Copyright © 1997, American Society for Microbiology
Structure-Function Analysis of the Triphosphatase Component
of Vaccinia Virus mRNA Capping Enzyme
LEI YU, ALEXANDRA MARTINS, LIANG DENG,ANDSTEWART SHUMAN*
Molecular Biology Program, Sloan-Kettering Institute, New York, New York 10021 Received 4 August 1997/Accepted 20 August 1997
The N-terminal 60 kDa (amino acids 1 to 545) of the D1 subunit of vaccinia virus mRNA capping enzyme is an autonomous bifunctional domain with triphosphatase and guanylyltransferase activities. We previously described two alanine cluster mutations, R77 to A (R77A)-K79A and E192A-E194A, which selectively inacti-vated the triphosphatase component. Here, we characterize the activities of 11 single alanine mutants—E37A, E39A, Q60A, E61A, T67A, T69A, K75A, R77A, K79A, E192A, and E194A—and a quadruple mutant in which four residues (R77, K79, E192, and E194) were replaced by alanine. We report that Glu-37, Glu-39, Arg-77,
Glu-192, and Glu-194 are essential forg-phosphate cleavage. The five essential residues are conserved in the
capping enzymes of Shope fibroma virus, molluscum contagiosum virus, and African swine fever virus. Probing the structure of D1(1-545) by limited V8 proteolysis suggested a bipartite subdomain structure. The essential residue Glu-192 is the principal site of V8 cleavage. Secondary cleavage by V8 occurs at the essential residue Glu-39. The triphosphatase-defective quadruple mutant transferred GMP to the triphosphate end of poly(A) to form a tetraphosphate cap structure, GppppA. We report that GppppA-capped RNA is a poor substrate for cap methylation by the vaccinia virus and Saccharomyces cerevisiae RNA (guanine-7) methyltransferases. The transcription termination factor activity of the D1-D12 capping enzyme heterodimer was not affected by mutations that abrogated ATPase activity. Thus, the capping enzyme is not responsible for the requirement for ATP hydrolysis during transcription termination.
Capping of vaccinia virus mRNAs occurs by a series of three
reactions in which the 59 triphosphate end of the primary
transcript is converted to a diphosphate end by RNA triphos-phatase, then capped with GMP by RNA guanylyltransferase, and finally methylated by RNA (guanine-7-) methyltransferase (25). All three steps are catalyzed by the vaccinia virus capping enzyme, a heterodimer composed of 95- and 33-kDa subunits encoded by the viral D1 and D12 genes, respectively (6, 15, 24, 31, 33). Catalytic domains are organized in a modular fashion. The amino-terminal 545-amino-acid segment of the 844-ami-no-acid D1 subunit catalyzes the triphosphatase and guanylyl-transferase reactions (9, 16, 29, 34). The methylguanylyl-transferase reaction is catalyzed by a distinct, nonoverlapping domain con-sisting of the carboxyl portion of the D1 subunit heterodimer-ized with the D12 protein (1, 10, 13).
The guanylyltransferase component of D1(1-545) performs two sequential nucleotidyl transfer reactions involving a cova-lent enzyme-guanylate intermediate in which GMP is linked to
the ε-amino group of Lys-260 (2, 18, 27). Lys-260 is situated
within a motif, KXDG, that is conserved among the cellular and DNA virus-encoded capping enzymes (2, 11, 18–20, 28). The capping enzymes share five other colinear sequence motifs downstream of the lysine nucleophile; these motifs comprise the GTP binding pocket and the active site of covalent catalysis (8, 30). Single alanine substitutions within the conserved motifs of the D1 protein elicit defects in transguanylylation but have no effect on the triphosphatase component (3).
Vaccinia virus capping enzyme hydrolyzes theg-phosphate
of triphosphate-terminated RNA and theg-phosphate of
nu-cleoside triphosphates (NTPs) (31, 33). Both reactions occur at a single active site, which is distinct from the active site of the guanylyltransferase (17, 34). The triphosphatase active site in-cludes essential constituents located within the N-terminal half
of the D1(1-545) protein. Myette and Niles showed by photo-cross-linking that the binding sites for ATP and RNA are in a region between amino acids 1 and 221 (17). We characterized two alanine cluster mutations within this region, R77 to A (R77A)-K79A and E192A-E194A, that inactivate the triphos-phatase but not the guanylyltransferase (34). A novel property of the R77A-K79A and E192A-E194A proteins was their abil-ity to catalyze GMP transfer to a virgin triphosphate end of RNA to generate the unusual dinucleoside tetraphosphate cap, GppppA (34).
In addition to its role in RNA 59 processing, the capping
enzyme acts as a transcription termination factor during early mRNA synthesis by vaccinia virus RNA polymerase (26). Both subunits of the capping enzyme are essential for termination factor activity (12). We showed previously that the guanylyl-transferase and methylguanylyl-transferase active sites are irrelevant to the termination event (12). We have since shown that termi-nation is tightly coupled to the hydrolysis of ATP (7). This raises the possibility that the triphosphatase activity of the capping enzyme is pertinent to transcription termination.
In the present study, we address five issues: (i) the individual contributions of residues Arg-77, Lys-79, Glu-192, and Glu-194 to enzyme activity, (ii) the identification of other catalytically essential residues, (iii) the subdomain structure of D1(1-545), (iv) the effects of tetraphosphate capping on the guanine meth-ylation step of RNA cap formation, and (v) the potential role of the triphosphatase activity of capping enzyme in transcrip-tion terminatranscrip-tion.
