temperature. Two hundred fifty microliters of chloroform was added to the TRIzol mixture, mixed by hand, and allowed to incubate for 15 min before centrifugation at 12,000⫻gfor 10 min at 4°C to separate the phases. The upper phase containing the RNA was removed, 1 volume of isopropanol was used to precipitate the RNA, and 1l of GlycoBlue (Ambion) was added to aid in visualizing the RNA pellet. After 15 min of incubation at room temperature, this was centrifuged at 12,000⫻gfor 15 min at 4°C and then washed with ice-cold 75% ethanol. The pellet was briefly allowed to air dry and then resuspended in 30l nuclease-free water. RNA samples were treated with Turbo DNA-Free reagent (Ambion) according to the manufactur-er’s instructions. Five microliters of the RNA was then used in a Qiagen OneStep reverse transcriptase PCR (RT-PCR) kit, using primers specific to an internal region within the PAN RNA locus (forward primer, TAA TGT GAA AGG AAA GCA GCG CCC; and reverse primer, TAA CAT TGA AAG AGC GCT CCC AGC), ORF45 (forward primer, ACG TCC GGA GAG TTG GAA CTG TCA T; and reverse primer, GGC GTC CAT GGG ATG GGT TAG TCA G), or U1 RNA (forward primer, ATA CTT ACC TGG CAG GGG AG; and reverse primer, CAG GGG AAA GCG CGA ACG CA). The no-RT control was sub-jected to PCR only, not to the reverse transcriptase step.
For qPCR of immunoprecipitated RNA, first we synthesized cDNA by using either random hexamers, dNTPs, and Superscript III reverse transcriptase (In-vitrogen) or an iScript kit (Bio-Rad) according to the manufacturer’s instruc-tions. The resulting cDNA was then used along with TaqMan Universal PCR master mix (Applied Biosystems) and specific primers and 6-carboxyfluorescein (FAM)-labeled probes (IDT) in an Eppendorf RealPlex thermal cycler. The following qPCR program was used: 1 cycle at 95°C (hot start) for 5 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Primers used for detection of specific gene expression were as follows: PAN Forward, TAA TGT GAA AGG AAA GCA GCG CCC; PAN Reverse, CAT TTA GGG CAA AGT GGC CCG ATT; PAN probe, 56-FAM-ACA GTG GTG-ZEN-CAC TAC CTA TCT GCT CA-3 IABkFQ; ORF50 Forward, ACC AAG GTG TGC CGT GTA GAG ATT; ORF50 Reverse, AGC CTT ACG CTT CTT TGA GCT CCT; ORF50 probe, 56-FAM-AGG CGA CAA-ZEN-CAC CCA AAC GAA AGC A-3IABkFQ; LANA Forward, AAC AAA TTG CCA GTA GCC CAC CAG; LANA Reverse, TAA CTG GAA CGC GCC TCA TAC GA; and LANA probe, 56-FAM-ATA CAC CAG-ZEN-ACG ATG ACC CAC AAC CT-3IABkFQ.
Antibodies to the following proteins were used for the ChIP assays: histone H2A (Active Motif), ORF59 (Advanced Biotechnologies Inc.), histone H1 (Ab-cam), RPA70 (Ab(Ab-cam), p53 (Santa Cruz),␣-actinin (Santa Cruz), and IRF-4 (Santa Cruz).
RESULTS
Identification of PAN RNA binding partners.As a first step toward understanding the role of PAN RNA in KSHV growth,
we wanted to develop a protocol to identify putative factors that bind to PAN RNA in infected cells. Binding partners for other noncoding RNAs were elucidated using an RNA-protein affinity purification strategy (43, 48). Hence, we also used this strategy for the identification of binding partners for PAN RNA. We synthesized PAN RNA in vitro and coupled the full-length transcript to CNBr agarose beads. The CNBr-PAN RNA complexes were loaded into affinity gravity columns. Nu-clear extracts were prepared from lytic cycle-induced BCBL-1 cells, which harbor KSHV genomes. As a control for nonspe-cific protein binding, nuclear extracts from BJAB cells were also prepared and subjected to the same experimental proto-col. These nuclear extracts were passed through the CNBr-PAN RNA column, the column was washed, and bound pro-tein was eluted using increasing NaCl concentrations. Eluted protein was resolved by 2D gel electrophoresis. 2D gels for BCBL-1 cell proteins eluted from PAN RNA-containing affin-ity columns were compared with BJAB cell-eluted proteins, and protein spots that were distinct to the BCBL-1 cell samples were selected and picked. Each protein spot was subjected to LC-MS analysis to identify specific proteins that interacted with PAN RNA.
