JOURNALOFVIROLOGY,Oct. 1994,p.6347-6362 0022-538X/94/$04.00+0
Copyright (C 1994, American Society for Microbiology
Improved Cell Survival
by the Reduction
of
Immediate-Early
Gene
Expression in Replication-Defective
Mutants
of
Herpes
Simplex Virus Type 1 but
Not
by Mutation of
the
Virion Host Shutoff
Function
PAULA. JOHNSON,* MING JINGWANG,ANDTHEODORE FRIEDMANN
Department of Pediatrics, CenterforMolecular Genetics, University ofCalifomia,
SanDiego, LaJolla, California 92093-0634
Received 4 March 1994/Accepted6 July 1994
Derivatives of herpes simplex virus type 1 (HSV-1) have elicited considerable interest as gene transfer
vectors because of theirability to infect a wide range of cell types efficiently, including fullydifferentiated
neurons. However, it has been found that infection of many types of cell with vectors derived from
replication-defectivemutantsof HSV-1 isassociated with cytopathic effects (CPE).We have previously shown
thatviralgeneexpression playedanimportant role intheinductionof CPE caused byanHSV-1mutantdeleted
for theessential immediate-early gene3 (IE 3) (P. A. Johnson,A. Miyanohara, F. Levine, T. Cahill, and T.
Friedmann,J.Virol. 66:2952-2965,1992). We have investigated which viralgenesmight be responsible for CPE
by comparing the ability of each of the individual genes expressed by an IE 3 deletion mutant during a
nonproductive infectiontoinhibit biochemical transformation after cotransfectionof BHKorCV-1 cells with
aselectablemarkergene.Transfection of IEgenes 1, 2, and 4individually allcaused amarkedinhibition of
colonyformation,whiletransfection ofIE 5 and the large subunit of ribonucleotide reductase had little effect.
These results suggested that itwould benecessaryto mutateorreduce the expression of nearly allHSV-1 IE
genestoreducevirus-induced CPE. Therefore,wehave used VP16mutants,whichareunabletotransinduce
IE gene expression (C. I. Ace, T. A. McKee, J. M. Ryan, J. M. Cameron, and C. M. Preston, J. Virol.
63:2260-2269, 1989),toderivetworeplication-defective strains: 14HA3,which is deleted for both copies of IE
3,andin1850A42,whichhasadeletionintheessential earlygeneUL42. The IE 3-VP16 doublemutant,14HA3,
is significantly less toxic than a single IE 3 deletion mutant over a range of multiplicities of infection, as
measured in a cell-killing assay, and has an enhanced ability to persist in infected cells in a biologically
retrievable form.Incontrast, theUL42-VP16doublemutant,in1850A42, showed reduced toxicity onlyatlow
multiplicities of infection. To test the roleofthevirion host shutofffunction as anadditional candidateto
influencevirus-inducedCPE,wehave introducedalarge insertion mutation intothevirionhost shutoffgene
ofanIE 3 deletionmutantandthe doublemutant14HA3. Mutation of thisgenedidnotreduce thecytotoxicity of eitherstrain. Theseresults demonstrate thatlong-term survival of cellsinfected withreplication-defective HSV-1 mutantscanbeenhancedthrough genetic manipulations that reduce viralgeneexpression.
Approaches to human gene therapy take advantageof the
highefficiencyof viralvectorsinintroducing foreigngenesinto
mammalian cells (56).To be useful inmostcases, theforeign
genes should be expressed stably and efficiently, and the
processofgenetransfer shouldnotproducecelldamage.Some vectors,suchasthose derivedfromretroviruses, satisfymostof theserequirements,butmostretroviralvectorsarenotcapable ofgenetransfertopostmitoticcells. Therearemanyinstances inwhichgenetransfertonondividingcells would bedesirable, whethertopermitstudies ofgenetic manipulationin differen-tiated cell types or for more applied therapeutic purposes.
Herpes simplexvirus type 1 (HSV-1) has attracted
consider-able interest for itspotentialas agenetransfer vector. Much is known about HSV-1atthe molecularbiological level,it is able to infectawide rangeofreplicatingand nonreplicating cells, and it can maintain a lifelong latent infection in sensory
neurons(for recentreviews onHSV-1vectors,see references 5 and35).
*Correspondingauthor.Mailingaddress:Dept.ofPediatrics,
Cen-terfor MolecularGenetics, UniversityofCalifornia at SanDiego,La Jolla, CA 92093-0634. Phone: (619) 534-1419. Fax: (619) 534-1422. Electronic mail address: [email protected].
Vectors derived fromreplication-defectivemutantsor atten-uated strains of HSV-1 haveprovedsuccessful for thetransfer
andexpressionofforeigngenesinthe mammalian braininvivo
(8, 11, 17) and in primarycentralnervous system neurons in
vitro(30). Thetargetcells for infection with HSV-1vectorsare
not limited to neurons, as demonstrated by the high but
transient levels of circulating transgene products in mice
following injectionof the liver with HSV-1 vectorsexpressing
canine factor IX or hepatitis B virus surface antigen (43).
However,inmostcases,attempts toachievelong-term
expres-sion offoreigngenes, at least in cellsotherthan those of the
peripheral nervous system in vivo, have been hampered by
cytotoxicity and/or shutoff oftransgeneexpression.
We have studied the cytopathic effects (CPE) caused by infection withvectors derivedfrom an HSV-1 mutant called
D30EBA (49),deletedfornearlythe entirecodingportionof
immediate-early gene 3 (IE 3), whose product, Vmwl75
(ICP4), is the major transcriptional activating protein of
HSV-1(51).Since Vmwl75iscritically requiredforexpression of viralearlyand lategenes,anIE3mutantexpressesveryfew viral genes and iscompletely replication defective (9, 24, 49,
51). However,wehave demonstratedsignificant CPE,
includ-ingfragmentationof cellularDNA andcytoplasmicblebbing,1
6347
Vol. 68,No. 10
on November 9, 2019 by guest
http://jvi.asm.org/
to 3 days after infection with an IE 3 mutant (29). We also
found thatwaspossible toreduceCPEbytwo methodswhich reduced viralgeneexpression: UVirradiation of thevirusand
pretreatment of cells with interferon (29). These results
sug-gest that one or more of thefew viral genesexpressed in the absence ofVmwl75were, at least in part, responsible for the CPE.
The viral genes efficiently expressed by an IE 3 deletion mutant include IE 1, whose product, VmwllO (ICPO), is a
general transactivator of transcription but is not absolutely
essentialfor virus replication (6, 12, 65);IE2,whoseproduct,
Vmw63 (ICP27),isrequired forexpression of truelategenes, full levelsofviral DNAsynthesis,andmodulationofearlygene
expression and which, in transfection experiments, can act as
an activatororrepressor oftranscription incombination with Vmwl75 and Vmwl10 (38, 55, 60) (Vmw63has recentlybeen
shown to affect mRNA splicing and stimulate viral mRNA 3'-end processing [42, 58]); IE 4, whose product, Vmw68
(ICP22), isrequired forefficientvirus replication insome cell
types(59); IE5,whose product,Vmwl2(ICP47), hasrecently
been shown to inhibit antigen presentation to CD8+ T
lym-phocytes in infected human fibroblasts (68); and the gene
encoding ICP6, which is the large subunit of ribonucleotide
reductase,whichis notessential forvirusreplication intissue
culture (20). Wehave previously tested the cytopathogenicity
ofHSV-1 mutantsbearing lesions in one or more of thefive individual IEgenesanddetermined that noneofthe IEgene
products alone areresponsibleforCPE (29). In addition, not
everycellinfectedwiththe IE3mutant issubjecttocelldeath
(29),afindingconsistentwith thenotionthatathresholdlevel
ofthe IEgene productsmay berequiredfortheinductionof
CPE and that cell survival may be enhanced by reducing or
turning offviral geneexpression.
The first part of the present study was aimed at the identificationof the HSV-1 IEgenesresponsiblefor inducing
CPE during infection with an IE 3 mutant. As an assay to
measure the potential for CPE, we have cotransfected cells
with the bacterial neomycin phosphotransferase gene (neo), which confers resistance to the neomycin analog G418, as a
selectable marker, together with HSV-1 sequences that may
interfere with stable biochemical transformation. We postu-latedthatfewerneo-transformed cells would beobtained from
cotransfections which include a gene encoding a
cytopatho-genic product. As a control for promoter competition or for
the presence of spurious inhibitory sequences, we have
in-cludedplasmids bearingmutationswithinthecoding regions of
theHSV-1 genesbeingtested. Ourresultsindicatethat IE 1,
IE 2, and IE 4 all encodecytopathogenic geneproducts. We havealsofoundthatIE3causesinhibitionoftransformation in
CV-1 cells butnot inBHKcells.The ICP6geneandthe IE5 gene didnotdemonstrateinhibition oftransformationin this assaysystem.