Expression and purification of alanine substitution mutants
of D1(1-545).The individual catalytic contributions of Arg-77,
Lys-79, Glu-192, and Glu-194 were gauged by replacing each residue by alanine. The mutations were introduced into the D1 (1-545) gene via the two-stage PCR overlap extension strategy (34). NdeI-BglII restriction fragments of the PCR-amplified mutated D1(1-545) DNAs were inserted into pET16b, thereby generating a series of pET-His-D1(1-545)-Ala expression
plas-* Corresponding author.
9837
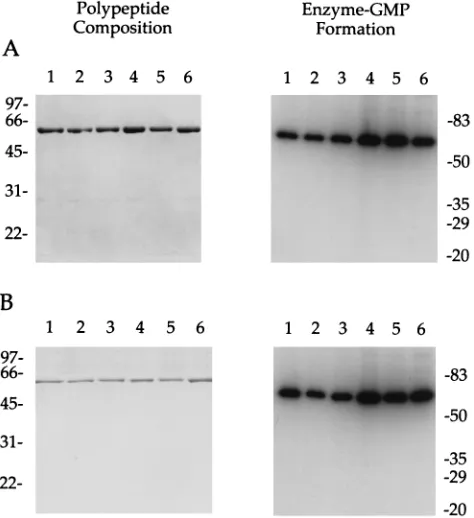
on November 9, 2019 by guest
http://jvi.asm.org/
mids. In addition, we engineered a quadruple mutant, D1 (1-545)-EERK, in which all four residues were replaced by alanine. The entire 1.7-kbp coding region of each expression plasmid was sequenced to exclude the occurrence of unwanted PCR-generated mutations. His-tagged versions of the wild-type and mutated D1(1-545) proteins were expressed in bac-teria. Purification was achieved by sequential Ni-nitrilotriacetic acid agarose and phosphocellulose chromatography steps, fol-lowed by glycerol gradient sedimentation (34). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) anal-ysis of the phosphocellulose fractions revealed a predominant 60-kDa polypeptide (Fig. 1A). Guanylyltransferase activity of all six phosphocellulose preparations was demonstrated by
la-bel transfer from [a-32P]GTP to form a 60-kDa covalent
en-zyme-GMP complex (Fig. 1A). The wild-type and mutant D1 (1-545) proteins sedimented as discrete peaks during glycerol gradient sedimentation (not shown). SDS-PAGE analysis of the peak glycerol gradient enzyme fractions showed them to be nearly homogeneous with respect to the 60-kDa polypeptide (Fig. 1B). Guanylyltransferase activity cosedimented with the 60-kDa protein during sedimentation (Fig. 1B and data not shown).
The specific guanylyltransferase activities of the mutant en-zymes were determined via titration experiments and were found to be within a factor of 2 of the specific activity of wild-type D1(1-545) (Table 1). It was expected that none of the single alanine mutations would affect transguanylylation, given the earlier findings that the R77A-K79A and E192A-E194A double mutants retained guanylyltransferase activity (34). We now find that side chain removal at all four residues was also without effect on the guanylyltransferase activity of D1(1-545)-EERK. Note that the extent of GMP labeling in vitro reflects the concentration of available GMP binding sites, which de-pends on the relative amounts of free versus GMP-bound D1 (1-545) in the purified enzyme fraction. The wild-type
D1(1-545) preparation was labeled with [32P]GMP in vitro to an
extent of 20% of the available binding sites. This calculation was based on the measured protein concentration and the assumption that all free enzyme molecules were catalytically active. We attribute activities of 51% (R77A) or 190% (EERK) of the wild-type value in enzyme-guanylate formation to variations in the relative amounts of preguanylated protein in the individual mutant enzyme fractions.
Arg-77, Glu-192, and Glu-194 are essential for
triphospha-tase activity.ATPase activities of the D1(1-545) preparations
were assayed by the release of32P
ifrom [g32P]ATP (1 mM)
during a 30-min incubation. Specific ATPase activities of wild-type and mutant D1(1-545) proteins were determined by
pro-tein titration. The wild-type D1(1-545) released 11 nmol of Pi
per min permg of protein. The relative specific activities of the
D1(1-545) mutants (normalized to the wild type) are shown in Table 1. K79A retained ATPase activity. The activities of the other three single mutants, R77A, E192A, and E194A, were
#1% of that of wild-type D1(1-545). A mixing experiment
established that the inactivity of these three proteins was not caused by inhibitors of ATP hydrolysis in the enzyme prepa-rations (not shown). We conclude that Arg-77, Glu-192, and
Glu-194 are essential forg-phosphate cleavage. The quadruple
mutant was defective for ATP hydrolysis, as expected from the effects of the single mutations.
Probing the subdomain structure of D1(1-545) by limited V8
proteolysis.Purified His-D1(1-545) was subjected to
[image:2.612.52.289.71.330.2]proteoly-sis with increasing amounts of V8 protease, which is specific for cleavage at glutamic acid. Although there are 41 potential V8
FIG. 1. Purification and guanylyltransferase activity of D1(1-545) mutants. (A) The polypeptide compositions of the phosphocellulose D1(1-545) fractions were analyzed by SDS-PAGE; 2mg of protein was applied to each lane. The gel was fixed and stained with Coomassie blue dye. The positions and sizes (in kilodaltons) of coelectrophoresed marker polypeptides are shown on the left. Guanylyltransferase activity was measured by formation of a covalent enzyme-GMP complex. Reaction mixtures (20ml) containing 50 mM Tris HCl (pH 8.0), 5 mM dithiothreitol, 5 mM MgCl2, 5mM [a-32P]GTP, and 20 ng of the indicated
[image:2.612.309.547.531.613.2]phosphocellulose fraction were incubated for 5 min at 37°C. Reactions were halted by the addition of EDTA to 10 mM and SDS to 1%. The samples were analyzed by electrophoresis through a 10% polyacrylamide gel containing 0.1% SDS. Radiolabeled polypeptides were visualized by autoradiography of the dried gel. The positions and sizes (in kilodaltons) of prestained marker polypeptides are indicated on the right. (B) The polypeptide compositions of the glycerol gradient D1(1-545) fractions were analyzed by SDS-PAGE; 0.5mg of protein was applied to each lane. Guanylyltransferase reaction mixtures contained 20 ng of the indicated glycerol gradient preparation. Lanes: 1, wild type; 2, R77A; 3, K79A; 4, E194A; 5, E192A; 6, E192A-E194A-R77A-K79A.