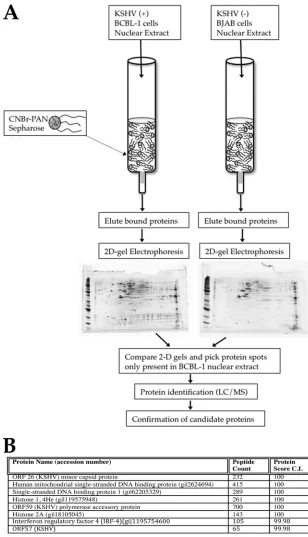
Figure 1A is a schematic showing the protocol used to elu-cidate the binding partners for PAN RNA in BCBL-1 cell nuclear extracts. Figure 1B shows the distinct PAN RNA bind-ing proteins that were identified by LC-MS. Several viral pro-teins were identified: ORF26, -57, and -59. ORF57 was previ-ously shown to interact with PAN RNA (30). However, ORF26 (minor capsid protein) and ORF59 (processivity factor) were not previously demonstrated to interact with PAN RNA. In-terestingly, two single-stranded DNA binding proteins were also shown to interact with PAN RNA. Human single-stranded binding protein 1 (SSBP1) and mitochondrial single-stranded binding protein (mtSSBP) were both identified in our screen. Additionally, several histone proteins were also identified, in-cluding histone 1 (4He) and histone 2A. We also identified IRF4 as a putative binding partner for PAN RNA. Taken together, these interactions suggest a role for PAN RNA in nuclear scaffolding and gene regulation, viral DNA replication, and regulation of immune responses.
Confirmation of protein-RNA interactions in an infected cell environment.Since thein vitrocolumn affinity assay iden-tified several cellular and viral factors that interacted with PAN RNA, we next wanted to determine if PAN RNA interacted with the identified proteins in an infected cell environment. We also wanted to confirm the identified interactions from the proteomic assay by using a method that preserved intact cells and more closely resembled the natural environment of PAN RNA. An rChIP assay with BCBL-1 cells, a cell line harboring KSHV latent genomes that can be induced to enter the lytic phase, takes advantage of a well-defined system for KSHV lytic reactivation. Additionally, this type of assay is able to deter-mine and confirm specific protein binding to PAN RNA in an intact cellular environment. BCBL-1 cells were treated with
n-butyrate to induce the lytic phase of virus replication (and expression of PAN RNA), and at 3 days posttreatment, rChIP assays were performed using antibodies specific for H2A, IRF4, SSBPs, and KSHV ORF59. We also immunoprecipi-tated two proteins that were not identified in our initial pro-teomic screen, namely, p53 and ␣-actinin. As a control for
on November 7, 2019 by guest
http://jvi.asm.org/
nonspecific binding of RNA, we used beads alone and primers specific to an unrelated KSHV-encoded transcript (ORF45) and to the nuclear U1 RNA. Immunoprecipitated RNA was subjected to reverse transcriptase PCR using random primers. cDNAs from samples were then evaluated by using PCR
[image:4.585.132.440.67.604.2]prim-ers specific for PAN RNA or control RNA. The rChIP assay showed detectable PCR bands for all tested proteRNA in-teractions, strongly suggesting that PAN RNA interacts with the identified factors in the infected cell environment and confirming the findings from the column affinity assay (Fig. 2, FIG. 1. Proteomic analysis of PAN RNA binding factors. (A) Experimental design.In vitro-transcribed PAN RNA was coupled to an activated CNBr Sepharose column. Nuclear extracts from KSHV-positive (⫹) (BCBL-1) or KSHV-negative (⫺) (BJAB) cells were flowed over PAN RNA columns, and proteins were eluted from the columns. Proteins were resolved by 2D gel electrophoresis, and gels were compared for distinct protein spots present only in the gel that contained the BCBL-1 cell eluates. Spots were excised from the gel and then subjected to LC-MS for protein identification. (B) LC-MS-identified proteins from lytically induced BCBL-1 cells that bound to PAN RNA.
on November 7, 2019 by guest
http://jvi.asm.org/
top panel). Control samples in which immunoprecipitated pro-tein-RNA complexes were subjected to PCR amplification us-ing primers specific for ORF45 or U1 RNA showed no amplification product (Fig. 2, bottom panels). Also, immuno-precipitations using p53- or␣-actinin-specific antibodies were negative for PAN RNA (Fig. 2, top panel).