Animportant regulatorofHSV-1 IEgeneexpression isthe
virion polypeptide VP16 (Vmw65), which functions to
stimu-lateIEgenetranscription atthe earlieststagesof infection (3,
7) through formation of a complex containing VP16 and
cellular transcription factors, including Oct-1, which together
specifically bind the IE promoter element TAATGARAT,
whereRisapurine (45,52,66).Aceetal. (2) constructedan
HSV-1mutant, in1814,whichhasa12-bpinsertionin theVP16 gene which abolishes the ability of VP16 to transinduce IE
gene expression. However, this virus is stillable toreplicate,
particularly at high multiplicities of infection (MOIs). These
resultssuggestthat itmightbepossibletogenerateavirus that
does not induce CPE during a nonproductive infection ifthe
VP16 mutantin1814were furthermutated to render it
repli-cation defective. For that reason, we haveconstructed viruses
with deletions in either IE 3 or UL42 aswell asthe insertion
mutation in
VP16,
designated
14HA3 andin1850A42,
respec-tively. In
comparison
to their VP16-intact counterparts, wehave found that 14HA3 has reduced
cytotoxicity
over a rangeof
MOIs,
whereas in1850A42 is less toxiconly
atlow MOIs.The virion host shutoff(vhs)function is acomponent of the
virion encoded bygeneUL41
(40),
which has beenthought
tocontribute to the
cytotoxicity
of HSV-1 vectors. The vhsfunction suppresses translation of
preexisting
mRNAs andincreases the turnover of both viral and cellular nascent
mRNA
(33, 47,
53), although
thestrength
of this function mayvarybetweenstrains and is weak in HSV-1 strain17+
(15).
Thevhsmutantvhs-1 isreplication competent (53), althoughinan
earlier
study,
it did not show reducedcytotoxicity
compared
with itsparentalwild-type strain
(KOS)
evenwhen viralDNAreplication was inhibited
(29).
In the presentstudy,
we havetested morerigorouslytheinvolvement of vhs in the induction
of CPE by replication-defective mutants of strain
17+
by
introducing alarge insertion mutation into the vhs gene ofan
IE 3 deletion mutant and alsointo the vhs gene of the
newly
constructed IE 3-VP16double mutant 14HA3.
Analysis
oftheresultingmutants indicated that vhs doesnotcontributetothe
cytotoxicity inducedbythis strain of HSV-1 duringa
nonpro-ductive infection. To determine whether a potential
involve-ment of vhs in virus-induced cytotoxicity was obscured by the
effectsof viral gene expression, we measured cellviability after
infection with UV-irradiatedvhsmutants of strains KOS and
17+ and their parental virus strains, but again, there was no
significant difference in cytotoxicitywhich correlated with the
vhsmutation.
MATERLILSANDMETHODS
Cells and viruses. All cell lines were propagated in
Dulbec-co'smodificationofEagle'sminimal essential medium
(DME)
containing 10% fetal calf serum. BHK TK- and CV-1 cells
were used to assay the inhibition of colony transformation.
TO-119 primary human diploid fibroblasts were used to assay
cellkilling and the ability of virus to persist in infected cells.
Cells used to support growth of IE 3 deletion mutants were
RR1(29) and E5 (obtained from N. A. DeLuca, University of
Pittsburgh,Penn.) (10);V9 cells were used to supportgrowth
of UL42 deletion mutants (27). RR1, E5, and V9 cellswere
passaged in medium containing 400 ,ug of G418 (geneticin;
GIBCO BRL, Grand Island, N.Y.) per ml.
All the viruses used in this study were originally derived
from HSV-1 strain 17+ except where stated below. The VP16
mutants in 1814 and in1850 were generously provided by C. M.
Preston (Medical Research Council Virology Unit, Glasgow,
Scotland). The mutant in1850 was derived from in1814(2) by
insertion of theEscherichia coli lacZ gene into the thymidine
kinase (TK)coding sequence(Sla). The IE 3 deletion mutant
D30EBA was constructed by Paterson and Everett (49). CgalA42 contains a deletion in the UL42 coding sequence (27).
Viruses containing the in1814-specific lesion in the VP16 gene
wereprepared and titrated by infecting cells in the presence of
5 mM hexamethylene bisacetamide (HMBA; Sigma) for the
first 24 h, as described before (39). This step significantly improves both the titer and the particle/PFU ratio of VP16 mutants. An ICP8 deletion mutant derived from strain KOS, d301 (19), was used in the superinfection rescue procedure and
was kindly provided by D. Knipe (Harvard Medical School,
Boston, Mass.). The virion host shutoff mutant vhsl (53) and wild-type strain KOS were kindly provided by G. S. Read (University of Missouri-Kansas City), and the IE 3 deletion
on November 9, 2019 by guest
http://jvi.asm.org/
CYTOTOXICITY OF REPLICATION-DEFECTIVE HSV-1 MUTANTS 6349
mutantD30EBA was kindly supplied by R. D. Everett
(Med-ical Research Council Virology Unit).
Plasmids. The plasmids used in the colony inhibition assay
areshown in Fig. 1. The IE 1 gene contained in plasmidplll,
theIE 1 deletion mutantpllOdel4,the IE 3 gene inpl75,and
two deletion mutants of IE 3, pD8 and pD9, were obtained
from R. D. Everett(48,50). PlasmidpKl-2 contains IE 3 under
the control of itsown promoterand was obtained from N. A.
DeLuca (10). The IE 2 gene is contained in a 2,448-bp
BamHI-HpaIfragment in plasmid pIE63 (29), from which the following derivatives were made: pIE63+ contains an
addi-tional 815-bpHincII-BamHI fragment adjoining the 5' end of
IE2; pXin63+ contains anXhoIlinker inserted at the SalI site
ofpIE63+ (disruptionof the IE 2 sequenceatthisSallsite has
been shownto impairthe activator and repressor functions of
Vmw63 in a transient assay system [22, 55]); and pX63.1
containsanXhoIlinker inserted atthe site ofa28-bpdeletion
betweentwoRsrIIsites inpIE63.The IE 4 gene is contained in
plasmid pIE68, and the derivation ofpIE68A,whichcontainsa
425-bp deletion between the SstI andHindlIl sites of pIE68
andaninsertionof 176 bpofMoloneymurine leukemia virus
DNA, has been described(29).PlasmidpZ4containsa4.6-kb
human cytomegalovirus (HCMV)-lacZ fragment inserted
be-tween the SstI andHindIIIsites ofpIE68, asshown in Fig. 1.
The IE 5 gene is contained in pIE12 as an EcoRI-BamHI
subfragmentof BamHI-x. This fragmentwasinserted between the HindlIl and BamHI sites of pl,
replacing
thefirefly
luciferase gene (luc) (28), so that IE 5 is terminated
by
thesimian virus 40
(SV40) polyadenylation signal
present in pl; pIE12doesnotcontaincompleteopenreading
frames for the overlapping3' coterminal genesUSl0andUS11(41).
PlasmidpZ5 was derived from BamHI-x as described before
(29).
Plasmids pKHF, which contains the large subunit of
ribonu-cleotide reductase (ICP6), and
pKHFA,
which contains adeletion within the ICP6
coding region,
were obtained fromS. K. Weller (Universityof
Connecticut)
(20).
All the HSV-1genes described above were derived from strain
17+
exceptthose onplasmids
pKl-2,
pKHF, andpKHFA.
The plasmids used to confer resistance to G418 were
pSV2neo
(62)
and pHneo(28),
which contain the SV40 enhancer-promoter and the humanhypoxanthine
phosphori-bosyltransferase
(HPRT)
gene promoter,respectively,
driving
neo.
Theplasmidusedto delete IE 3
coding
sequencesfromtheviral genomewaspIllH.Thiswasderived from
pdelI11
(49)
by
insertion of a
587-bp
HaeIIIfragment
frompUC19
into theEcoRI site,
marking
the IE 3 deletionby
blunt-endligation
(see Fig.
3).
Theplasmid
used todisrupt
thecoding
sequenceof the vhs genewas
pUL41-lacZ.
Thisplasmid
containsthe5'portionof the vhs gene from
pBam-i-Sst,
into theNrulI siteofwhich was
inserted, by
blunt-endligation,
a 4.6-kb BamHIfragment
containing
an HCMV-lacZ cassette frompON249
(63) (see
Fig.
3b).
Other
plasmids
used in thisstudy
werepBaml,
whichcontains the HSV-1
BamHI-l
fragment
inpUC19,
andpUL42A,
which contains a1,258-bp
deletion of the UL42coding
sequencewhichis markedby
an insertion ofa280-bp
PstI-PvuII fragment from
pBluescript
KSII(27). pMC1
con-tainsthe
coding
sequenceof VP16andwaskindly
provided
by
C. M. Preston
(7).
Transfections and
colony
inhibition assay. We have usedthecalcium
phosphate
method of Graham andvander Eb(21)
for all transfections. For the
colony
inhibition assay,freshly
trypsinized cells were seeded on six-well cluster dishes at a densityof
105
cells per 35-mm well 1day
prior
totransfection.The cells were cotransfected in
triplicate
with 0.25 ,ug ofUL us
a) - | IRL I IA ITRS
UL5f].f:[UL55 (Fjt¶- (E:Z1J JED)CUS2 (.&L
IE2 IEl IE3 IE4 IE5
b) pIE63 BSaHp HcB
pIE63+ i Hc Xh pXin63+ r
B Sa
pX63.1 6
(AsRs)
Xh
HpSa Xh S R SH Ml
pp 4plp111 I- --- plE68 1Pp~~~~~~~~~~6pl11Odel4 p Z4
lacZHCMV
pKl-21
-S B
p175 i
-= SV40
Dg DB En.Pr
c)
Vmwl75
4
N~~~~~~~~~~~~~~~~~~~~~~~~~~~
_.
