0022-538X/88/082799-09$02.00/0
Copyright© 1988, AmericanSocietyforMicrobiology
Kinetic,
Quantitative, and
Functional Analysis of Multiple
Forms
of
the
Vesicular
Stomatitis Virus
Nucleocapsid
Protein in Infected Cells
RICHARD W. PELUSODepartmentof Microbiology, UniversityofMinnesota, Minneapolis, Minnesota55455 Received 31 December 1987/Accepted 15 April 1988
Multiple forms of the vesicularstomatitis virus nucleocapsid protein N have beendetectedin infectedcells. One form iscomplexedwiththeviralNS protein ina 1:1 molar ratio, and the other formsaredistinguished by theirmorerapid sedimentationratesonglycerolgradients. I performedaseries of experimentsdesignedto
analyze therelationships between these forms of the N protein. Pulse-chase experimentsdemonstrate that the Nproteinismade firstas the form which bindstotheNS protein, forminga 1-to-1molar complex, and that
with increasing times of chase it is either assembled into nucleocapsids or converted to the two higher sedimenting forms. Usinga newly developed quantitativeimmunoblotting procedure, I have quantitated the threedifferentially sedimentingspeciesofthe N protein and have shown thatatlatertimespostinfection (6to
7h), thefaster-sedimenting forms ofthe N protein accountforasmuchas50% of the soluble N protein in the
cell.The activity of these forms has been assessed,with only the1-to-1molarN-NScomplex demonstrating the abilityto supportthereplicationandencapsidation of viral genomic RNA. A model for the conversion of the Nprotein from the active N-NS complex into the other forms of the protein is presented, and the possible function oftheN-protein self-complexes is discussed.
The rhabdovirus vesicular stomatitis virus (VSV) pos-sesses a nonsegmented negative-stranded RNAgenome in the form of a helical nucleocapsid. Two distinct types of
RNA-synthetic events are directed by this structure:
tran-scription and replication (2). Transcription of the genome leads to the synthesis of a short leader RNA followed by the
synthesis of five mRNAs, whereas replication leads to the
synthesis of full-lengthcopies of the viral genome.
Replicat-ing molecules, but not mRNAs, are encapsidated. The nucleocapsid (N) protein of vesicular stomatitis virus plays a
crucial role in controlling these processes. This protein
associates tightly with replicating genomic RNA to form a nucleocapsid with helical symmetry, rendering the RNA in this structure resistant to degradation by ribonucleases (27).
Evidence for multiple forms ofthe nucleocapsid protein in
VSV-infectedcells hasbeen presented(23, 24).One formof
the protein is complexed withthe viral NS protein in a 1:1
molar ratio, and the complexofthese twoproteinsexhibits
the ability to support genomic RNA replication in vitro. Other forms of the N protein have been detected which appearedtobe complexes ofN with itself. TheseN-protein complexes have demonstrated little activity in supporting genomicRNAreplicationinvitro.Weperformedaseriesof experimentstodeterminetherelationshipbetween the forms
ofthe N protein, to quantitate the relative levels ofeach
form, and to assess their role in
replication
of the viral genome.Weprovide evidenceforperhapsthreeforms of the N protein, distinguished by their rate of sedimentationthrough glycerol gradients. However, only the complex consisting ofonemolecule each of N and NS has the
ability
to supportreplication of genomicRNAin vitro. MATERIALS ANDMETHODS
Cells and viruses.
Monolayer
cultures ofbaby
hamsterkidney (BHK) cells were used for all of the
experiments
in this paper. The HR strain of the Indiana serotype of VSVand the MS-T defective-interfering (DI) particlewere
prop-agated aspreviously described (22).