TABLE 1. Mutational effects on the guanylyltransferase and ATPase activities of D1(1-545)a
Mutant Sp act (% of wild type)
Guanylyltransferase ATPase
R77A 51 1
K79A 76 91
E194A 180 0.5
E192A 130 ,0.1
EERK 190 ,0.1
aThe glycerol gradient preparations of the wild-type and mutant D1(1-545) proteins were titrated for guanylyltransferase and ATPase activities. Guanylyl-transferase activity was assayed as described in the legend to Fig. 1. The extent of label transfer to the enzyme-GMP complex was quantitated by scanning the dried gels with a Fujix BAS1000 phosphorimager. ATPase reaction mixtures (10
ml) containing 50 mM Tris HCl (pH 8.0), 2 mM dithiothreitol, 10 mM MgCl2,
1 mM [g-32P]ATP, and various amounts of the glycerol gradient enzyme
prep-aration were incubated for 30 min at 37°C. Reactions were terminated by addi-tion of EDTA to 20 mM. An aliquot (6ml) of the reaction mixture was spotted on a PEI-cellulose TLC plate, which was developed in 0.75 M LiCl–1 M formic acid. The release of32P
ifrom ATP was quantitated by scanning the TLC plate
with a phosphorimager. Specific activities of each glycerol gradient preparation were determined in the linear range of enzyme dependence and are expressed as percent values relative to those of the wild-type D1(1-545).
on November 9, 2019 by guest
http://jvi.asm.org/
cleavage sites in D1(1-545), V8 cleaved preferentially at a
single site yielding two polypeptide fragments of;37 and;27
kDa (Fig. 2). Sequencing of these cleavage products by auto-mated Edman chemistry after transfer to a polyvinylidene di-fluoride membrane revealed that the smaller species originat-ed from the amino terminus of His-D1(1-545) and the larger polypeptide arose via cleavage between amino acids Glu-192 and Ile-193. These two species predominated at a level of V8 sufficient to cleave all the input His-D1(1-545) (Fig. 2). The amino-terminal V8 fragment was resistant to a higher level of protease that was sufficient to degrade the carboxyl domain
(Fig. 2). An;18-kDa polypeptide detected at higher V8 levels
arose by cleavage between residues Glu-39 and Leu-40 (Fig. 2). We surmise from the proteolysis experiment that D1(1-545) has a bipartite subdomain structure. The principal site of
pro-tease accessibility, the Glu-192–Ile-193 dipeptide, includes and flanks the two acidic residues that we have shown are essential for RNA triphosphatase activity. Treatment of the purified E194A mutant with V8 protease yielded the same pattern of proteolytic fragments detected with the wild-type D1(1-545) (data not shown). Thus, the elimination of the side chain of Glu-194 had no apparent impact on V8 cleavage at Glu-192. We infer from this result that the E194A mutation had no global effects on D1(1-545) tertiary structure. The 37- and 27-kDa fragments were absent from a V8 digest of the E192A mutant, as expected (data not shown).
Glu-38 and Glu-40 are essential for triphosphatase activity. Additional single alanine substitutions were introduced at seven new positions of D1(1-545). These were Glu-37, Glu-39, Gln-60, Glu-61, Thr-67, Thr-69, and Lys-75. The choice of residues to be mutated was guided by sequence conservation between the amino portion of the vaccinia virus D1(1-545) polypeptide and related proteins encoded by Shope fibroma virus, molluscum contagiosum virus, and African swine fever virus (Fig. 3) (19, 22, 32). The E37A, E39A, Q60A, E61A, T67A, T69A, and K75A proteins were expressed in bacteria as His-tagged derivatives and purified by Ni-agarose and phocellulose column chromatography. The purity of the phos-phocellulose enzyme preparations was comparable to that seen in Fig. 1 (data not shown). The specific activities of the mutant enzymes in enzyme-GMP formation and ATP hydrolysis were determined by protein titration and are expressed in Table 2 as the percent of the wild-type D1(1-545) specific activity. None of the mutations had a significant effect on the guanylyltrans-ferase component. Mutants Q60A, E61A, T67A, T69A, and K75A retained ATPase activity. The activity of the other two
single mutants, E37A and E39A, was,0.1% of that of
wild-type D1(1-545).