VmwllO0
B,SApR
I plEl12
IacZ HCMV
K\G) N.(3b c4.' S
9 .99.
(N
.P9
c.1
9\ V.l.
9 9
Vmw63 _
BHK
cv-1
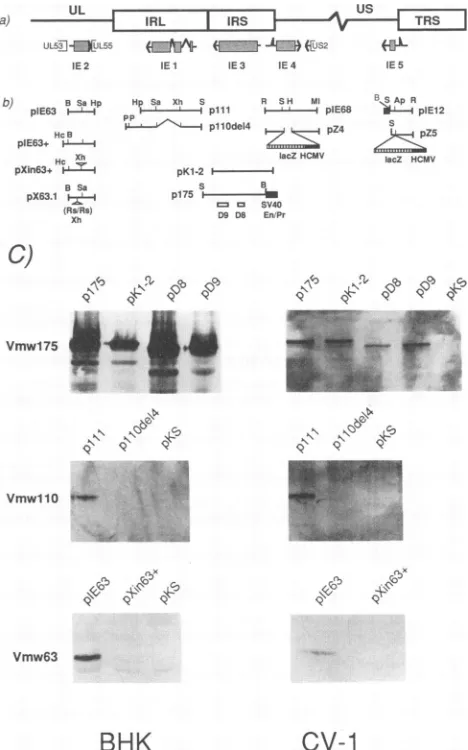
FIG. 1. Location ofthe HSV-1 IEgenesin the viral genome and the structures of IE
gene-containing
plasmids. (a)
Theright-hand
portion
oftheviral genome isshown,including
partof thelong
unique
region
(UL),
the internallong
repeat(IRL),
and the shortunique
region
(Us)
boundedby
internaland terminal shortrepeats(IRS
andTRS,
respectively).
The IEtranscripts (arrows)
and theircoding
regions (shaded
boxes)
andadditional openreading
frames thatmap closetoIE2(UL53
andUL55)
andIE 4(US2)
aredepicted
belowthe genomestructure.Notethatcopies
ofIE 1and3arepresentineach repeat(not shown). (b)
Structuresofthe IEgene-containing plasmids
and their mutant derivatives. Theorigins
and derivations of theseplasmids
aredescribedinMaterials and Methods. D8 andD9refertodeletions within
p175,
contained inplasmids pD8
andpD9,
respec-tively.
Ap, ApaLI;
B, BamHI; H,HindIII;
Hc, HincII;Hp, HpaI;
P, PstI; R, EcoRI; Rs, RsrII;S,SstI; Sa,Sall;
Xh,XhoI;
En/Pr,enhancer-promoter.
(c)
Immunoblot detection of theproducts
ofHSV-1 IE genes1,2,and 3.BHKand CV-1 cellsweretransfected withplasmids
carrying
theindividualIEgenesasindicated.After48h,thecellswere collectedby
scraping,
andtotal-celllysates
wereprepared
by boiling
in sodiumdodecyl
sulfate(SDS) gel loading
buffer(36).
Fractionsof the cell extracts were run onSDS-polyacrylamide gel electrophoresis
(PAGE),
andtheproteins
were electrotransferredontonitrocellulose membranes. The IE geneproducts
were detectedby
monoclonal antibodiesagainst
HSV-1 Vmwl75, VmwllO,andVmw63,obtained fromAdvancedBiotechnology,
withthe enhancedchemiluminescence kit fromAmersham.VOL.68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.316.550.95.470.2]pSV2neo
as aselectable markerto confer resistance togene-ticin and 2.5 ,ug of test
plasmid
DNAcontaining
HSV-1sequences. Sixteen hours after
transfection,
the medium wasreplaced,
and afterafurther 24 h ofincubation,
the cellsweresplit by
dilution factors of 1:4 and 1:50 and seeded onto100-mm
plates
in selection mediumcontaining
G418(400
,ug/ml).
Colonieswere counted 10to 14days
later.Generationof recombinant viruses.
(i)
Isolation ofD30HA3,
14HA3,
and14HRA3. RR1 cells(2
x105)
werecotransfectedwith
(i)
2,ug ofa1:1mixture ofintact andBamHI-SstI-cleaved
pIllH
and(ii)
2 jig ofD30EBA
or in1814genomic
DNA.After24
h,
the cotransfection mixturewasremoved,
and freshmedium
containing
5 mM HMBA was added to the cells tostimulate HSV-1 IE gene
expression
intheabsence oftransin-duction
by
VP16(39).
The cells werereplenished
with freshmedium
(without HMBA)
after an additional 24 h. Afterseveralmore
days
of incubationat37°C,
whendiscreteareasofCPE had
formed,
the cellswereharvested andfreeze-thawed,
and virus progeny in the
supernatant
wereplated
out atappropriate
dilutionsonto10-cmdishes ofE5
cellmonolayers
inthe presence of 5 mM HMBA. Thenext
day,
thecellswereoverlaidwithmedium
containing
0.8%agaroseinthe absenceofHMBAandincubated foranadditional 3to4
days
toallowtheformationof
plaques.
Recombinant virus which hadincor-porated
the IE 3 deletion mutation contained inpIllH
(marked by
thepUC19
HaeIIIDNAfragment)
were detectedandisolated
by
several rounds of in situplaque
hybridization
asdescribed
previously
(27), using
the587-bp
HaeIIIfragment
from
pUC19
as aprobe,
radiolabeledby
the method ofFeinberg
andVogelstein (14).
Thegenomic
structure ofvi-ruses was determined
by
Southernblotanalysis
of total DNAprepared
from 16-mm-well cell cultures infected withplaque
isolates,
picked
after each round ofplaque
hybridization.
In this manner, theincorporation
ofthepUC19
DNAfragment
into the correct location of both short repeats of the HSV-1
genomein
place
ofIE3 sequenceswasmonitoredtoallow theisolation and
purification
of recombinants derived fromD30EBA
andin1814,
which weredesignated
D30HA3
and14HA3,
respectively.
Markerrescueof the mutated VP16 genein 14HA3was
performed
by
cotransfection of RR1 cells with14HA3
genomic
DNAandpMC1,
which contains thewild-type
VP16 gene.
Larger
plaques
which grew in the absence ofHMBAwere
picked
andpurified,
andtheirgenomic
structurewas
analyzed
asdescribedabove for loss of the BamHI linkerinthe VP16 gene. Three suchisolatesallexhibited thecorrect
BamHI
restrictionprofile,
andalarge
preparation
made fromthe firstofthesewas
designated
14HRA3.(ii)
Isolation ofinl850A42.
BHK TK- cells(106)
wereco-transfected with 10
pLg
ofin1850DNA and 10 ,ug(total)
ofanequal
mixture of intact and BamHI-cleavedpUL42A
DNA(27).
Progeny
viruseswereplated
ontoV9 cellmonolayers,
and theresulting plaques
were screened for the presence of the280-bp fragment
ofplasmid
vectorDNAmarking
thedeletionofthe UL42 gene, as described above. Stockswere prepared
from a
plaque-purified
isolatewhich couldonly
formplaques
on V9 cells andwhich exhibited thecorrectrestriction digest
profiles corresponding
to the UL42 gene deletion and theVP16 gene insertion mutation
(26).
This virus was namedinl850A42.
(iii)
IsolationofA3vhsZand 14HA3vhsZ.Disruption
of the vhscoding
region
ofD30EBA
and 14HA3 was achieved by cotransfectionof RR1 cells withpUIA1-lacZ
DNA,
linearizedby digestion
withXhoI
and theappropriate
viral genomic DNA. Recombinantvirus in theresulting
progeny werede-tected
by
the appearance of blueplaques
afterplating
onmonolayers
ofE5
cells andstaining
with X-Gal(5-bromo-4-chloro-3-indolyl-,-D-galactopyranoside) as
previously
de-scribed (29). The genomicstructuresof virus isolates
carrying
the intended insertion mutation in the vhs genes ofD30EBA
and 14HA3 were confirmed
by
Southern blotanalysis,
andthese isolates were designated A3vhsZ and
14HA3vhsZ,
re-spectively.
Yield of progeny virus.E5orV9 cells
(as
appropriate)
wereseededat adensityof 3 x
105
cellsper 35-mm dish. Afteranassumeddoubling
by
24h,the cellswereinfected with virusatan MOI of 1 or 0.01 PFU per cell. Unabsorbed virus was
removed by washing 1 h
later,
and themonolayers
wereincubated foranadditional 19 hat37°C.Infected cultureswere
maintained continuously for the 24-h
period
in either thepresence or absence of 5 mM HMBA. The cellswere then
scraped into the medium anddisrupted
by freeze-thawing,
andvirus in theresultingsupernatantswastitrated
by plaque
assayonpermissive cells in the presence of 5 mM HBMA and 10%
pooledhuman serum.
Cell
viability
assays. The effect of virus infection on cellsurvival and growth,as measuredby thenumber of adherent
cells at 3 days postinfection, was determined as follows.