Pulse-chase analysis of soluble proteins. Subconfluent monolayers ofBHKcells in 100-mmplastic petri disheswere infectedwith VSVat amultiplicity of infection of10. At 4 h
postinfection (p.i.) the medium was removed and replaced
with fresh medium lacking leucine. This was removed after 15 min andreplaced with medium lackingleucine but con-taining[3H]leucine (200 ,uCiperdish). Aftera10-minpulse, onedishofcellswas harvestedand theotherswerewashed withcomplete mediumcontaining cycloheximide(100,ug/ml) topreventfurtherincorporation oftheisotope and incubated
in this medium for various periods as indicatedbelow. For harvest,cellswerepermeabilized withasolution of
lysolec-ithinasdescribedpreviously (22),scraped intoasolution of
0.2 M
NH4Cl,
0.1 M N-2-hydroxyethylpiperazine-N'-2-etha-nesulfonic acid(HEPES; pH 7.4), 7 mM KCI, and 4.5 mMmagnesium
acetate, andcentrifuged
at 800 x g for 5 min. Thesupernatant fluidwascollectedandsubjectedto centrif-ugation inaBeckman SW55rotor at50,000rpmfor65 min. The resulting supernatant fluid is designated the soluble protein fraction. Solubleprotein was analyzed by centrifu-gation through5to20%(wt/vol) glycerol gradients (made
inscrapingsaltsmix)inan SW41rotorfor22hat32,000rpm. The gradients were collected from the top into 1-ml frac-tions. Aportionof each fraction was
analyzed
by
immuno-precipitation with an excess ofrabbit anti-VSV serum to precipitateallofthe viral
proteins.
Theprecipitated
proteins
wereanalyzedby sodiumdodecyl
sulfate-polyacrylamide gel
electrophoresis
(SDS-PAGE)
andfluorography.
Individual proteinspecieswerequantitated
by
excising
theappropriate
regions from the dried
gel
with theautoradiogram
as atemplate,
dissolving
thegel
piece
in30%H202
at60°C
for 12 h, andperforming
scintillationcounting.
Quantitative electroblotting
analysis
of solubleproteins.
Subconfluent
monolayers
of BHK cells were infected with VSVat10 PFU percell. At successive 1-hintervalsstarting
2799
on November 10, 2019 by guest
http://jvi.asm.org/
at2 h
p.i.,
the cellsfrom oneplate
werepermeabilized
andscraped
inthesaltsmixasdescribed above. Solubleproteins
were
prepared
andanalyzed
by
glycerol
gradient
sedimen-tation and SDS-PAGE as
above,
except thatsamples
were notimmunoprecipitated prior
togel
analysis.
Theproteinspresentin each
gel
werequantitatively
transferredto asingle
sheetof Zetabind(AMF)
membraneasdescribedpreviously
(25).
Theresulting blots
werethenprocessed
tovisualize theviral
proteins by using
rabbit anti-VSV serum and12511
labeled
protein
Aasdescribedpreviously (25). Quantitation
was
performed by
excising
theappropriate
regions
of theblotsand
measuring
radioactivity
in agammacounter.Activity
ofglycerolgradient-fractionated
solubleproteins
in support of RNAreplication
andencapsidation.
Subconfluentmonolayers
ofBHKcellswereinfected with10PFU ofVSV percell. Solubleproteins
wereprepared
at4or7hp.i.
andanalyzed
by
glycerol gradient
fractionation as describedabove.
Samples
ofeachgradient
fraction were mixed withnucleocapsids
from cellsthatwerecoinfectedwithwild-type
VSV and the MS-T
defective-interfering
particle
asde-scribed
previously
(22).
Allsamples
were incubatedat30°C
for 90 min in a solutioncontaining
thefollowing:
0.13 MNH4Cl,
0.1 M HEPES(pH
8.0),
7 mMKCl,
8 mMmagne-siumacetate, 1 mM
dithiothreitol,
2 mMATP,
1 mMCTP,
1 mMGTP,
50 ,umUTP,
0.1 mMS-adenosyl
methionine,
1mM
spermidine,
40 U of creatinephosphokinase
perml,
10mMcreatine
phosphate,
2 ,ugofactinomycin
Dperml,
and 100,uCi
of[3H]UTP (35
Ci/mmol).