We also performed real-time PCR (qPCR) analysis of RNAs precipitated in the rChIP assay. This was done to show that our rChIP assay was specific for PAN RNA and that very little non-PAN RNA was present in samples immunoprecipi-tated with each specific antibody (Fig. 2). We measured the amounts of LANA, K-Rta, and PAN RNAs in input and im-munoprecipitated samples, and the averageCTvalues are
re-ported for each rChIP assay performed. The percentage of input RNA corresponding to the LANA or K-Rta mRNA found in each rChIP assay indicated that very little of the mRNA species was present in the immunoprecipitated sam-ples for each specific antibody used (Fig. 2, bottom panel, LANA and K-Rta). However, the percentage of input corre-sponding to PAN RNA indicated that a high percentage of input PAN RNA was immunoprecipitated with each specific antibody used in the rChIP assay (Fig. 2, bottom panel, PAN). Taken together, these data strongly suggest that the rChIP assay accurately confirmed the PAN RNA binding partners that were identified in our initial proteomic screen.
PAN RNA expression interferes with IRF4-mediated pro-moter activation.IRF4 is a transcription factor that interacts with PU.1 to upregulate gene expression for several immune response factors (8). IRF4 plays a critical role in the matura-tion of B and T cells and the activity of dendritic cells (18, 38). Since our proteomic analysis suggested that PAN RNA inter-acts with IRF4, we wanted to investigate if the activity of IRF4, specifically its ability to activate the interleukin-4 (IL-4) pro-moter, was affected by the presence of PAN RNA. It was previously shown that IRF4, along with PU.1, binds to and transactivates the IL-4 promoter (2). Hence, if PAN RNA interacts with IRF4 and this interaction in turn inhibits the
ability of IRF4 to bind to its DNA target and/or PU.1, we would observe a decrease in IL-4 promoter activation. To this end, we evaluated the ability of IRF4/PU.1 to transactivate IL-4 in the presence of PAN RNA in HEK293 cells cotrans-fected with plasmids expressing IRF4, PU.1, and PAN RNA or antisense PAN RNA along with a plasmid containing the IL-4 promoter upstream of the luciferase gene.
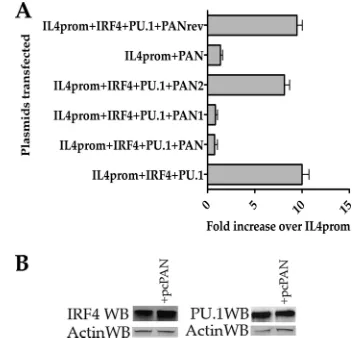
[image:5.585.58.265.68.185.2]Cotransfection of the IRF4 and PU.1 expression plasmids along with the IL-4 promoter–luciferase reporter plasmid showed an approximately 10-fold increase in promoter ac-tivity compared to transfection of the IL-4 promoter alone (Fig. 3A, IRF4⫹PU.1⫹IL4prom). The addition of the PAN RNA expression plasmid to the transfection mixture re-duced the level of luciferase significantly (Fig. 3A, compare IRF4⫹PU.1⫹IL4prom to IRF4⫹PU.1⫹IL4prom⫹PAN RNA). The addition of the reverse PAN RNA expression plasmid to the transfection mixture had little effect on promoter activation (Fig. 3A, IRF4⫹PU.1⫹IL4prom⫹ PANrev). We also generated PAN RNA expression plas-mids that produced nt 1 to 563 (PAN1) and nt 559 to 1121 (PAN2) of the PAN RNA transcript. These expression plas-mids were transfected along with IRF4 and PU.1 expression plasmids and the IL-4 promoter–luciferase reporter plas-mid. Only the PAN1 expression plasmid was able to reduce the IL-4 promoter activity, whereas the PAN2 expression plasmid had only a modest effect on promoter activation (Fig. 3A, IRF4⫹PU.1⫹IL4prom⫹PAN1). Transfection of the PAN RNA expression plasmid alone had no effect on FIG. 2. Interaction of PAN RNA with viral and cellular factors in
BCBL-1 cells. (Top) ChIP assay performed using lytically induced BCBL-1 cells. Immunoprecipitations were performed using the indi-cated specific antibodies. Beads alone were used as an immunopre-cipitation control, along with immunopreimmunopre-cipitations using p53- or␣ -ac-tinin-specific antibodies, followed by amplification of PAN RNA. Amplification using ORF45- or U1 RNA-specific PCR primers was used as a control to detect contaminating RNA. (Bottom) qPCR eval-uation of precipitated RNA. RNA precipitated in the rChIP assay with each antibody was subjected to qPCR analysis using primers and probes specific for LANA, K-Rta, or PAN RNA. Data shown are percentages of input and are averages for 3 replicates.