Human fibroblastswereseededat adensityof 5x 104cells per
16-mm well. The cellswereinfected in
triplicate
16 hlaterwithvirusatMOIs of0.1, 0.25,0.5, 1.0, 5.0,7.5,10,and 25 PFU per
cell. Aftera1-habsorptionperiod, the cellswerewashed and
replenished with fresh medium. At 3
days postinfection,
loosecells and debris were removed
by washing
withphosphate-buffered saline(PBS), and the adherent cellswereharvested
by
trypsinization and counted witha Coultercounter.The titers
of the virus stocks used in this cell viabilityexperiment were
also determined intermsofinfectious genomeunits(IGU)per
ml, as follows. Human fibroblasts (106) were infected with
either a standard volume of virus stock(25
,ul)
or a constantMOI (e.g., 5 PFU per cell). The cellswerewashedthreetimes
aftera 1-habsorptionperiodand thenincubated for 1 hmore
at37°C. The cellswerethenharvested inPBS,and cell nuclei
were isolated from outer cell membranes and
cytoplasmic
components following Nonidet P-40lysis. Nuclear DNAwas
prepared, and 2.5-,ug aliquots were digested with BamHI
alongside uninfected human fibroblast DNA which had been
spiked with 3.3to825pg ofpBam 1,correspondingto1 to250
HSV-1 genomecopies per cell in 2.5 ,ug of human fibroblast
DNA. The digested DNAs were separated by agarose gel
electrophoresis and analyzed by Southern blotting with
BamHI-lasaprobe. An example of this is shown in Fig. 5c. In
some cases, the blot was reprobed with a human DNA
sequence (human 1-galactosidase cDNA) to normalize the
totalamountof DNA loadedand transferred in each lane,but
itwasusually found tobe quite constant. Titers in PFU had
been determined in the presence of HMBA, which has been
shown previously by McFarlane et al. to help overcome the
replication defect in in1814, so that in1814 can be titrated
almost as efficientlyaswild-type virus (39). The ratios of IGU
to PFU in one experiment were as follows: D30HA3, 5.4;
14HA3,20.3; and 14HRA3, 3.2. Although the difference in the IGU/PFU ratio between D30HA3 and 14HA3 was fourfold in thisexperiment,thedifference was only twofold in an indepen-dent experiment. Some degree of variability in virus titer is inevitable, in part because of differences in the growth state
and passage number of permissive cell lines. Since MOI is a
verycritical parameterfor studying cytotoxicity, we have tried
tominimize the effects of variation in virus titer by comparing
the effects of different viruses using titer values that had been
determined at the same time. In some of the experiments
described in this paper, we have compared the toxicity of
different virusesusing PFU rather than IGU as a measure of
on November 9, 2019 by guest
http://jvi.asm.org/
CYTOTOXICITY OF REPLICATION-DEFECTIVE HSV-1 MUTANTS 6351 virus titer, but we bear in mind that this places more stringent
conditions on the VP16 mutants because of at least a two- to fourfold impairment in plaquing efficiency.
Cell viability measurements were also determined by trypan blue exclusion, as follows. Human primary fibroblasts were
seeded onto 24-well plates at a density of 5x 104cells per well
and infected in triplicate the next day with mutant virus strains
at an
MOI
of 10 PFU per cell. Viruses that were inactivated byUV irradiation were exposed to a constant UV source (1,200
[LW/cm2)
for 5min.
We had previously determined that thisUV dose would reduce
,-galactosidase
expression in CgalA3by more than 30-fold and reduce virus titer by more than 100,000-fold (29). One hour after infection, the virus inoculum was removed and replaced with fresh medium containing full
serum, and the cells were incubated for 3 days at
37°C.
Thecells were then harvested by trypinization and stained with 0.5% trypan blue in PBS. The number of viable cells which excluded trypan blue were counted with a hemacytometer.
Superinfection rescue procedure. The ability of different mutant virus strains to persist in infected primary fibroblasts and be "rescued" by a complementing mutant strain 11 days later was measured by a semiquantitative superinfection rescue procedure similar to that described previously (29). Briefly, human fibroblasts in 10% fetal calf serum were seeded onto a
24-well plate at a density of 6x
104
cells per 16-mm well. Afterhaving reached confluency (about 2.5x
105
cells per well) in 3days, cultures on duplicate plates were infected in duplicate
with each mutant virus strain, at an
MOI
ranging from 0.005 to2.5 PFU per cell. After 1 h, the cells were washed three times
and either maintained in 0.2% serum for 11 days (with fresh
medium added every 2 to 4 days) or processed directly for the superinfection rescue. The rescue was performed by
superin-fecting cells with the
ICP8
mutant d301 at anMOI
of 5 PFUper cell in the presence of 5 mM HMBA for 4 h. The superinfected cells were then harvested by trypsinization, and appropriate dilutions were plated onto 10-cm plates preseeded
with
E5
cells (to titerIE 3 deletion mutants) or V9 cells (totiter UL42 deletion mutants), in 5 ml of medium containing 5 mM HMBA and 10% pooled human serum. After 16 h, 5 ml of additional medium was added, and the cells were incubated at
37°C
for 3 to 4 days until the resulting plaques could becounted. In control experiments, the highest number of plaques for a replication-defective mutant obtained on day 0 without superinfection was 3.4% of the number of plaques obtained with superinfection, indicating that the vast majority
of plaques which do arise do so through complementationwith
the superinfecting virus and not because of persistence of viable intact viral particles (26). By day 11, no plaques were
observed without superinfection. Also, no more than one or
two plaques have been detected following superinfection of mock-infected cells; these few plaques probably represent
occasional rescue of the
ICP8
deletion in d301 duringprepa-ration of virus stocks. Previous analysis has shown that the
plaques which arise from the superinfection rescue procedure
represent a mixture of the initial virus persisting in the cells
and the superinfecting virus (29).
RESULTS
Inhibition of colony formation with transfected HSV-1
se-quences. To determine whether we could identify potential
virus genes responsible for cytopathogenicity, we cotransfected
BHKTK- cells and CV-1 cells with plasmids containing each
of the candidate viral genes expressed by an IE 3 deletion
mutant (Fig. 1) together with pSV2neo in a 10:1 ratio (wt/wt).
The cells were then split at various dilutions and placed under
selection for neomycin resistance, and the number of colonies obtained was counted 10 to 14 days later. For the sake of completeness, we also examined the effect of IE gene 3 in these experiments. The plasmids containing the IE genes were chosen or constructed so that the presence of additional open
reading frames or the presence of extraneous sequenceswould
be very limited. However, the frequency of colony formation could be influenced by promoter competition, the presence of
"poison"
sequences, or additional open reading frames in thecotransfected test plasmids. To control for these
possibilities,
we also examined the frequency of colony formation with
controlplasmids containing mutations in the coding regions of
each of the respective IE genes. To ensure that the plasmids
encoding IE genes 1, 2, and 3 expressed the expected IE
proteins in transfected cells, we also performed Western
(immunoblot) analysis with specific monoclonal antibodies
raised against these IE proteins (Fig.1C).In additionalcontrol
experiments with luciferase as a reporter gene, we have
determined that the promoters of IE genes 1, 2, 3, and 4/5were
highly active in transfected BHK TK- and CV-1 cells (26).
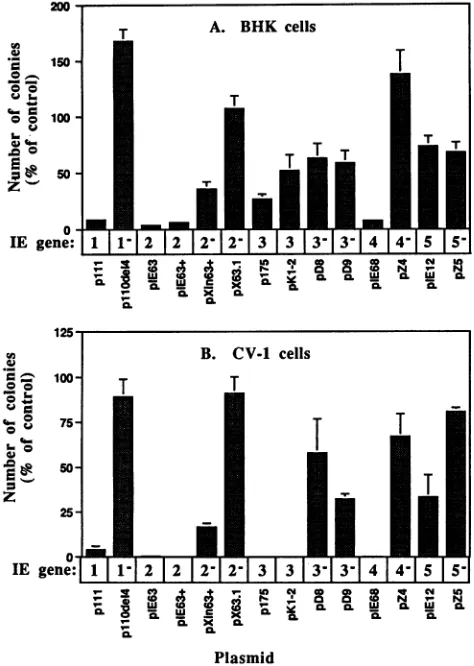
As shown in Fig. 2, plasmids encoding IE 1 (plll), IE 2
(pIE63
and pIE63+), and IE 4 (pIE68) all strongly inhibitedthe frequency of colony formation in both BHK TK- andCV-1
cells, in comparison to the number of coloniesobtained by the
control cotransfection with pBluescript KS. Cotransfection
with control plasmids bearing lesions in thecoding portions of
IE genes 1 (pllOdel4), IE 2 (pXin63+ and pX63.1),and IE 4
(pZ4) reduced the inhibitory effects observed with the intact IE
genes. The inhibitory effect of IE 2 was more completely
abolished by pX63.1, which has a 28-bp deletion in the IE 2
coding region, than by pXin63+, which has a singleXhol linker
insertion mutation. Unexpectedly, both plasmids encoding IE
3 (p175 and pKl-2) strongly inhibited colony formation in
CV-1 cells but not in BHK TK- cells. Theinhibition of colony
formation in CV-1 cells cotransfected with IE 3sequences was
more effectively abolished by pD8, which encodes an IE 3
deletion product retaining transactivation activity butdefective
for repression (48), than by pD9, which encodes an IE 3
product with full repression activity but only 4% of the
transactivation activity of the wild-typeIE 3 protein (48). The
plasmid encoding IE 5 (pIE12) did not cause inhibition of
colony formation in BHK cells, although amoderate inhibition
of transformation frequency (50%) was observed inCV-1 cells.