Eachsample
was thendilutedwithan
equal
volume ofwater,CaCl2
wasaddedto1mM,
and micrococcal nuclease was added to 10,ug/ml
todegrade
allnonencapsidated
RNA molecules. Afterincuba-tion at
37°C
for 30 min,ethylene
glycol-bis
(P-aminoethyl
ether)-N,N,N',N'-tetracetic
acid(EGTA)
was added to 10mM. The
samples
were then treated with protease K andSDS and
phenol-chloroform
extracted,
and the RNA wasanalyzed by electrophoresis
on 1.5%agarose-7
Mureagels
(22).
Sedimentation
analysis
ofnucleocapsid-derived
Nprotein.
Seven confluent flasks of VSV-infected BHK cells were labeled with
[35S]methionine (200 ,uCi
perflask)
from 2to6 hp.i.
N protein was isolated from the intracellularnucleo-capsid
exactly
as describedby Blumberg
et al.(4)
anddialyzed against
1 M NaCl-50 mM Tris(pH
7.8)-i
mMEDTAat
-3°C.
Theresulting
Nprotein
was thenanalyzed
by glycerol
gradient
sedimentation in thefollowing
ways.One half of the
sample
wascentrifuged
for22 hat32,000
rpm inanSW41rotor at0°C through
a5to20%
glycerolgradientin the
dialysis
bufferdescribed above. The other half of thesample
wasdiluted withdialysis
bufferlacking
NaCltoyield
a solution of 0.2 M
NaCl,
50 mM Tris (pH 7.8), and 1 mM EDTA and thenincubatedat37°C
for30mintopromote theself-assembly
ofthe Nprotein
intodisk-shaped
structures(3).
Theresulting protein
wasanalyzed by
glycerol gradient sedimentation under identical conditions as the nontreatedsample. Samples
of each gradient were then analyzed bySDS-PAGEand
autoradiography.
RESULTS
Pulse-chaseanalysisofsolubleproteins. Ourprevious stud-ies have demonstrated that several different forms ofthe
nucleocapsid
protein
Nof VSV weredetectedwhensolubleprotein
extracts of infected cells that were labeled with[3H]leucine for
relatively
long periods(1h)wereanalyzed byglycerol
gradient
sedimentation (23, 24). One species of Nproteincosedimentedwith the NSprotein near the top of the
gradient,
forming
a 1-to-1 molarcomplex;
a second hetero-genousspecies
sedimented in the bottom third of the gradi-ent; and a thirdspecies
was present in thepellet.
To establish therelationships
amongtheN-protein
forms and todetermine whether any
postsynthetic processing
or matura-tion processeswereresponsible
for the appearance ofmul-tiple N-protein species,
Iperformed
aseries of short pulse-chaseexperiments.
Cells were labeled with[3H]leucine
for 10min,
and theproteins
wereanalyzed
eitherdirectly
after thepulse
orafter variousperiods
ofchase in the presence ofcycloheximide.
The results ofthisexperimentarepresented
inFig.
1.Thedata inFig.
1Ademonstratethatthe Nprotein
is
synthesized
as the species that cosediments onglycerol
gradientswithNSproteinina 1-to-1complex,
i.e.,
theform of Nprotein capable
ofsupporting
replication
ofnucleocap-sidsinvitro(23,24). With increasing timesofchase,upto2
h, there is a dramatic shift in the sedimentation ofthe N
protein to more rapidly sedimenting forms, with two new
species
present, oneinfractions6to12,
and anotherinthe pellet fraction(fraction
13). Note that cycloheximide was present in the cultures after the pulse-labeling period to ensure that the changes we observed in the sedimentation properties of the N protein were due to changes in theprotein and not to new synthesis. However, essentially
identical results have been obtained in experiments in which
cycloheximide was
replaced
with a1,000-fold
excess of unlabeled leucineduringthechaseperiods (datanotshown).