FIG. 3. PAN RNA expression reduces the ability of IRF4/PU.1 to activate the IL-4 promoter. (A) HEK293L cells were cotransfected with plasmids expressing IRF4, PU.1, and PAN RNA or antisense PAN RNA along with an IL-4 promoter–luciferase reporter plasmid. Cells were incubated for 48 h posttransfection, cell lysates were pre-pared, and luciferase expression was measured using a luminometer. Each experiment was done in triplicate and repeated 3 times. Error bars are standard deviations for three experiments. The data are shown as fold increases over the luciferase activity obtained from transfection of the IL-4 promoter-luciferase reporter plasmid alone. The PAN RNA expression plasmids produced nt 1 to 563 (PAN1) or nt 559 to 1121 (PAN2) of the PAN RNA transcript. (B) PAN RNA expression does not affect the level of IRF4 or PU.1 protein. Cotransfections were performed as described for the luciferase assay, except that protein extracts were prepared and lysates were separated using SDS-PAGE, transferred to a PVDF membrane, and incubated with IRF4-, PU.1-, or actin-specific antibodies.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.329.505.70.239.2]IL-4 promoter activation (Fig. 3, IL4prom⫹PAN). These data suggest that the interaction domain and/or functional region of PAN RNA with respect to the ability to interfere with IRF4-mediated transactivation activity lies within nt 559 to 1121 of the transcript.
As a control for expression, we evaluated the protein expres-sion levels of IRF4 and PU.1 in the presence of PAN RNA expression. The presence of the PAN RNA expression plasmid pcPAN did not affect the level of IRF4 or PU.1 protein ex-pression in the transient transfection assay, indicating that PAN RNA did not influence protein expression (Fig. 3B). These results strongly suggested that the expression of PAN RNA interfered with the ability of IRF4 to transactivate the IL-4 promoter in a transient reporter protocol.
PAN RNA expression results in a decrease in expression of specific human interferon signaling and response mRNAs.
Our initial screen identified several putative binding partners for PAN RNA. Previous studies involving ncRNAs also sug-gested that these nucleic acids could repress transcription in
trans(28). In some cases, ncRNAs can physically interact with
regulatory proteins that interact with repressive complexes and downregulate the activity of genes (13, 15).
Since we already established an interaction of PAN RNA with IRF4 and determined that this interaction could repress the activation of an IRF4-responsive promoter, we chose to examine expression profiles for some of the genes encoding human interferon signaling and response factors in human cells in the presence of PAN RNA expression. We reasoned that since PAN RNA interacted with a transcription factor that modulated the immune response, perhaps PAN RNA has a wider role in regulating immune responses in general.
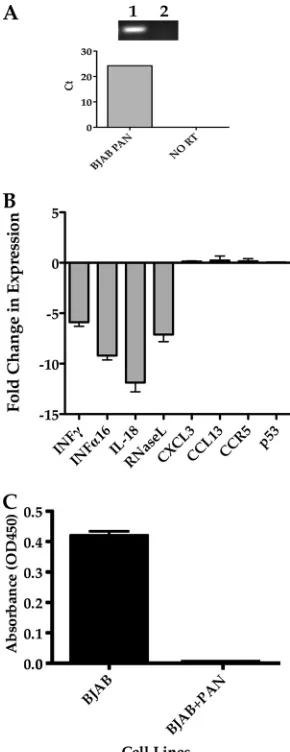
To test our hypothesis that PAN RNA could impact the ex-pression profiles of specific human interferon signaling and re-sponse genes, we generated a BJAB cell line that constitutively expressed PAN RNA. We transfected a PAN RNA expression plasmid into BJAB cells and selected PAN RNA-expressing cell lines by using G418. BJAB-PAN cell populations were expanded, and the cell line was shown to express PAN RNA by RT-PCR and qPCR (Fig. 4A). Lane 1 in Fig. 4A shows the presence of a PAN RNA cDNA-specific band, whereas an RNA sample in which reverse transcriptase was omitted failed to amplify a spe-cific PCR product (Fig. 4A, lane 2). qPCR analysis also confirmed the expression of PAN RNA in the BJAB cell line (Fig. 4A, bottom panel).