Inhibition of colony formation following cotransfection with
plasmids encoding IE genes 1, 2, and 4 has alsobeenobserved
with a different selectable markerconstruct, in which theneo
gene was driven by the human HPRT promoter instead of the
SV40 promoter (data not shown). We have also determined
that neither plasmid pKHF, which encodes thelarge subunit of
ribonucleotide reductase (ICP6), which is alsoexpressed asan
IE gene, nor pKHFA, which harbors adeletionwithin the ICP6
coding region, causes inhibition of colony formation (26).
Finally, since the products ofIE genes 1, 2, and 3areknownto
modulate transcriptional activity, it was possible that they
might directly affect the level ofneomycinphosphotransferase
produced by the selectable markerplasmid, therebyproviding
a trivial explanation for their effect onthefrequencyofcolony
formation. We therefore measured the effect of
cotransfecting
IE genes 1, 2, and 3 with the neo gene on theresultinglevel of
neomycin phosphotransferase activity and determined that
while plasmids bearing IE 2 or IE 3 had little effect,IE 1
caused a moderate (twofold) stimulation in activity (data not
shown).
Isolation of mutant viruses. (i)IE 3-VP16 double mutant.
To reduce the expression of HSV-1IE genesduringa
nonpro-ductive infection, we have constructed double mutant viruses
VOL. 68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
(U
0
Ist
IE gene: 1 1-1 2
1
212-1
3 | 31
3-1 3-1 41
4-1 51
5-|o-
_X N 0.v-0 0
4(
0. " CL06
CL~~~~0
ffi $ CL 3R^Ssa f S ^
Plasmid
FIG. 2. Effect of HSV-1 IE gene-carrying plasmids on the
fre-quencyofcolonyformation in cotransfected BHKTK-andCV-1 cells.
Approximately 105cellsper35-mm wellwerecotransfected in
tripli-cate with 0.25 ,ugofpSV2neoand 2.5 ,ugof the indicated testplasmid.
Cellswerewashed after24 handsplitthefollowing dayat 1:4 and 1:50 dilutions into selectionmediumcontaining G418(400 ,ug/ml). Colo-nieswerecounted 10 to 14daysafter transfection. Anaverage of 933
and 2,916 colonies per 0.25 ,ug of pSV2neo were obtained in the control transfections(with2.5 ,ugofpBluescript KS)of BHKTK-and CV-1 cells, respectively. No colonies developed in the absence of pSV2neo.Errorbars indicate standard deviations.
withamutation inanessentialgene toprevent lytic replication
and with theinsertionmutation in the VP16genedescribedby
Preston and colleagues (2) to abolish the ability of VP16 to transinduce IE gene expression. The double mutant 14HA3
wasgenerated by introducingadeletioninto bothcopiesofthe
IE 3gene of the VP16 mutant in1814. As shown in Fig. 3c,
plasmid pIllH,usedtogeneratethe IE3deletion,containsa
DNAfragment from pUC19 (a 587-bpHaeIIIfragment
con-tainingthe 5' portionof the ampicillinresistancegene) atthe
site ofthedeletiontoserve as atagto aid theidentification of viral recombinants by plaque hybridization. This plasmid is otherwise identicaltopdelIll,whichwasused togeneratethe IE 3 deletion mutant D30EBA (49). Since initially we were
unsure how well an IE3-VP16 mutantwould grow, we also isolated a derivative ofD30EBA from plasmid pIllH,
desig-natedD30HA3,toprovide apositivecontrol for identification
ofrecombinantsby plaque hybridizationwith thepUC19DNA
fragment and to ensurethatrecombinants could beidentified
under conditionsthatdid not requireselectionagainstthe IE
3 gene. Southern blot analysis of the genomic structure of
14HA3 isolates is shown in Fig. 3d (leftmost panel). The
presence of the 0.52-kb band in BamHI-EcoRI-digested
14HA3 DNA, which has been probedwith a DNAfragment
from the 5' end of IE3,indicates that theanticipateddeletion
ofthe IE 3 gene has takenplace, althoughoverexposureof this
blotdid reveal the minor presence ofa1.8-kbband,indicative
of thewild-type IE3BamHIfragment. The viral stock from
which this DNAwas prepared was also able to form a few
plaquesonVerocells,whicharenoncomplementaryfor IE 3 mutants. Therefore, this 14HA3 virus isolate was further plaque purified until no such IE 3-independent plaques ap-pearedinan inoculumcontainingat least 105PFU.
Toensurethatphenotypicdifferences between 14HA3 and
anIE3 deletionmutant wereduetothe mutation in the VP16
gene and not to a second mutation, the VP16 lesion was
repaired byrecombining14HA3 DNA with
plasmid
pMC1 (7),which contains an intact VP16 gene. Rescued virus could be
obtained readily, since it was able to grow much more
effi-ciently than the 14HA3 double mutant in the absence of
HMBA.The genotypes of three such isolates wereexamined
by Southern blot
analysis
for the presence ofan intact 8-kbBamHI-f fragmentin
place
of the 3- and 5-kb bands associatedwiththe BamHI linkerinsertion present in in1814 and 14HA3
(Fig. 3d,secondpanel),andastockpreparedfromoneof these purifiedisolateswas
designated
14HRA3.(ii) UL42-VP16 doublemutant.Toexamine the effect of the
VP16 mutation on virus-induced
cytotoxicity
in theback-groundofa mutantblocked for
replication
at astepin thelytic cycle different from that ofthe IE 3 deletion mutant,wealsointroduced a deletion into an essential early geneofa VP16
mutant(seeMaterialsand
Methods).
Theearlygenewechose to mutate was UL42, which encodes a 65-kDa accessoryfunction for the HSV-1 DNA polymerase
(18),
essential forvirus replication
(27,
37). The purified UL42-VP16 doublemutantisolatewas designated inl850A42.
(iii) IE 3-vhs double mutant and IE 3-vhs-VP16 triple mutant.To assessthe role ofthe vhs function in the cytotox-icity of replication-defective mutants, we introduced an
HCMV-lacZcassetteinto the UL41coding regionof D30EBA
and14HA3(seeMaterials andMethods).Thegenomic struc-tures of the recombinants, designated A3vhsZ and 14HA3 vhsZ, respectively, were examined by Southern blot analysis (Fig. 3d). DigestionofA3vhsZand14HA3vhsZ DNA with SstI
gave rise to a 3.2-kb band detectable with a probe from a
BamHI-SstI subfragment ofBamHI-i, which is the size
ex-pectedfromcorrectinsertionofthelacZgene,instead ofthe
2.2-kbfragmentfromanintact UL41 5'region (Fig.3b, andd).
When this blot was reprobed with lacZ, 3.2- and 3.5-kb
fragments were detected, indicating correct insertion of the
lacZ gene into theUL41 codingregion. We also checked for
the presence of the BamHI linker in the VP16 gene of 14HA3vhsZ (but not
A3vhsZ),
indicated by cleavage of the 8-kbBamHI-f fragmentinto 3-kband 5-kb subfragments (Fig.3d).
Smibert and Smiley have shown that insertion of anICP6-lacZcassetteinto theBamHIsite of the UL41 gene of an
HSV-1 KOS strain resulted in inactivation of the vhs function
(61). Since their mutation disrupted the UL41 open reading
frame afterresidue342, and in ours it is disrupted after residue
237
(see
Fig. 3b),it isprobable that insertion of the lacZ geneat the NruIsite will also result ininactivationofthe vhs
func-tion.
Growth characteristics of mutant viruses. The insertion
mutation inin1814preventsVP16 fromforming the
multipro-tein complex that normally transactivates transcription of
HSV-1 IE genes uponinfection, thereby impairing the ability
on November 9, 2019 by guest
http://jvi.asm.org/
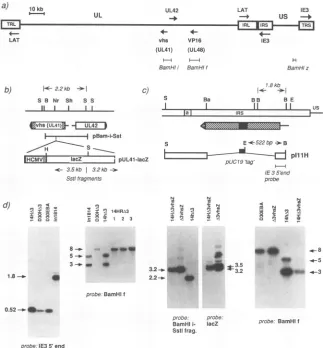
[image:6.612.59.296.74.407.2]CYTOTOXICITY OF REPLICATION-DEFECTIVE HSV-1 MUTANTS 6353 a)
LAT 10 kb
UL UL42
vhs VP1 6
(UL41) (UL48)
Bamf s Ift
BamHti BamHIf
b) 1<-2.2kb --I
S B Nr Sh S S
11 I I I
C)
S Ba
LAT IE3
-*+ us -+
IE3
H
BamHlz 7.8kb
1- -1j
BB BE
II I I i,
4Evhs(iJLgq MM2
i j pBam-i-Sst
--- S E*522bp.-B
| t
--iig
lacZ L pUL41-lacZ t i a, i pIllHc 3.5kb 3.2kb
-Sstl fragments probe
8--s4.0
5-4-4_ __
1.8-..