The amount of N protein in each fraction of eachgradient
wasquantitatedby excisingtheband from the driedgelsand
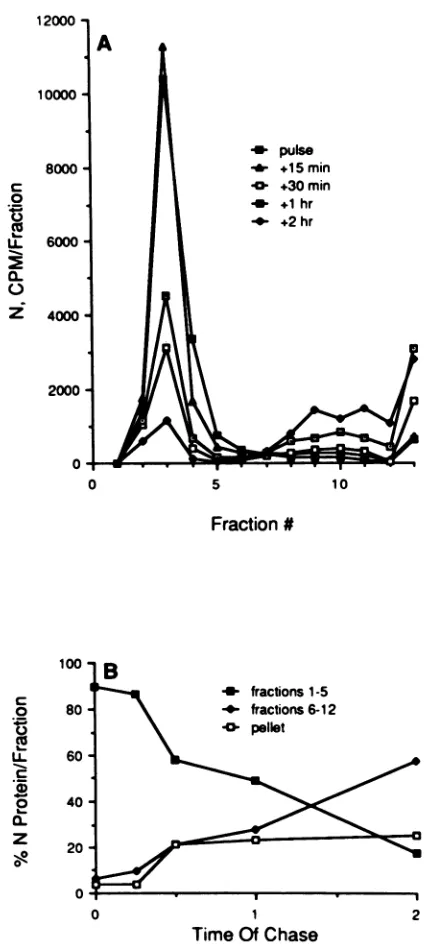
countingin ascintillation counter.These results are shown in Fig. 2. These data demonstrate several points.
First,
approximately 60% of the soluble N protein labeled
during
the 10-minpulsewas still in the soluble fraction aftera2-h
chase, indicatingthattherearepoolsofNproteininthe cell that may be increasing as infection proceeds. The presence of solublepoolsof Nproteinhas beenreportedbyothers
(1,
13,19, 23, 26, 28). Second,inthe pulse-labeled sample, 90% of the N protein sedimented near the top of the
gradient,
where most ofthe NSproteinalso sedimented, butby2 h of chase, 80% of the N protein that remained in the cell was present asthe morerapidly sedimenting forms (fractions 6to 13). Third, there was a sixfold increase in the faster-sedi-menting species of N protein after 2 h of chase relative to the pulse.Fourth, 50% of the N protein in the pulsed samplewas converted to more rapidly sedimenting species by 2 h of
chase,with the remainder either assembledinto
nucleocap-sids (which were pelleted before the soluble proteins were analyzed) or remaining near the top of the gradients. The percentage of N protein which sedimented in the pellet
fraction(fraction 13) reached a plateau atapproximately23% of the total by 30 min of chase, but the percentage in fractions 6 to 12 of the species of N protein exhibiting an intermediate sedimentation rate continued to increase. These results suggest that the N protein is complexed with the NS protein shortly after synthesis and that it can be
converted into morerapidly sedimentingN-N complexesas afunction of time aftersynthesis.