Tetraphosphate cap formation by D1(1-545)-EERK.We
[image:3.612.63.278.67.192.2]showed previously that capping of triphosphate-terminated poly(A) by the R77A-K79A and E192A-E194A double mu-tants resulted in the synthesis of a novel tetraphosphate cap structure, GppppA. Tetraphosphate cap constituted 16 and 25%, respectively, of the total cap dinucleotide formed by the R77A-K79A and E192A-E194A proteins, with the rest being the standard triphosphate cap, GpppA (34). In the present study, we examined the GMP-labeled poly(A) products syn-thesized by the EERK quadruple mutant of D1(1-545). Diges-tion of the RNA capped by D1(1-545)-EERK with nuclease P1 yielded two species of cap dinucleotide that were resolved by
FIG. 2. Limited proteolysis of D1(1-545). Reaction mixtures (20ml) contain-ing 1mg of His-D1(1-545) and 3.3, 10, 33, or 100 ng of V8 protease (increasing from left to right as indicated) were incubated on ice for 5 min. A control sample was incubated without protease (lane2). The reactions were quenched by addition of EDTA to 10 mM and SDS to 1%. The samples were electrophoresed through a 12% polyacrylamide gel containing 0.1% SDS. A photograph of the Coomassie blue-stained gel is shown. The positions and sizes (in kilodaltons) of coelectrophoresed marker proteins are indicated at the left. N-terminal sequenc-ing of D1(1-545) and the polypeptide products of partial V8 digestion was performed as described previously (21). In brief, a duplicate set of V8 digests was resolved by SDS-PAGE. Polypeptides were transferred electrophoretically to a polyvinylidene difluoride membrane and then stained with Coomassie blue. Slices containing individual proteolytic products were excised. Automated se-quencing of the immobilized polypeptide was performed with a modified Applied Biosystems model 477A microsequencer. The structures of the proteolytic frag-ments, inferred from their N-terminal sequences, are illustrated in cartoon form on the right. Amino acid coordinates of the N termini of the V8 fragments refer to the sequence of the native (untagged) D1 polypeptide. The shaded box at the N terminus of His-D1(1-545) denotes the His tag.
FIG. 3. Amino acid residues essential for the triphosphatase activity of D1(1-545) are conserved in other viral capping enzymes. The amino acid sequence of the D1 subunit of the vaccinia virus capping enzyme (Vac) from residues 13 to 213 is aligned with those of the homologous portions of the capping enzyme large subunits of Shope fibroma virus (SFV), molluscum contagiosum virus (MCV), and African swine fever virus (ASF). Conserved residues in D1(1-545) that were targeted for mutagenesis is this study are indicated by dots. The five amino acids that are essential for triphosphatase activity are shown in boxes. The sites of V8 protease cleavage at Glu-39 and Glu-172 are indicated by arrows.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.133.461.547.683.2]polyethyleneimine (PEI)-cellulose thin-layer chromatography (TLC) (Fig. 4, lane 5). The more rapidly migrating species corresponded to the standard GpppA cap, whereas the slower, more negatively charged species was the tetraphosphate cap GppppA. Both caps were resistant to digestion with alkaline phosphatase (not shown). GppppA constituted 33% of the to-tal cap dinucleotide product synthesized by D1(1-545)-EERK. A control experiment showed that nuclease P1 digestion of the GMP-labeled poly(A) product synthesized by wild-type D1 (1-545) yielded only the standard GpppA cap dinucleotide (Fig. 4, lane 1).
The mechanism of GppppA synthesis entails GMP transfer
to the 59triphosphate end of the poly(A) substrate. The
ca-pacity of vaccinia virus capping enzyme to guanylate a triphos-phate end is normally masked by the fact that the hydrolysis
of triphosphate-terminated poly(A) is;100-fold faster than
GMP transfer to the poly(A) 59terminus (33). Capping
en-zyme mutations like EERK affect the capping reaction by
rendering theg-phosphate cleavage step rate-limiting. The
quadruple mutant synthesized a higher proportion of GppppA than did either of the double mutants, presumably because the EERK mutant had a lower level of residual triphosphatase activity. We suspect that the synthesis of standard caps by triphosphatase-defective capping enzyme mutants is a function of residual triphosphatase activity plus the inevitable presence of some diphosphate-terminated poly(A) in the RNA substrate preparation.
Methylation of the tetraphosphate cap.In order to evaluate
the consequences of tetraphosphate capping, we tested wheth-er RNAs containing a GppppN cap wwheth-ere effective substrates for cap methylation. In the experiment shown in Fig. 4, the cap-labeled poly(A) synthesized by D1(1-545) or D1(1-545)-EERK was reacted with purified recombinant Saccharomyces
cerevisiae cap methyltransferase, ABD1 (14). In the presence of S-adenosylmethionine (AdoMet), ABD1 quantitatively
meth-ylated the GpppA RNA terminus produced by wild-type D1(1-545). Nuclease P1 digestion of the methylated capped poly(A) yielded the dinucleotide m7GpppA, which migrated more rap-idly than GpppA during thin-layer chromatography (Fig. 4, lanes 2 to 5). The population of GpppA caps synthesized by D1 (1-545)-EERK were methylated by ABD1 with the same effi-ciency as GpppA caps made by wild-type D1(1-545), as gauged by the dependence of the extent of methylation on the con-centration of input ABD1 (Fig. 4, lanes 6 to 8). Hence, the presence of tetraphosphate-capped RNAs did not interfere with methylation of standard caps by ABD1.