> N > N
" 0 ±7 C)~ I ¶2 0r
.. 3s 2
2.2-..
probe-BamHI
i-Sstifrag.
probe.
acZ
[image:7.612.154.477.75.423.2]probe. I1E3 5' end
FIG. 3. Construction and analysis of genetic modificationstothe HSV-1genome.(a)Mapof the HSV-1genome,consisting of the unique long
(UL)andunique short (Us)regions, bound by terminal and internal long (TRL and IRL) and short (TRS and IRS) repeats. Shownarethe
locationsofgenesUL41 (vhs), UL42, UL48 (VP16), IE 1,IE3, and theLATtranscripts, and the locationsof BamHI fragments i,f, andzused
intheconstructionand/oranalysis ofmutants.(b)Construction of vhsmutants.AnHCMV-lacZcassettewasinsertedinto the NruI site of UL41, containedonplasmid pBam-i-Sst, creating pUL41-lacZ. The sizes of novel and wild-type SstI fragments detected by BamHI-iorlacZprobesare
given. (c) Deletion of the IE 3gene.Theposition of the IRScopyof theIE3transcript (line) and coding region (hatched box) isshown,asis the
extentoftheIE3deletionwithinplasmid pIllH. pIllHwasderived from pdelIll (49) by insertion ofa522-bp HaeII fragment from pUC19,to
serveas atagforthe detection of recombinantsbyinsitu plaquehybridization. OnanEcoRI-BamHI digest of unpurified recombinant viral DNAs,
theindicatedIE35' probewilldetecta 1.8-kbwild-type fragmentor a522-bp novel virus fragment. (d) Southern blot analysis ofrecombinant viruses. Firstpanel: 14HA3, D30HA3, D30EBA, and in1814 viral genomic DNAsweredigested with EcoRI and BamHI and probed with theIE 35'-end fragmentshown inpanelc.Thepresenceof the0.52-kbbandin14HA3,D30HA3, and D30EBAindicatesthatdeletion of theIE3gene
hasoccurred.Secondpanel:ViralDNAs fromin1814,D30HA3, 14HA3,and threeisolates of 14HA3 which had beenrescuedwithanintact VP16 gene(14HRA3)weredigestedwith BamHI andprobedwith theBamHI-ffragment.Thepresence of the BamHIlinkerinthe VP16generesults incleavageof the8-kbBamHI-f fragmentinto5-kband3-kb subfragments. Thirdpanel: 14hA3vhsZ, A3vhsZ,and 14HA3 DNAsweredigested with SstI.Thepresenceof theHCMV-lacZcassetteisindicatedbyanovel3.2-kbbandinplaceofthewild-type2.2-kbband whenprobedwith theBamHI-i-SstIfragmentshown inpanelb.Reprobingwith lacZgaverisetoanadditional 3.5-kb bandinthe vhsmutants.Fourthpanel:The indicatedviral DNAsweredigestedwithBamHI andprobedwithBamHI-ftoconfirm thepresenceofthe VP16 mutation in14HA3vhsZ,indicated bythe5- and 3-kbsubfragmentsofBamHI-f.Restriction sites: S,SstI; B, BamHI; Sh, SphI; Ba, BalI; E, EcoRI; Nr, NnrI.
of in1814toinitiate infectionatlowMOI(2).In thepresence
ofHMBA,however, in1814 is abletoinitiateinfection almost
as efficientlyaswild-type HSV-1 (39). To determine whether
thenewVP16mutantvirusesexhibit asimilar phenotype, we
measured theyieldofprogenyvirusafter24h ofinfectionof
permissive cells at a low MOI (0.01 PFU per cell), in the
presenceandabsence of HMBA. Asshown in Table 1,viruses with the VP16mutation(14HA3,14HA3vhsZ,andin1850A42)
grew very poorly in the absence of HMBA, but their yields
were improved by30- to 60-fold in itspresence. HMBA had
little effect on the yield of viruses without the VP16 lesion
(D30HA3, 14HRA3, A3vhsZ, andCgalA42). Viruseswith the
UL41 mutation were further impaired for growth, since the
yield of A3vhsZ was 10-fold below that of D30HA3, and
although in this experiment the yield of 14HA3vhsZ with
HMBA was only 2-fold below that of 14HA3, the titers of stocks of14HA3vhsZwere usually5- to 10-fold below thatof 14HA3 (26). These observations are consistent with those of other studiesreporting that large insertion or deletion muta-tions in the vhsgenecause a5-to10-folddropin viral titer(15,
61). Togetherwith the confirmation ofgenomic structure by
Southern analysis,we conclude from these observations that d) ) ,n< <
tx0
- I 14HR.A3
C :1 1 2 3
.l
-C C to 1.V.m>
J=
9 A 4 x
0 < - 11-1
0.52-.*11..M
probe:BamHlf
*e **O-m
.4-8 -5 -4-3
probe. BamHIf
VOL.68, 1994
uscIC
on November 9, 2019 by guest
http://jvi.asm.org/
TABLE 1. Yield ofprogeny
Yieldb Ratio, YieldtoD30HA3orCgalA42d
-HMBA +HMBA +HMBA/-HMBAC -HMBA +HMBA
D30HA3 IE 3- 1.0x 106 0.9x 106 0.9 1.0 1.0
D30EBA IE 3- 1.1 x 106 1.2x 106 1.1 1.1 1.3
14HA3 IE 3-,VP16in 3.8 x 103 2.3x 105 60.5 0.004 0.25
14HRA3 IE3- 1.5x 106 2.0 x 106 1.2 1.5 2.0
A3vhsZ IE3-,vhs-lacZ 1.0X 105 1.2X 105 1.2 0.1 0.13
14HA3vhsZ IE3-,VP16'n,vhs-lacZ 2.8 x 103 1.0 X 105 35.7 0.003 0.11
CgalA42 UL42-, BamHI-z-lacZ 1.2 x106 1.5 x 106 1.3 1.0 1.0
inl850A42 UL42-,tk-lacZ,VP16in 2.5 x 103 1.0 X 105 40.0 0.002 0.067
aGenotype indicateswhetherviruseshavedeletions inIE3 orUL42,thepresenceof the insertion mutation in theVP16genedescribedbyAceetal.(2),and the presenceof a lacZ geneinsertedinto theBamHI-zregionor alacZinsertional mutationinthe vhsortkgene.
bPermissivecells thatcomplementgrowth ofmutants(E5 forIE3mutants,V9 forUL42mutants)wereinfectedat anMOIof 0.01 PFU per cell for 20 h ineither theabsenceorcontinuouspresence of 5 mMHMBA,asindicated.Thecellswerethenscrapedinto the medium anddisruptedbyfreeze-thawing,and the titers ofvirus intheresultingsupernatants were determined on theappropriatepermissivecells in the presenceof 5mM HMBA. Valuesrepresenttheyieldof progenydetermined fromduplicate infected cultures and arerepresentativeofrepeatedexperiments.
cValuesrepresent theyieldofvirusdeterminedinthepresenceofHMBAdividedbytheyieldof virusdeterminedinthe absence ofHMBA.
dValues represent theyield of eachIE 3 mutantdividedbytheyieldof
D30HA3
ortheyieldofCgalA42
and theUL42-VP16mutantin1850A42
dividedbytheyield ofCgalA42, determinedin the presence and absenceofHMBA.wehavesuccessfullygenerated virus strains with defects in the
virion components vhs and/or VP16 whicharealsocompletely
defective forreplication.
Comparison ofcellviabilityfollowinginfection withmutant
virus strains. Wehave examined the effect of infection with
the different mutant virus strainsonthemorphology of human
primary fibroblast cultures. Confluent monolayers of
hu-man fibroblasts were either mock infected or infected with
D30HA3, 14HA3, A3vhsZ, 14HA3vhsZ, CgalA42, orinl850A
42 at an MOI of 2.5 PFU per cell, and the cultures were
examineddailyfor the appearanceof CPE(Fig.4).The results
show that cultures infected with 14HA3 (Fig. 4c) remained
almostashealthyasthemock-infectedcontrol(Fig. 4a),while
those infected with D30HA3 (Fig. 4b) exhibited increasingly
severe CPE withincreased timepostinfection. Therefore,as a
result of the VP16lesion in14HA3,thecytopathogenicity of an
IE 3 deletion mutant has been significantly reduced. In
con-trast, A3vhsZ (Fig. 4d),which has a defective vhs function in
addition to the IE 3 deletion, did not appear to be any less
cytopathogenic thanD30HA3.Cultures infected with the triple
mutant 14HA3vhsZ (Fig. 4e), which has a VP16 lesion as well
asthe IE 3 and UL41mutations, also remained healthy for the
duration of the experiment. However, inl850A42 (Fig. 4g),
which has aVP16lesion and isdeleted for the essential early
geneUL42, didcausegeneralized CPE in infected cultures at thisMOI,asdidCgalA42(Fig. 4f), which is deleted forUL42
buthasanintact VP16 gene.Inthese experiments, the cultures
wereinfected with the same number of PFU, even though the
plaque-forming ability of the VP16 mutant strains (determined
in the presence ofHMBA) was likely to represent a two- to
fourfold underestimate of the relative number of infectious viral genomes (see Materials and Methods) and thereby place
amorestringent test on the ability of the VP16 mutants not to
causeCPE.Nevertheless, these results show that as a result of the VP16 mutation, the cytopathogenicity of an IE 3 deletion
mutanthas been sufficiently reduced so that an infected culture
does not develop extensive CPE when infected at anMOI of
2.5, whereascultures infected with mutantscarrying mutations
in IE 3 aloneorUL42alone,an IE 3-vhs double mutant,ora
UL42-VP16 doublemutantall developCPEatthisMOI. We
note,however, thatcells infected with14HA3and14HA3vhsZ
did exhibitsomegranularity,although this experimentwas not
designed to quantify the effects of viral infection on cell
division and/or survival.