Little ifany NS protein wasdetected cosedimenting with the N protein infractions 7 to 13 of the gradients (Fig. 1). To
investigate this more thoroughly, cells were labeled with [3H]leucine for 5 h beginning at 2 h after infection, rather thanpulsed for 10 min, as in theexperimentinFig. 1. When solubleproteinpreparationsfrom these cells were separated onglycerol gradients and analyzed for the presence of NS protein in the bottom two-thirds of the gradient, small amounts ofthe NSprotein were foundto coprecipitate with
on November 10, 2019 by guest
http://jvi.asm.org/
N
i. _
13
N
.."- __0_ q_ _~
--13 B
G
NS
N
Ml
13
L
40G NS
_ _-_0 N
~~ N~~
13
FIG. 1. Pulse-chase analysisof differentially sedimenting forms of the VSV N proteinpresentinasolubleformin infected cells. BHKcells
wereinfected with VSV and pulse-labeled with[3H]leucinefor10minat4.25hp.i.Thecontentsofoneplatewereharvested after thepulse,
and theremainingfourplateswerewashed and incubated further in mediumcontainingcycloheximide (100 ,ug/ml)beforeharvest.Cellswere
harvestedbypermeabilizingwithlysolecithin, and soluble proteinswerepreparedandanalyzed bysedimentationthrough20to40%glycerol gradients, whichwerethenfractionatedinto 13 1-ml fractions. Theproteins in each fractionwere precipitatedwithanti-VSV serumand
electrophoresedonSDS-polyacrylamide gels. A, pulse; B, 15-min chase;C,30-minchase;D, 1-hchase; E, 2-h chase.Thepositionsof the
viralproteinsareindicated. Fraction1 is thetop,and fraction13is the bottom (pellet) of eachgradient. Markerproteinswere analyzedin aseparategradient,withbovineserumalbumin(molecular weight 68,000)infraction3, ,3-galactosidase(molecular weight 116,000)infraction 5,andphosphorylase (B)(molecular weight 185,000)infraction 10. rRNA markers(18Sand28S)werefound in thepelletfractions. the Nprotein. Quantitation of these species revealed that the
NS proteinwasgreatly reduced inamount relativetothe N protein andwaspresentinaratio of between four and eight N proteinstoeach NSprotein in fractions 8to 12(data not shown).
The G protein of VSV was not detected in the soluble fraction of cells after the 10-min pulse, but by 15 min, and certainly by 30min,ofchasetwospecies of Gweredetected.
Oneformsedimented infractions 2to 4, and the otherwas found in the pellet fraction. Careful inspection of the gels revealed that the G protein in fractions 2 to 4 exhibited a fasterelectrophoretic mobilitythanthat ofthe G proteinin thepellet. The G proteinnearthe topof thegradientmaybe analogous tosolubleG protein,apreviously describedform of theglycoprotein whichlacksamembrane anchor domain (7, 15, 16, 20). The failuretodetecttheaccumulation of the A
_mm
N
13 C
L1
I Ila
E
L
I
4
rz
s N
0
L
m
I
I
q
L
A
0
I I
1 I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.144.469.59.534.2]12000
10000
8000
A
W puke
-_-+15min
U +30 min
U +1 hr
_-
+2hr0 5 10
Fraction
#0 1 2
Time OfChase
FIG. 2. Quantitative analysis of the N protein during a
pulse-chaseexperiment.The Nproteinineach fraction of eachgradientin
the pulse-chase experiment shown in Fig. 1 was quantitated by
excising the appropriate region of each gel and counting it in a
scintillation counter. (A) The total counts in each band. (B)The
percentageof Nprotein presentasthe N-NScomplex (fractions1to
5),astheintermediately sedimentingform(fractions6 to12),and in
thepelletfraction(fraction13).Fraction 1 is thetopof thegradient. slowly sedimenting form of the Gproteinin cellsby
quanti-tativeelectroblotting (see Fig. 3) isconsistent with itsbeing
shed into themedium.
The NS and L proteins were barely detectable in the
soluble fraction of cells after a 10-min pulse, but were
detectableby15 and60minofchase, respectively, suggest-ingthattheseproteinsmay enterasolublepoolmoreslowly
thanthe N protein.
Quantitationofsolubleproteinsininfected cells. Theresults
thatIobtained from thepulse-chase experimentsdescribed
above ledme toinvestigate thepossibilitythat levels of the two more
rapidly sedimenting
forms of the Nprotein
would increaseasinfection with VSVproceeded,
since60% of thepulse-labeledNproteinwasstill in the soluble fraction of the cell even after 2 h of chase and 80% of it was rapidly
sedimenting.
I determined the relative levels of the NSproteinand each of theN-protein speciespresent in cells as afunctionof time
p.i. by using
arecently developed
quan-titativeelectroblotting procedure (25). Usingthis method ofanalysis,
I could avoidcomplications
and artifacts associ-atedwithmetaboliclabeling,
suchasvariability
intransportand
varying pool
sizes of amino acidsowing
toviralinfec-tion. I
prepared
solubleproteins
from VSV-infected cellsat successive 1-h intervalsstarting
at 2 hp.i., separated
theproteins by glycerol
gradient centrifugation,
andanalyzed
the
proteins
in each fractionby gel electrophoresis
and Westernimmunoblotting.