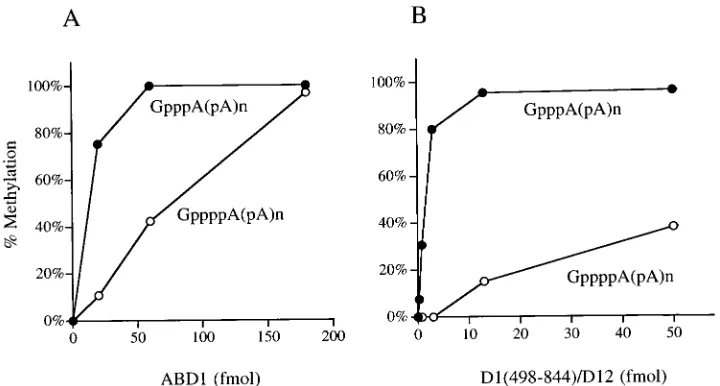
ABD1 was capable of methylating the tetraphosphate RNA cap, as evinced by conversion of the GppppA dinucleotide into
a novel species migrating behind m7GpppA (Fig. 4, lanes 6 to 8). Formation of this species by ABD1 was inhibited complete-ly when S-adenosylhomocysteine was added in place of AdoMet in the reaction mixture (not shown). Note that the tetraphos-phate-capped RNA ends were less effective than triphosphate caps as methyl acceptors for ABD1, insofar as the ABD1 concentration dependence of the conversion of GppppA to m7GppppA was shifted to the right compared to the methyl-ation of GpppA in the same reaction mixture (Fig. 4, lanes 6 to 8). The results are expressed quantitatively in Fig. 5A. We es-timate that ABD1 methylated tetraphosphate caps with about
;12% of the efficiency of triphosphate caps.
Similar experiments were performed with the methyltrans-ferase domain of the vaccinia virus mRNA capping enzyme (9). Recombinant vaccinia virus methyltransferase [a hetero-dimer of D1(498-844) and the full-length D12 subunit] was more fastidious than yeast ABD1 in its preference for standard caps over tetraphosphate caps as methyl acceptors (Fig. 5B). At limiting enzyme concentrations, the vaccinia virus enzyme exclusively methylated the triphosphate caps.
Transcription termination by triphosphatase-defective
cap-ping enzyme.To determine whether the triphosphatase
func-tion has any bearing on the transcripfunc-tion terminafunc-tion activity of vaccinia virus capping enzyme, we introduced the R77A-K79A and E192A-E194A cluster mutations into the full-length D1 protein. Wild-type D1, D1(R77A-K79A), and His-D1(E192A-E194A) were each coexpressed in bacteria with the D12 polypeptide and the heterodimers were purified by Ni-agarose and phosphocellulose chromatography (12). The poly-peptide compositions of the phosphocellulose preparations were examined by SDS-PAGE. All three preparations were highly enriched with respect to the large and small capping enzyme subunits (Fig. 6). The specific activities of the D1 (R77A-K79A)-D12 and D1(E192A-E194A)-D12 proteins in enzyme-GMP formation were 36 and 115%, respectively, of
FIG. 4. Synthesis of a GppppA RNA cap by His-D1(1-545)-EERK and tet-raphosphate cap methylation by S. cerevisiae ABD1. RNA capping reaction mixtures (20ml) containing 50 mM Tris HCl (pH 8.0), 1.25 mM MgCl2, 5 mM
dithiothreitol, 10mM [a-32P]GTP, 23 pmol (of ends) of triphosphate-terminated
poly(A), and 20 ng of D1(1-545) or D1(1-545)-EERK were incubated for 30 min at 37°C. Cap-labeled poly(A) was recovered by three rounds of trichloroacetic acid precipitation. The acid-insoluble material was then ethanol precipitated and resuspended in 10 mM Tris HCl (pH 8.0)–1 mM EDTA. Cap methylation reaction mixtures (10ml) containing 50 mM Tris HCl (pH 7.5), 5 mM dithio-threitol, 50mM AdoMet, [32P]GMP-labeled poly(A) synthesized by D1(1-545)
[image:4.612.311.546.443.570.2](lanes 1 to 4) or D1(1-545)-EERK (lanes 5 to 8), and either 20 fmol (lanes 2 and 6), 60 fmol (lanes 3 and 7), or 180 fmol (lanes 4 and 8) of purified recombinant ABD1 protein were incubated for 5 min at 37°C. ABD1 was omitted from control reaction mixtures (lanes 1 and 5). The reactions were halted by heating the mixtures at 95°C for 3 min. The samples were adjusted to 50 mM sodium acetate (pH 5.2) and then digested with 5mg of nuclease P1 (Boehringer) for 60 min at 37°C. The products were analyzed by TLC on PEI-cellulose plates developed with 0.35 M ammonium sulfate. An autoradiogram of the TLC plate is shown. The identities of the32P-labeled cap dinucleotides are indicated on the right.
TABLE 2. Effects of alanine substitutions on the guanylyltransferase and ATPase activities of D1(1-545)a
Mutant Sp act (% of wild type)
Guanylyltransferase ATPase
E37A 45 ,0.1
E39A 83 ,0.1
Q60A 42 44
E61A 60 61
T67A 63 86
T69A 57 89
K75A 100 140
aThe phosphocellulose preparations of the wild-type and mutant D1(1-545) proteins were titrated for guanylyltransferase and ATPase activities. Specific activities of each were determined in the linear range of enzyme dependence and are expressed as percent values relative to those of the wild-type D1(1-545).
on November 9, 2019 by guest
http://jvi.asm.org/
the wild-type D1-D12 activity. With respect to ATPase, the specific activities of D1(R77A-K79A)-D12 and
D1(E192A-E194A)-D12 were 0.4 and,0.1%, respectively, of the activity
of wild-type D1-D12. Hence, the two alanine cluster mutations inactivated the triphosphatase component of the capping en-zyme heterodimer, just as they inactivated the triphosphatase of the isolated D1(1-545) domain.
Transcription termination by vaccinia virus RNA polymer-ase was assayed in an in vitro system that is dependent on added capping enzyme (7). Transcription was programmed by a linear G21(TER59)A78 template linked to paramagnetic beads (5). The transcription unit consists of a synthetic early promoter fused to a 20-nucleotide G-less cassette. A run of three G residues immediately following the G-less cassette serves as an elongation block when GTP is omitted from the reaction mixtures (5). Situated 59 nucleotide downstream of the transcription initiation site is a transcription termination signal TTTTTTTTT. The strategy for analysis of termination during a single round of transcription entails pulse-labeling of nascent chains within ternary complexes arrested prior to the termination signal and purification of the complexes free of unbound proteins and labeled NTP precursor, followed by resumption of elongation upon provision of unlabeled NTPs. capping enzyme is included or omitted during the chase as desired.