We therefore set out to quantify the effect of the VP16
lesion on the toxicity of the IE 3 and UL42
replication-defectivemutantsas afunction ofMOI. In thisexperiment,we
used IGU as the measure of virus titer to compensate for
differences in theefficiencyofplaquingbetweenVP16mutants
andVP16-intact virus. The determination of IGU titer for the
stocks of IE 3mutantsused inthisexperimentis shown inFig.
Sc and Table 2.To assaytoxicity,wecountedcell numberat3
days postinfection, to allow sufficient time forvirus-induced
cytotoxicity to occur. We used a mitotic fibroblast cell line
because CPE occurs faster and there is less ambiguity in
distinguishing livingcells than ifwehad usednondividingcells.
Cell numbertherefore reflects both the survival andreplication
of cellsoverthe3-dayperiod after the infection. Subconfluent
monolayersof humanfibroblastswereinfectedwithD30HA3,
14HA3, 14RHA3, CgalA42, and inl850A42 over a range of
MOIs from0.1to25 PFU percell. Thetiters of the virus stocks
used inthisexperimentweredeterminedatthe same time in
termsof IGU per mltocompensatefor the reducedplaquing
efficiency of the VP16 mutants(asshownfor the IE 3 mutants
in Fig. 5c and Table 2). As shown in Fig. 5a, the curve
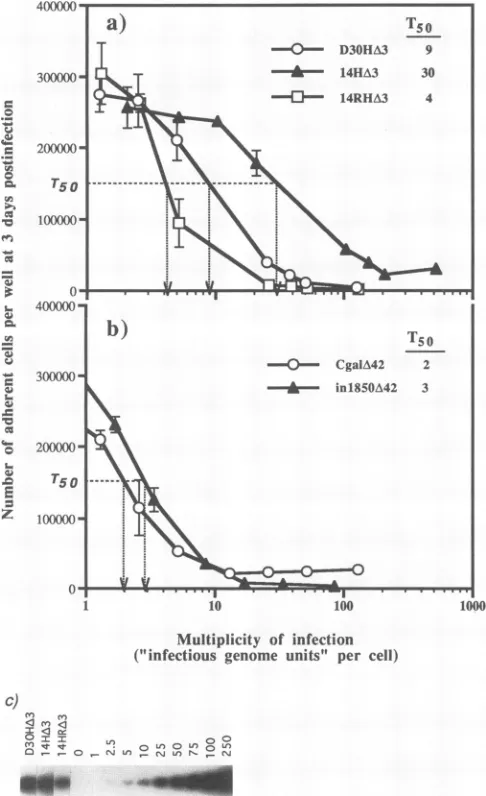
produced from the number ofadherent cells at 3 days
postin-fection with increasing amounts of 14HA3 is shifted
signifi-cantlytothe right of the curves corresponding toD30HA3and 14HRA3, indicatingthat14HA3is less toxic over a wide range
ofMOIs.Inthecaseof14HA3, the curve was steepest after an
MOI of 10 IGU per cell,which indicates the point at which
virustoxicity is measurably affecting cell growth and survival,
whereas for infections with 14HRA3 and D30HA3, significant
loss in cell number occurred after an MOI of approximately 3
FIG. 4. Infection ofhumanprimaryfibroblastswithHSV-1 mutants. Confluent monolayers of human fibroblasts were either mock infected (a) orinfected withD30HA3(b),
14HA3
(c),A3vhsZ(d), 14HA3vhsZ(e),CgalA42(f), orin1850A42(g) at anMOIof2.5PFU per cell. Monolayers wereobserveddailyfortheappearanceofCPE andphotographed.on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.61.553.86.203.2]~~~~~~~
_2 day
3das
4 ay
6355
1
day
on November 9, 2019 by guest
http://jvi.asm.org/
-u!
44-1
0
lL
(-0.
LC.
la
Cu
en
'a-co
w
S.
a6
Multiplicity of infection ("infectious genome units" per cell)
c)
re
LIC]c
et US)0L 0LO
FIG. 5. Effect of the VP16 mutationon cell survival and growth
after infection with mutant viruses. Subconfluentmonolayersof
hu-manfibroblasts (5 x 104per16-mmwell)were infectedin triplicate
with(a)D30HA3,14HA3,or14HRA3or(b) CgalA42orin1850A42 at increasingMOIs.The MOI is shownhere in terms ofIGU per cell.
TheIGU titersof virus stocksweredeterminedasdescribed forpanel c.Theaverage number of adherentcellsremainingat 3days postin-fection,determined with aCoultercounter,are plotted againstMOI (errorbarsindicate standarddeviations).T50is definedasthe MOI at
which the number of adherent cells was reduced by 50% of the
maximumnumberof cellson acontrol uninfectedplate(IGUpercell). (c)Southernblotshowingtheamount of viral DNAdetected inhuman
fibroblasts infected with 25-pAl aliquots of D30HA3, 14HA3, and
14HRA3. Theinfected-cell DNAs and cellular DNAspikedwith the equivalent of 0 to 250 copiesofpBaml plasmidDNAper cellwere digestedwith BamHI and probed with radiolabeled BamHI-l
frag-ment. Thecalculationof IGUfromdensitometricanalysisof thisblot
is shown inTable 2.
IGU per cell. Takingthe maximum number ofcells from a
controluninfected wellatday3 tobe300,000,the titer(inIGU
percell)atwhichcellnumberwasdecreasedby50%(T50)was
30 for 14HA3, 9for D30HA3, and4for 14HRA3. Therefore,
using theT50 valuesdeterminedfrom this assayas ameasure
[image:10.612.63.306.76.474.2]of virus toxicity, the IE 3-VP16 double mutant 14HA3 is
TABLE 2. Infectious genome titer
Virus copies/celraNo. of IGUb PFUc IGU/PFU ratio
D30HA3 45.3 1.8 x 109 3.5 x108 5.1
14HA3 32.3 1.3 x109 6.4 x 107 20.3
14HRA3 20.0 8.0 x 108 2.5 x 108 3.2
aThe number of viral genome copies percell was determined following infectionof106 TO-119 fibroblastswith 25 ,ul ofeach virus stock and from densitometry of the Southern blot showninFig. 5c,asdescribed in Materials and Methods.
b The titer of virus in infectious genome units (IGU) per milliliter was calculatedfrom thecopy numberof viralgenomespercell(N)multipliedbythe total numberof cells(106)and dividedbythevolume ofthevirus inoculum(V, inmilliliters)asfollows:(Nx 106)/K
cThetiter ofvirus in PFU per ml wasdetermined from thesamevirus stocks atthe same timefollowinginfectionofE5cells in thepresenceofHMBA.
betweenthree- and sevenfold less toxic than either of the IE 3
mutantswhich haveanintact VP16 gene. Since intwoseparate
determinations 14HA3 had a two- to fourfold-higher IGU/
PFUratio thanD30HA3,itfollows that if PFUwasusedasthe
measure of virus titer in this cell-killing experiment, the
reductionintoxicityasaresult of the VP16mutationinthe IE
3 mutantbackgroundwould be lesspronouncedor not
appar-ent.
Incontrast,cell survivalfollowing infection with inl850A42
was not markedly improved over that after infection with CgalA42 (Fig. 5b). A steep reduction in cell number was
apparentat anMOI of 2 IGU per cell for both UL42mutants.
The titers atwhich the maximum cell number (300,000) was
reducedby50%were2IGU percell forCgalA42and 3 IGU
per cell for inl850A42. Therefore, the VP16 lesion in
in1850A42appearedtohave little benefitover aUL42 deletion
alone in reducing viral toxicityin this assay. In addition, the
UL42mutants were moretoxic than the IE 3deletion mutants, irrespectiveof the VP16 lesion. However, these results donot
rule out the possibility that inl850A42 is less toxic than
CgalA42 at much lower MOIs, as examined in the following experiment.
Rescue of virus from persistently infected cells. We have
previouslyusedanin vitrolatency system to show that at a low
MOI,asmall percentage of cells infected withanIE3deletion
mutantmaintain the virus inabiologically retrievable form for
at least2 weeks (29). However, it was notpossible to
signifi-cantlyincrease the number of cells which could maintain virus by increasingthe MOI,presumably because of increased viral toxicityathigherMOIs. We reasoned that if any of the mutant virus strains described in this study have reduced toxicity, cells
should be abletosustaininfection at a higher MOI and more
virus should be available for rescue a week or later after
infection. To test this hypothesis, we infected human
fibro-blasts with D30HA3, 14HA3, CgalA42, inl850A42, A3vhsZ,
and 14H3vhsA3 at increasing MOIs. We used PFU as the
measure of virus titer inthis experiment since the method of
titer determination will not ultimately affect the maximum
number of virus that can be rescued over a wide range of
MOIs. The amountofvirus that could be recovered
immedi-ately(1 hpostadsorption) or 11 days after infection by rescue
with a complementing HSV-1 strain deleted for ICP8, d301
(19),is shown inFig.6. Superinfection with d301 provides the
IE 1 geneproduct, VmwllO,which is important for
reactiva-tion oflatent virus(34, 57),aswellasthe IE 3 and UL42 gene
products, which are needed for complementation of viruses
with mutations in these genes. Since the plaques from the
rescueprocedure have arisen from infected human fibroblasts
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.319.563.83.141.2]CYTOTOXICITY OF REPLICATION-DEFECTIVE HSV-1 MUTANTS 6357 6
5 4 3
I
0.L
0
a
I-z 2 1 5 4
3 2
5
5 4 3 2
2 3 4 5 6/2 3 4 5 6
Number of
infecting virus
(log
PFU per
well)
FIG. 6. Recovery of virus mutants from infectedfibroblasts. Confluent monolayers of human fibroblasts (approximately 2.5 x 105cellsper 16-mmwell)wereinfected at MOIs of from 1 X 103to5 x 105PFU perwell with each of the mutantsD30HA3, 14HA3, CgalA42,in1850A42, A3vhsZ, and14HA3vhsZ,asindicated. Graphs show the average number of plaques from duplicate cultures that could be recovered 1 h (open symbols)or11days(solid symbols) postinfection.Recovery wasperformed by superinfection with theICP8mutantd301(19). Superinfected cells wereplatedoutatvariousdilutionsontomonolayers ofE5cells (for IE 3mutants)or V9cells(forUL42mutants) as appropriate and incubated at37°Cinthepresenceof5%humanserumuntil plaquescould be counted.