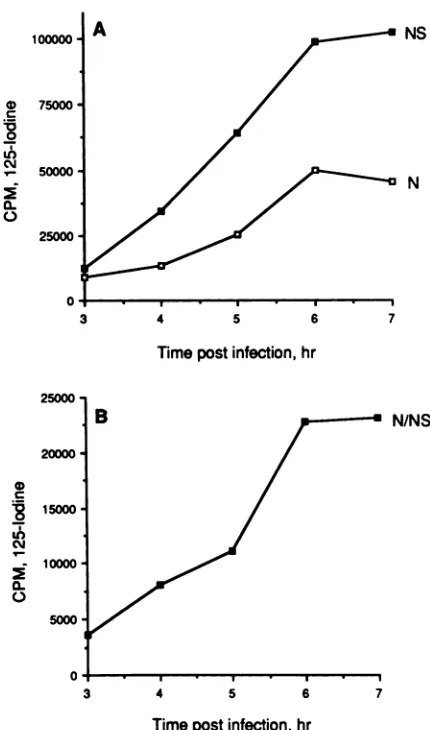
These results are shown inFig.
3 and 4. At all timepoints studied, approximately
95% of the NSprotein
sedimented near the top of thegradients
in anincreasing pool.
Pools of NSprotein
in cells have beenreported by others (6,
8,
10,13, 17, 18, 29).
Avery differentpicture
emerged
fromtheanalysis
of theNprotein.
Atearly
times
p.i. (up
to 5h)
themajority
of the Nprotein
cosedi-mented with the NSprotein,
butatlater times(6
to7 hp.i.)
asmuchas50% of the N
protein
in the soluble fraction of the cellsedimented fasteras twospecies (one
in fractions9to12 andasecond inthepellet
fraction[fraction 13]).
Thesedata are consistent with thepulse-chase experiments
inFig. 1,
which demonstratedaconversion ofthe
slowly
sedimenting
form of N
protein
to theother,
morerapidly
sedimenting
forms of N
protein.
The Nprotein
isapparently
stableas a solublenon-nucleocapsid-bound species,
asevidencedby
itsincreasing
presence in a soluble formas the infectionpro-ceeds.
The data obtained from the
quantitative
electroblotting
analysis
ofsolubleVSVprotein
in cellsas afunction of timep.i.
have beenanalyzed
forrelative abundances of total Nand NS
proteins
andfor the Nprotein
present as a 1-to-1N-NS
protein complex.
Theseresultsareshown inFig.
5,
inwhichthe totalcountsof
1251
boundtoallforms ofthe Nand NSprotein
ateachtimepoint
havebeentotaled. Thereisasteady
increase in the NSprotein
level between 3 and 6 hp.i.,
aslight
increase between 6 and 7 hp.i.,
andanoveralleightfold
increase from 3 to 7 hp.i. (Fig. 5A).
A similarpattern
emerges for totalNprotein,
withasixfold increase from3to6 hp.i.
andaslight
decrease from6to7hp.i.
Thisdecrease, however,
is notalways
observed,
and in thisexperiment
it was due to a decrease in the Nprotein
recovered from the
pellet
fraction.Whenextracareis taken to ensure total recovery ofthepellet
fraction,
the solubleN-protein
level isslightly higher
at7h than at6hp.i. (data
not
shown).
A
slightly
differentpattern
isseenwhenthe accumulation of the Nprotein species
presentasthe 1-to-1N-NSprotein
complex
(fractions
1to5)
isanalyzed (Fig.
5B). Between 3 and 5 hp.i.
thereisarelatively
linearincrease inthelevel of this form of the N protein, with a doubling of the level between 5 and 6 hp.i
and onlya slight increase between 6 and 7 hp.i.