After confirming the expression of PAN RNA in the BJAB cell line, we evaluated the accumulation of IFN-␥, IFN-␣16, IL-18, and RNase L mRNAs in cells constitutively expressing PAN RNA and compared the mRNA levels to those in a BJAB cell line transfected with empty vector. The expression levels for all of the immune response genes tested showed a decrease in mRNA accumulation in cells expressing PAN RNA (Fig. 4B). Gene ex-pression levels for CXCL3, CCL13, CCR5, and p53 were affected
⬍1-fold compared to controls, suggesting that only a specific set of genes were altered by PAN RNA expression (Fig. 4B). We next chose to evaluate the protein expression level of IFN-␥by mea-suring the amount of IFN-␥in the supernatant of BJAB cells expressing PAN RNA compared to that for control BJAB cells. Cells were stimulated with PMA and ionomycin, cellular super-natants were collected, and the concentration of secreted IFN-␥ was measured by ELISA. PAN RNA expression resulted in the
inhibition of secretion of IFN-␥compared to that in control cells (Fig. 4C). These results suggest that PAN RNA can suppress the expression of immune response genes.
Taken together, these findings strongly argue that PAN RNA mediates cellular gene expression levels, and for the first time, we establish at least one function for this very abundant virus-encoded RNA.
DISCUSSION
[image:6.585.347.492.69.445.2]Long noncoding RNAs are classified as RNAs that are lon-ger than 200 bases and apparently do not encode a protein FIG. 4. PAN RNA expression in BJAB cells mediates changes in expression levels of specific immune modulation factors. (A) (Top) RT-PCR showing the expression of PAN RNA in BJAB cells. Lanes: 1, BJAB cell PAN RNA PCR; 2, no-RT control BJAB cell PAN RNA RT-PCR. (Bottom) qPCR analysis of PAN RNA-expressing BJAB cell line. (B) Graph of qPCR results for accumulation of IFN-␥, IFN-␣16, IL-18, RNase L, chemokine ligand 3 (CXCL3), chemokine ligand 13 (CCL13), chemokine receptor 5 (CCR5), and p53 mRNAs. Data are expressed as fold reductions in expression compared to a BJAB cell line transfected with empty vector. Each experiment was done in triplicate, and error bars are standard deviations for three separate experiments. (C) Graph of ELISA measurements of secreted IFN-␥ from BJAB cells expressing PAN RNA and control cells stimulated with PMA and ionomycin.
on November 7, 2019 by guest
http://jvi.asm.org/
product (48). ncRNAs are emerging as major regulatory fac-tors of cellular functions (20, 21, 24, 37, 44). Although the proposed mechanisms of action for ncRNAs vary, there are reports of this class of nucleic acids associating with chromatin-modifying complexes to effect gene expression (13, 15, 28). In some cases, ncRNAs have been demonstrated to mediate global gene expression pathways (13). ncRNAs have the ca-pacity to directly interact with cellular proteins and alter func-tion or subcellular localizafunc-tion (44). Hence, it is likely that interacting proteins tether or link ncRNAs to their target loci. Long ncRNAs may also act via a sequence recognition mech-anism. The secondary structure of the RNA, i.e., base pairing and looping within the RNA molecule, may allow for an inter-action of distant sequences that results in a binding molecule that is not apparent from the primary sequence. Alternatively, it could be the RNA structure itself that mediates an interac-tion with proteins and/or DNA loci. Interestingly, KSHV PAN RNA was recently shown to have a distinct secondary structure that forms a triple helix (26).