platedonpermissive cellmonolayers andnotfrom freevirus,
theyshouldnotexceed the total number of cells available for
infection, which was approximately 1 x 105 to 2 x 105, as
determined by counting with a Coulter counter. Therefore,
since cells whichwereinfectedbymorethanoneviruscanstill
give rise to only one plaque, theactual amount ofpersistent
virus in this experimentcannotbe determined above a
maxi-mumof1PFU percell. In somecases, the number of rescued
plaquesatday0 exceeded the number ofPFUin theinfecting
inoculum (for instance, 1 x 103 PFU ofD30HA3 resulted in
2.5 x 103 rescued plaques), which probablyreflects the fact
that theactual titer of infectious genomes abletocomplement
d301 isseveralfold higherthan the PFU titer.
Asshown inFig. 6, infection with eitherD30HA3or 14HA3
at increasing MOIs led to a maximum number of plaques
obtainable on day 0 of approximately 105, which correlates
reasonably well with the total number of cells available for
infection. Striking differences were observed between the
numbers ofD30HA3and14HA3which could be rescuedatday
11.First,atlowMOIs(103to104PFUper105cells),lessthan
10% oftheinput D30HA3 could berecovered,comparedwith
45 to 50% for 14HA3. Second, the maximum number of
rescuableD30HA3 reachedasharppeakof 3.5 x 103 PFUat
an MOI of 104 PFU per well and then rapidly declined at
higher MOIs. In contrast, the number of rescuable 14HA3
began toplateau at 9 x
103
PFU at an MOI of104
PFUper VOL.68, 1994on November 9, 2019 by guest
http://jvi.asm.org/
[image:11.612.132.496.76.516.2]well and reached a
peak
of 1.2 x104
PFU at an MOI of 105PFU perwell. The number of rescuable 14HA3 declined above
an
infecting
inoculum of105
PFU perwell.Therefore,
by
thefollowing
twocriteria,
(i)
theproportion
ofinfecting
virus thatcan be recovered at a later time
point
and(ii)
the maximumamountof virus thatcanbe recovered after sustained infection
of
cells,
14HA3 has aconsiderably
less toxicprofile
thanD30HA3.
Comparison
of the UL42 mutantsCgalA42
andin1850A42 revealed that a
greater
proportion
of in1850A42than of
CgalA42
could be recovered at low MOIs.Also,
themaximumamountofin1850A42 that could be recovered
(1
x104
PFU)
wassubstantially higher
than the maximumamount ofCgalA42 (6
x 102PFU).
However,
above aninfecting
inoculum of
104
PFU perwell,
the recovery of both virusesdropped precipitously, indicating
that both UL42 mutantsweremore toxic thantheir
corresponding
IE 3 mutantcoun-terparts,
inagreement
withthe results described earlier.Com-parison
of the IE 3-vhs double mutants A3vhsZ and14HA3vhsZ
showedagain
that inclusionof the VP16 mutation(in 14HA3vhsZ)
allowed an increased recoveryof virus at 11days postinfection,
intermsof both theproportion
ofinfecting
virus recovered
(less
than 10% for A3vhsZversus 30 to 50%for
14HA3vhsZ)
and themaximum sustainable virusload(3
x102 PFU for A3vhsZ versus 1 x 104PFU for
14HA3vhsZ).
Interestingly,
A3vhsZappeared
tobemoretoxicthanD30HA3
by
this assay. The recovery ofA3vhsZwas less efficient thanthat of
D30HA3
onday
0,
and thepeak
amountofA3vhsZthatcould be recoveredwas 10-fold below that for D30HA3. In
terms of virus recovery and maximum sustainable virus
load,
the
triple
mutant14HA3vhsZ
wassimilartothe doublemutant14HA3,
indicating
that inclusion of the vhs mutation in this mutantbackground
had littleor no benefit inreducing
virustoxicity.
Finally,
wetestedthepossibility
that thevhs function doescontribute to CPE but its effects are
usually
maskedby
agreater
level ofcytopathogenicity
causedby
viral geneexpres-sion. We therefore infected human
primary
fibroblasts at anMOI of 10 PFU per cell with
D30EBA,
A3vhsZ,
and14HA3vhsZ
whichhad either been untreatedorirradiated with UVlight
toabolish viral geneexpression.
Ithas been shownpreviously
thatthe vhs function isnotinactivatedby
suchUV treatment(16, 44).
Cellswereharvestedby
trypsinization
at3days
postinfection,
and cellviability
wasdeterminedby
count-ing
cells which excluded trypan bluedye (Fig. 7).
In theabsence of UV treatment, the number of viable cells in
comparison
tothat inamock-infectedculturewasreducedtoless than 3% for A3vhsZ and D30EBA and to 15% for
14HA3vhsZ.
After UV irradiation ofthese mutants, cellvia-bility following
infectionwas muchimproved
but very similar for each mutant(about
42%), indicating
that factors other thanvhsarecausing
cytotoxic
effects in the absence of geneexpression.
In aprevious
study,
we found that cells infectedwithstrain KOS and the vhsmutantof strainKOS called vhsl
(53)
sufferedcytopathic
effects at an MOI of2, even in thepresence of
phosphonoacetic
acidtopreventviral DNArepli-cation,
whereas infection with UV-irradiated viruses at thesame
MOI
abolished CPE(29).
We therefore infected cellswith UV-irradiated KOS and vhsl at an MOI of 10 to determinewhetherdifferencesin theinduction of CPE can be detected at a
higher
MOI.
Cellviability
following infectionwiththese viruses intwoseparate
experiments
wasreduced tobetween 52 and 62%
(Fig. 7),
and therewasnot a significantdifference between vhsl and
KOS, indicating
that the vhs function of strainKOS,
like that of strain 17+, does not contributein amajor
waytothe induction of CPE.0
-4a0
0 v
.0 DrI.
m -I .< N N N N
0 ciO r i E a A s s
Exp.I Exp. 2 i = _
Virus strain
FIG. 7. Effectof vhs mutationoncellviabilityafter infection with
UV-irradiated viruses. Humanprimary fibroblastswere seeded at a
densityof 5 x 104cellsperwell and infected intriplicatethefollowing
day with KOS, vhsl, D30EBA, A3vhsZ,or 14HA3vhsZat anMOI of
10 PFUpercell. At 3 days postinfection,the cellswereharvestedby
trypsinization, and the number of viable cells in each culture was
estimated after staining with 0.5% trypan blue and counting the
unstained cells. The average numberof viable cells is shownas the
percentageofviablecells inamock-infectedculture,determinedfrom
counting in eight fields ofahemacytometerfor each of threeinfected
cultures. Error bars indicate standarddeviations.
DISCUSSION
Replication-defective mutants of HSV-1 retain a strong
potential to induce CPE andcausecelldeathduringinfection
of nonpermissive cells. The results of a previous study had
indicated that viral gene expressionwas amajor factor
causing
CPE (29). In an attempt to construct rather more
benign
vectors for gene transfer, we have in this study examined
individual HSV-1 genes and virion components which mightbe
responsiblefor CPE. We have shown thatatleastthreeHSV-1
genes, which are efficiently expressed by an IE 3 deletion
mutantduringinfection ofnonpermissive cells, cause a marked
inhibition ofcolony formation in cotransfection experiments.
As a means to reduce IE gene expression globally in a
replication-defective
mutant,andasan alternative to trying to produce a mutant defective for three or four IE genes, weconstructed viruses which were deleted for either of the
essential genes IE 3orUL42 andincluded the mutation inthe
VP16 gene ofin1814 describedby Ace et al. (2). This mutation
abolishes the ability of VP16 to transinduce IE gene
expres-sion. The IE3-VP16 double mutant 14HA3 is significantly less
cytotoxicthan a single mutant deleted for IE 3, whereas the
reduction intoxicityof theUL42-VP16 mutant is less marked.
The vhs function has also been considered a potentially
cytotoxiccomponentof the HSV-1 virion. However,inclusion
ofamutation in the vhs gene does not reduce the
cytopatho-genicity of either an IE 3 deletion mutant or the double IE
3-VP16 mutant.
Virally encoded cytopathogenic gene products. Plasmids
bearing
HSV-1 IE genes 1, 2, and 4 had a marked inhibitoryeffect on the
ability
to obtain colonies from a cotransfectedselectable marker gene (Fig. 2). Plasmids bearing IE 3 also