Pulse-labeling transcription reaction mixtures containing
1 mM ATP, 0.1 mM UTP, 1mM [a32P]CTP, and 0.1 mM
39-O-methyl-GTP (39OMeGTP) yielded a 39
OMeGMP-ar-rested 21-mer nascent RNA (Fig. 7, lane 1). Template-en-gaged ternary complexes were recovered by centrifugation and immobilization of the beads with an external magnet. Upon provision of unlabeled NTPs and magnesium, the blocking
39OMeGMP moiety in the nascent RNA is removed by a
hydrolytic activity intrinsic to the vaccinia RNA polymerase and the bead-bound ternary complexes resume elongation (7). Elongation proceeded to the end of the linear DNA, as evinced by conversion of the pulse-labeled 21-mer RNA into a 349-nucleotide runoff RNA product (Fig. 7, lane 2). Runoff tran-scripts predominated even though the template contained a functional termination signal. The addition of recombinant
wild-type D1-D12 capping enzyme to the purified pulse-la-beled ternary complexes just prior to the start of the NTP chase resulted in the appearance of a heterogeneous array of terminated transcripts (Fig. 7, lane 5). It was established pre-viously that these terminated RNAs were released from the DNA template (5, 7). The size distribution of terminated
chains indicated that most 39ends were formed at a distance of
60 to 140 nucleotides downstream of the first U residue in the termination signal. The triphosphatase-defective D1(R77A-K79A)-D12 and D1(E192A-E194A)-D12 mutant proteins re-tained termination factor activity (Fig. 7, lanes 3 and 4). Titra-tion experiments indicated that the specific terminaTitra-tion factor activity of the mutant capping enzymes was comparable to that of the wild-type enzyme (not shown). This result suggests that
[image:5.612.119.479.68.261.2]FIG. 5. Efficiency of methylation of triphosphate versus tetraphosphate caps. GMP-labeled poly(A) synthesized by D1(1-545)-EERK was incubated with recom-binant yeast (A) or vaccinia virus (B) cap methyltransferases (13, 14) in the presence of 50mM AdoMet. The products were digested with nuclease P1 and analyzed by PEI-cellulose TLC as described in the legend to Fig. 4. The TLC plates were scanned with a Fujix BAS1000 phosphorimager. The extents of methylation of the triphosphate-capped poly(A), expressed as [m7GpppA/(m7GpppA1GpppA)] 3 100, and the tetraphosphate-capped poly(A), expressed as [m7GppppA/ (m7GppppA1GppppA)]3100, were plotted as a function of the amount of input yeast ABD1 (A) or vaccinia virus D1(498-844)-D12 (B).
FIG. 6. Purification of triphosphatase-defective heterodimeric capping en-zyme mutants. Expression and purification of His-tagged D1-D12 heterodimers were performed as described previously (12). The polypeptide compositions of the phosphocellulose fraction of wild-type His-D1-D12 (lane WT), His-D1 (R77A-K79A)-D12 (lane RK), and His-D1(E192A-E194A)-D12 (lane EE) were analyzed by SDS-PAGE; 3mg of protein was applied to each lane. The gel was fixed and stained with Coomassie blue dye. The positions of the D1 and D12 polypeptides are shown on the left.
on November 9, 2019 by guest
http://jvi.asm.org/
the capping enzyme does not perform the essential ATP hy-drolysis step during transcription termination.
Conclusions and implications.We have identified five
indi-vidual amino acid residues in the N-terminal half of D1(1-545) that are essential for the triphosphatase activity of vaccinia virus mRNA capping enzyme. Side chain removal at Glu-37, Glu-39, Arg-77, Glu-192, and Glu-194 reduced ATPase activity by at least 2 orders of magnitude without impacting signifi-cantly on the guanylyltransferase component. It is likely that the five essential side chains are situated at the triphosphatase active site. We speculate that Arg-77 interacts directly with the
negatively charged 59triphosphate moiety to promote
nucleo-philic attack by water on theg-phosphorus. The role of Glu-37,
Glu-39, Glu-192, and Glu-194 may be to coordinate the essen-tial divalent cation cofactor(s). The observation that ATP hy-drolysis by D1(1-545) requires divalent cation in excess over the available ATP (17, 34, 35) is consistent with the existence of a metal binding site(s) on the enzyme. Glu-37, Glu-39, Arg-77, Glu-192, and Glu-194 are conserved in the D1-ho-mologs of Shope fibroma virus, molluscum contagiosum virus, and African swine fever virus.
The 60-kDa triphosphatase-guanylyltransferase domain of vaccinia virus capping enzyme was defined initially by limited proteolysis of the native D1-D12 heterodimer with trypsin (23). In the present study, we showed that the D1(1-545) polypeptide consists of two V8 protease-resistant segments. The protease accessibility of Glu-192 suggests that this residue is located either on the surface of the protein or in a flexible hinge separating the two structural subdomains. It appears from the mutational data that the active site of the triphos-phatase is composed of constituents from both subdomains. Glu-39 is also V8 accessible, albeit less so than Glu-192. The
finding that all four glutamate residues that are essential for triphosphatase activity are positioned at or near sites of V8 cleavage (Fig. 3) is consistent with the idea of a surface loca-tion for the triphosphatase active site.