Since PAN RNA had no known function, we took the initial approach of investigating the putative binding partners present in lytically infected nuclear extracts. Similar methods have been used to identify other DNA/RNA binding partners (31, 41, 43). For our experiments, we minimized false-positive re-sults by subtracting proteins eluted from an affinity column subjected to uninfected BJAB cell nuclear extract. Data col-lected from these experiments suggested that PAN RNA in-teracts with proteins that bind directly to chromatin (histones H1 and H2A). We confirmed many of these interactions by using an RNA ChIP assay of infected BCBL-1 cells and showed that PAN RNA does interact with histones and other cellular factors in an infected cellular environment. We also confirmed the interaction of PAN RNA with the virally en-coded processivity factor ORF59. Additionally, since our screen also identified interactions with a transcription factor involved in immune modulation (IRF4), we also speculated that PAN RNA might have a wider role in influencing specific gene expression with respect to the interferon signaling and response pathway.
Among DNA viruses, human cytomegalovirus (HCMV) provides the best example of a long regulatory ncRNA. HCMV produces an ncRNA that regulates the apoptotic pathway (27). The interaction of KSHV PAN RNA with human mtSSBP and SSBP1 is very intriguing. mtSSBP is involved in mitochondrial DNA replication, and like all SSBPs, it is thought to maintain the integrity of single-stranded DNA intermediates generated during DNA synthesis and to protect them from refolding or nucleolytic attack. Also, despite its name, mtSSBP is found within the cell nucleus. Recently, mtSSBP was shown to inter-act with p53 (45). Interestingly, the Epstein-Barr virus-en-coded protein ZTA interacts with mtSSBP and apparently uses the mtSSBP function to promote viral DNA synthesis (42). The interaction of SSBPs with PAN RNA may act to protect the transcript from degradation or implicate PAN RNA as having a role in DNA synthesis. Although SSBPs do have a low affinity for RNA, they primarily bind to single-stranded DNA; hence, the interaction of these proteins with PAN RNA may be unique to PAN RNA and suggests a novel and as yet unchar-acterized role for mtSSBP/SSBP in KSHV growth. Likewise, the interaction of PAN RNA with histones (also typically
in-volved in DNA binding) may suggest that these proteins act to protect PAN RNA or influence its structure. The interaction with histone proteins may facilitate the ability of PAN RNA to regulate gene transcription or help to stabilize the structure of PAN RNA in the nucleus. PAN RNA could affect gene ex-pression patterns by interacting with specific genomic regions via tethering with histone proteins. Another alternative is that PAN RNA could facilitate gene silencing or upregulation by interacting with protein complexes that alter histone methyl-ation states. It has been reported that noncoding RNAs inter-act with chromatin-modifying complexes (11, 28). These types of interactions will be explored further.
Our proteomic screen identified KSHV-encoded proteins ORF26, -59, and -57 as putative binding partners for PAN RNA. ORF26 is the minor capsid protein, and the interaction of this protein with PAN RNA is interesting because PAN RNA was shown to be a component of the virion (4). Hence, it is possible that PAN RNA contains an interaction domain that allows for tethering to a viral structural protein. The in-teraction of PAN RNA with a virus structural protein and the fact that PAN RNA is part of the virion allow for the possibility that the immunosuppressive activity of PAN RNA can occur before the onset of the lytic virus program.
ORF59 is the DNA polymerase processivity factor (PPF). Although ORF59 is associated with DNA polymerase, herpes-virus PPFs are multifunctional and are implicated in having a wider role in herpesvirus initiation of DNA replication, partic-ularly in the regulation of DNA synthesis. Our recent studies demonstrated that ORF59 interacts with ORF50 and that this complex possibly recruits the replication complex to oriLyt (29). ORF59 may serve to direct PAN RNA to regions of the KSHV genome. PAN RNA may mediate or facilitate lytic DNA replication in an as yet unknown capacity. Alternatively, ORF59 may serve to stabilize PAN RNA or help with the formation of a structural conformation that aids in regulation of host cell gene expression.
ORF57 was recently shown to interact with PAN RNA and apparently protects PAN RNA from degradation (30). Our proteomic screen did identify ORF57 as a binding partner for PAN RNA, again lending a level of confidence to the affinity column methodology used. The observation that PAN RNA interacts with several proteins of viral origin suggests a multi-functional role for this ncRNA.
This study identified a novel pathway targeted by KSHV by the expression of an abundant ncRNA. Experiments are in progress to address the roles of the viral and cellular binding partners in the function of PAN RNA. The scope of the pres-ent study was to establish PAN RNA as a functional transcript in infected cells. Identification of IRF4, a factor involved in immune responses, also led to the discovery that PAN RNA could function as a regulatory ncRNA that downregulates the expression of immune response modulators.
We have clearly established that PAN RNA interacts with several virus- and host cell-encoded proteins and is a modula-tor of gene expression, affecting at least some of the genes involved in immune responses and regulation. Other gene ex-pression programs may also be affected and are currently being studied. Although we have not yet identified the exact mech-anism(s) by which PAN RNA regulates cellular gene expres-sion, the focus of this study was to establish a functional role
on November 7, 2019 by guest
http://jvi.asm.org/
for PAN RNA. Since PAN RNA was shown to interact with histone proteins, this suggests a targeted or direct chromatin reorganization or modification as a possible mechanism. PAN RNA accumulation in KSHV-infected cells is extremely high, so we predict that it has other functions and an even more pronounced effect on gene regulation than that observed in our expression cell lines. Immune response suppression by KSHV has been shown previously and involves various virus-encoded proteins (1, 5, 14, 17, 22, 23, 47). Interestingly, PAN RNA expression also resulted in a knockdown of expression of RNase L. Other viruses have evolved mechanisms to neutralize the function of RNase L during virus infection (12, 39, 40).
The data presented here establish another highly efficient mechanism used by KSHV to specifically downregulate the immune response pathway.
ACKNOWLEDGMENTS
We thank M. Kaplan (University of Indiana) and N. Conrad (Uni-versity of Texas) for expression and reporter plasmids.
REFERENCES
1.Ahmad, H., et al.2011. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing be-ta-interferon (TRIF). J. Biol. Chem.286:7865–7872.
2.Ahyi, A. N., H. C. Chang, A. L. Dent, S. L. Nutt, and M. H. Kaplan.2009. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J. Immunol.183:1598–1606.
3.AuCoin, D. P., et al.2004. Amplification of the Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology318:542–555. 4.Bechtel, J., A. Grundhoff, and D. Ganem.2005. RNAs in the virion of
Kaposi’s sarcoma-associated herpesvirus. J. Virol.79:10138–10146. 5.Bi, X., L. Yang, M. E. Mancl, and B. J. Barnes.2011. Modulation of
inter-feron regulatory factor 5 activities by the Kaposi sarcoma-associated herpes-virus-encoded viral interferon regulatory factor 3 contributes to immune evasion and lytic induction. J. Interferon Cytokine Res.31:373–382. 6.Chang, P. J., et al.2002. Open reading frame 50 protein of Kaposi’s
sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol.76:3168–3178.
7.Conrad, N. K., and J. A. Steitz. 2005. A Kaposi’s sarcoma virus RNA element that increases the nuclear abundance of intronless transcripts. EMBO J.24:1831–1841.
8.Eisenbeis, C. F., H. Singh, and U. Storb.1995. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev.9:1377–1387.
9.Gao, Y., K. Colletti, and G. S. Pari.2008. Identification of human cytomeg-alovirus UL84 virus- and cell-encoded binding partners by using proteomics analysis. J. Virol.82:96–104.
10.Gradoville, L., et al.2000. Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol.74:6207–6212. 11.Gupta, R. A., et al. 2010. Long non-coding RNA HOTAIR reprograms
chromatin state to promote cancer metastasis. Nature464:1071–1076. 12.Han, J. Q., et al.2007. A phylogenetically conserved RNA structure in the
poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol.81:5561–5572.
13.Huarte, M., et al.2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell142:409–419. 14.Joo, C. H., et al.2007. Inhibition of interferon regulatory factor 7
(IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol.81:8282–8292.
15.Khalil, A. M., et al.2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U. S. A.106:11667–11672.
16.Khalil, A. M., and J. L. Rinn.2011. RNA-protein interactions in human health and disease. Semin. Cell Dev. Biol.22:359–365.
17.Li, Q., R. Means, S. Lang, and J. U. Jung.2007. Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol.81:2117–2127.
18.Lu, R., K. L. Medina, D. W. Lancki, and H. Singh.2003. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev.17:1703–1708. 19.Lukac, D. M., J. R. Kirshner, and D. Ganem.1999. Transcriptional activation by
the product of open reading frame 50 of Kaposi’s sarcoma-associated herpes-virus is required for lytic viral reactivation in B cells. J. Virol.73:9348–9361.
20.Mattick, J. S., P. P. Amaral, M. E. Dinger, T. R. Mercer, and M. F. Mehler.