Although the six conserved motifs that comprise the active site of the guanylyltransferase are all located in the distal V8 fragment from residues 193 to 545, two lines of evidence sug-gest that this fragment is not sufficient for guanylyltransferase activity: (i) recombinant proteins D1(184-545) and D1(193-545) expressed in bacteria had no detectable guanylyltrans-ferase activity (16, 35) and (ii) an alanine cluster mutation, F160A-K161A, located upstream of the V8-sensitive domain boundary, caused inactivation of the guanylyltransferase but not the triphosphatase (34). We surmise that the guanylyltrans-ferase and triphosphatase functional domains overlap physi-cally. Resolving D1(1-545) into autonomous enzymatic com-ponents is therefore an unlikely prospect.
Our finding that the tetraphosphate caps synthesized by triphosphatase-defective mutants of D1(1-545) are poor sub-strates for cap methylation provides some rationale for the tight physical association of the triphosphatase and guanylyl-transferase activities. This arrangement would ensure that all primary transcripts are rapidly converted to diphosphate ends and thus preclude the guanylylation of unmodified triphos-phate termini, which may be viewed as wasteful, insofar as the GppppN ends would not be efficiently methylated and any GppppN-capped RNAs would presumably be inefficiently translated. An interesting question is whether RNAs contain-ing methylated tetraphosphate caps would be recognized by the translation machinery.
Finally, we provide evidence that the capping enzyme is not the energy coupling factor for transcription termination during early mRNA synthesis by vaccinia virus RNA polymerase. Capping enzyme mutants that were severely defective for ATP hydrolysis were fully competent in promoting transcription ter-mination in vitro. We recently identified a second viral protein that is required along with the capping enzyme to elicit tran-scription termination in vitro (4). That protein, designated factor X, has an associated ATPase activity. Factor X is nor-mally tightly associated with the polymerase elongation com-plex (4). In light of the present data, we posit that factor X is the sole energy coupling factor during transcription termina-tion. We hypothesize that the capping enzyme may recognize the UUUUUNU termination signal in the nascent RNA and trigger the activation of the factor X ATPase.
Lei Yu’s contribution to the present work was submitted to the faculty of the Cornell University Graduate School of Medical Sciences in fulfillment of the requirements for the degree of Master of Science.
REFERENCES
1. Cong, P., and S. Shuman. 1992. Methyltransferase and subunit association domains of vaccinia virus mRNA capping enzyme. J. Biol. Chem. 267:16424– 16429.
2. Cong, P., and S. Shuman. 1993. Covalent catalysis in nucleotidyl transfer: a KTDG motif essential for enzyme-GMP complex formation by mRNA cap-ping enzyme is conserved at the active sites of RNA and DNA ligases. J. Biol. Chem. 268:7256–7260.
3. Cong, P., and S. Shuman. 1995. Mutational analysis of mRNA capping enzyme identifies amino acids involved in GTP binding, enzyme-guanylate complex formation, and GMP transfer to RNA. Mol. Cell. Biol. 15:6222– 6231.
4. Deng, L., and S. Shuman. 1996. An ATPase component of the transcription elongation complex is required for factor-dependent transcription termina-tion by vaccinia RNA polymerase. J. Biol. Chem. 271:29386–29392. 5. Deng, L., J. Hagler, and S. Shuman. 1996. Factor-dependent release of
nascent RNA by ternary complexes of vaccinia RNA polymerase. J. Biol. Chem. 271:19556–19562.
[image:6.612.137.202.67.268.2]6. Guo, P., and B. Moss. 1990. Interaction and mutual stabilization of the two subunits of vaccinia virus mRNA capping enzyme coexpressed in Escherichia FIG. 7. Transcription termination. Pulse-labeling transcription reactions and
isolation of bead-bound G21 ternary complexes were carried out as described previously (5). The beads were treated transiently with heparin (0.5mg/ml) prior to isolation (4). Elongation reactions entailed incubation of the isolated ternary complexes for 10 min at 30°C in 20-ml mixtures containing 20 mM Tris HCl (pH 8.0), 6 mM MgCl2, 2 mM DTT, 1 mM ATP, 1 mM GTP, 1 mM CTP, 1 mM UTP,
and capping enzyme as specified. Capping enzyme was added prior to NTPs. [32P]CMP-labeled transcription products were recovered and analyzed by
elec-trophoresis through a 17% denaturing polyacrylamide gel. An autoradiograph of the gel is shown. Lane 1 shows the products of the pulse-labeling reaction. Elongation reaction products are shown in lanes 2 to 5. Lane 2, no added capping enzyme; lane 3, 200 fmol of His-D1(R77A-K79A)-D12; lane 4, 200 fmol of His-D1(E192A-E194A)-D12; lane 5, 200 fmol of wild-type His-D1-D12. RT, runoff transcript; T, terminated transcript.
on November 9, 2019 by guest
http://jvi.asm.org/
coli. Proc. Natl. Acad. Sci. USA 87:4023–4027.
7. Hagler, J., Y. Luo, and S. Shuman. 1994. Mechanism of factor-dependent transcription termination by vaccinia RNA polymerase: kinetic coupling and requirement for ATP hydrolysis. J. Biol Chem. 269:10050–10060. 8. Hakansson, K., A. J. Doherty, S. Shuman, and D. B. Wigley. 1997. X-ray
crystallography reveals a large conformational change during guanyl transfer by mRNA capping enzymes. Cell 89:545–553.