2009. RNA regulation of epigenetic processes. BioEssays31:51–59. 21.Mattick, J. S., and I. V. Makunin.2006. Non-coding RNA. Hum. Mol.
Genet.15(Spec. No. 1):R17–R29.
22.Means, R. E., S. M. Lang, Y. H. Chung, and J. U. Jung.2002. Kaposi’s sarcoma associated herpesvirus immune evasion strategies. Front. Biosci.
7:e185–e203.
23.Means, R. E., S. M. Lang, and J. U. Jung.2007. Human gammaherpesvirus immune evasion strategies, p. 559–586.InA. Arvin et al. (ed.), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge Univer-sity Press, Cambridge, United Kingdom.
24.Mercer, T. R., M. E. Dinger, and J. S. Mattick.2009. Long non-coding RNAs: insights into functions. Nat. Rev. Genet.10:155–159.
25.Mesri, E. A., et al.1996. Human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J. Exp. Med.
183:2385–2390.
26.Mitton-Fry, R. M., S. J. DeGregorio, J. Wang, T. A. Steitz, and J. A. Steitz.
2010. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science330:1244–1247.
27.Reeves, M. B., A. A. Davies, B. P. McSharry, G. W. Wilkinson, and J. H. Sinclair.2007. Complex I binding by a virally encoded RNA regulates mi-tochondria-induced cell death. Science316:1345–1348.
28.Rinn, J. L., et al.2007. Functional demarcation of active and silent chroma-tin domains in human HOX loci by noncoding RNAs. Cell129:1311–1323. 29.Rossetto, C. C., N. K. Susilarini, and G. S. Pari.2011. Interaction of Kaposi’s sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J. Virol.85:3833–3841.
30.Sahin, B. B., D. Patel, and N. K. Conrad.2010. Kaposi’s sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog.6:e1000799.
31.Si, H., S. C. Verma, and E. S. Robertson.2006. Proteomic analysis of the Kaposi’s sarcoma-associated herpesvirus terminal repeat element binding proteins. J. Virol.80:9017–9030.
32.Song, M. J., X. Li, H. J. Brown, and R. Sun.2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol.76:5000–5013.
33.Soulier, J., et al.1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood86:1276–1280. 34.Sun, R., S. F. Lin, L. Gradoville, and G. Miller.1996. Polyadenylylated
nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A.93:11883–11888.
35.Sun, R., et al.1998. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A.
95:10866–10871.
36.Sun, R., et al.1999. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol.73:2232–2242.
37.Taft, R. J., K. C. Pang, T. R. Mercer, M. Dinger, and J. S. Mattick.2010. Non-coding RNAs: regulators of disease. J. Pathol.220:126–139. 38.Tamura, T., et al.2005. IFN regulatory factor-4 and -8 govern dendritic cell
subset development and their functional diversity. J. Immunol.174:2573– 2581.
39.Townsend, H. L., et al.2008. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA14:1026–1036.
40.Townsend, H. L., B. K. Jha, R. H. Silverman, and D. J. Barton.2008. A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol.5:263–272.
41.Wang, Y., H. Li, Q. Tang, G. G. Maul, and Y. Yuan.2008. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: involvement of host cellular factors. J. Virol.82:2867–2882.
42.Wiedmer, A., et al.2008. Epstein-Barr virus immediate-early protein Zta co-opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. J. Virol.82:4647–4655. 43.Willingham, A. T., et al.2005. A strategy for probing the function of
non-coding RNAs finds a repressor of NFAT. Science309:1570–1573. 44.Wilusz, J. E., H. Sunwoo, and D. L. Spector.2009. Long noncoding RNAs:
functional surprises from the RNA world. Genes Dev.23:1494–1504. 45.Wong, T. S., et al.2009. Physical and functional interactions between human
mitochondrial single-stranded DNA-binding protein and tumour suppressor p53. Nucleic Acids Res.37:568–581.
46.Xu, Y., et al.2005. A Kaposi’s sarcoma-associated herpesvirus/human her-pesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol.79:3479–3487.
47.Zhang, T., et al.2011. Lysine residues of interferon regulatory factor 7 affect the replication and transcription activator-mediated lytic replication of Ka-posi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Gen. Virol.
92:181–187.
48.Zhou, H., H. Hu, and M. Lai.2010. Non-coding RNAs and their epigenetic regulatory mechanisms. Biol. Cell102:645–655.