9. Higman, M. A., N. Bourgeois, and E. G. Niles. 1992. The vaccinia virus mRNA (guanine-N7-) methyltransferase requires both subunits of the mRNA capping enzyme for activity. J. Biol. Chem. 267:16430–16437. 10. Higman, M. A., L. A. Christen, and E. G. Niles. 1994. The mRNA
(guanine-7-) methyltransferase domain of the vaccinia virus mRNA capping enzyme: expression in Escherichia coli and structural and kinetic comparison to the intact capping enzyme. J. Biol. Chem. 269:14974–14981.
11. Ho, C. K., J. L. Van Etten, and S. Shuman. 1996. Expression and charac-terization of an RNA capping enzyme encoded by Chlorella virus PBCV-1. J. Virol. 70:6658–6664.
12. Luo, Y., X. Mao, L. Deng, P. Cong, and S. Shuman. 1995. The D1 and D12 subunits are both essential for the transcription termination factor activity of vaccinia virus capping enzyme. J. Virol. 69:3852–3856.
13. Mao, X., and S. Shuman. 1994. Intrinsic RNA (guanine-7) methyltransferase activity of the vaccinia virus capping enzyme D1 subunit is stimulated by the D12 subunit: identification of amino acid residues in the D1 protein required for subunit association and methyl group transfer. J. Biol. Chem. 269:24472– 24479.
14. Mao, X., B. Schwer, and S. Shuman. 1995. Yeast mRNA cap methyltrans-ferase is a 50-kDa protein encoded by an essential gene. Mol. Cell. Biol. 15:4167–4174.
15. Martin, S. A., E. Paoletti, and B. Moss. 1975. Purification of mRNA gua-nylyltransferase and mRNA (guanine-7-) methyltransferase from vaccinia virions. J. Biol. Chem. 250:9322–9329.
16. Myette, J., and E. G. Niles. 1996. Domain structure of the vaccinia virus mRNA capping enzyme: expression in Escherichia coli of a subdomain pos-sessing the RNA 59-triphosphatase and guanylyltransferase activities and a kinetic comparison to the full-size enzyme. J. Biol. Chem. 271:11936–11944. 17. Myette, J., and E. G. Niles. 1996. Characterization of the vaccinia virus RNA 59-triphosphatase and nucleoside triphosphate phosphohydrolase activities: demonstration that both activities are carried out at the same active site. J. Biol. Chem. 271:11945–11952.
18. Niles, E. G., and L. Christen. 1993. Identification of the vaccinia virus mRNA guanylyltransferase active site lysine. J. Biol. Chem. 268:24986– 24989.
19. Pena, L., R. Yanez, Y. Revilla, E. Vinuela, and M. L. Salas. 1992. African
swine fever virus guanylyltransferase. Virology 193:319–328.
20. Schwer, B., and S. Shuman. 1994. Mutational analysis of yeast mRNA capping enzyme. Proc. Natl. Acad. Sci. USA 91:4328–4332.
21. Sekiguchi, J., and S. Shuman. 1995. Proteolytic footprinting of vaccinia topoisomerase I bound to DNA. J. Biol. Chem. 270:11636–11645. 22. Senkevich, T. G., J. J. Bugert, J. R. Sisler, E. V. Koonin, G. Darai, and B.
Moss.1996. Genomic sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273:813–816.
23. Shuman, S. 1989. Functional domains of vaccinia virus mRNA capping enzyme: analysis by limited tryptic digestion. J. Biol. Chem. 264:9690–9695. 24. Shuman, S. 1990. Catalytic activity of vaccinia mRNA capping enzyme
sub-units coexpressed in Escherichia coli. J. Biol. Chem. 265:11960–11966. 25. Shuman, S. 1995. Capping enzyme in eukaryotic mRNA synthesis. Prog.
Nucleic Acid Res. Mol. Biol. 50:101–129.
26. Shuman, S., S. Broyles, and B. Moss. 1987. Purification and characterization of a transcription termination factor from vaccinia virions. J. Biol. Chem. 262:12372–12380.
27. Shuman, S., and J. Hurwitz. 1981. Mechanism of mRNA capping by vaccinia virus guanylyltransferase: characterization of an enzyme-guanylate interme-diate. Proc. Natl. Acad. Sci. USA 78:187–191.
28. Shuman, S., Y. Liu, and B. Schwer. 1994. Covalent catalysis in nucleotidyl transfer reactions: essential motifs in Saccharomyces cerevisiae capping zyme are conserved in Schizosaccharomyces pombe and viral capping en-zymes and among polynucleotide ligases. Proc. Natl. Acad. Sci. USA 91: 12046–12050.
29. Shuman, S., and S. G. Morham. 1990. Domain structure of vaccinia virus mRNA capping enzyme: activity of the Mr 95,000 subunit expressed in Escherichia coli. J. Biol. Chem. 265:11967–11972.
30. Shuman, S., and B. Schwer. 1995. RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Mol. Microbiol. 17:405–410. 31. Shuman, S., M. Surks, H. Furneaux, and J. Hurwitz. 1980. Purification and characterization of a GTP-pyrophosphate exchange activity from vaccinia virions. J. Biol. Chem. 255:11588–11598.
32. Upton, C., D. Stuart, and G. McFadden. 1991. Identification and DNA sequence of the large subunit of the capping enzyme from Shope fibroma virus. Virology 183:773–777.
33. Venkatesan, S., A. Gershowitz, and B. Moss. 1980. Modification of the 59end of mRNA: association of RNA triphosphatase with the RNA guanylyltrans-ferase-RNA (guanine-7-) methyltransferase complex from vaccinia virus. J. Biol. Chem. 255:903–908.
34. Yu, L., and S. Shuman. 1996. Mutational analysis of the triphosphatase domain of vaccinia virus mRNA capping enzyme. J. Virol. 70:6162–6168.
35. Yu, L., and S. Shuman. Unpublished data.