There isasixfold increase in the amountofthe N-NScomplexfrom 3to7 hp.i.Invitroactivity oftheN-protein species. Previous reports from our laboratory concerning the ability ofthe various
forms of the N
protein
to supportreplication of nucleocap-sids demonstratedthat alloftheactivity
wasassociatedwith theN-NScomplex
(23, 24). The morerapidly sedimenting forms of the Nprotein
hadessentially
no activity in this c0
U-0
C.)
z~
2000
0
c 0
0-cL
0
z
0IR
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.68.283.67.542.2]2h < 3h
.~~~~Ns-
#4Ns1 13 1 13
4h 5h
G q
~~~~N#4
us
1 13 1 13
6h 7h_
Q w
_ 1
[image:5.612.150.481.74.383.2]1 13 1 13
FIG. 3. Quantitative analysis of soluble viral proteins in VSV-infected cells. BHK cells were infected with VSV, and soluble proteins were preparedfrom2to 7 hp.i. as indicated. Soluble proteins were sedimented on glycerol gradients, the gradients were fractionated, and the proteins in each fraction were analyzed by SDS-PAGE. The proteins in each gel were then quantitatively transferred to Zetabind membranes and reacted with anti-VSVserumfollowed by incubation with "25I-labeledprotein A. Autoradiograms were prepared from each blot. The positions of the viral proteinsareindicated on the right side of each blot. Fraction 1 is the top, and fraction 13 is the bottom (pellet) of each gradient.
process. Theseprevious experiments werecarried out with
proteinspresentincells at 4 h after infection (23, 24). In light
of the data inFig. 3 and4,demonstratingthat the abundance
ofthefaster-sedimentingN-proteinspeciesis low at 4 h p.i.
but high at 7 h p.i., I assessed the differently sedimenting
forms of solubleNproteinpresentin cellsat4and7 hp.i. for
the ability tosupport replication of nucleocapsids invitro. This result is shown in Fig. 6. At both 4 and 7 h after infection, all oftheactivitywasfound withtheN-NSprotein complex, and not with either of the faster-sedimenting N-proteinspecies,eventhough thesespecies madeupabout one-half of the total soluble N protein in the cells at that
time. Note that theactivity oftheN-NScomplexwashigher
at 7 h p.i. than at 4 h p.i., perhaps reflecting its greater
concentrationatthattime.
Sedimentation analysis ofnucleocapsid-derived N protein.
Several years ago, Blumberg et al. (3)
reported
that Nprotein could be isolated from assembled
nucleocapsids
free from other viral proteins and that this Nprotein
would interact with the leader RNA to form aribonucleoprotein
structurewithpropertiesnotunlikethoseofa
nucleocapsid.
Inview ofourfindings aboveand elsewhere(23, 24)that the
abilitytosupport
genomic
RNAreplication
andnucleocap-sidassembly isapropertyof theN-NS
protein
complex
and notofthe Nprotein alone, Iinvestigated
thesedimentation properties ofthenucleocapsid-derived
Nprotein
todeter-minewhether it hadpropertiessimilartoany of the
species
of soluble N proteinwhich I found in the cell. Theseresults
are shown in Fig. 7. When conditions reported to prevent
self-assembly of the nucleocapsid-derived N protein (high ionicstrength, pH 7.8 and0°C)wereused,theresultinFig.
7A was obtained. The N protein wasdispersedthroughout thegradient, with 43% beingin thepellet fraction.When the same
preparation
ofNprotein
wasincubated under condi-tions which promote its self-assembly into disklike struc-tures(lower ionicstrengthand37°C),
nearlyall(96%)
of theNproteinwaspresentinthe
pellet
fraction(Fig.
7B), which isnotabletosupport RNAreplication
invitro(Fig. 6), withtheremainder
being
foundnearthetopofthegradient.
The fact that most of the Nprotein
derived from assembled nucleocapsids pelleted throughtheglycerolgradient
whenit was warmed innear-physiological salt solution is consistent with my inability to demonstrate that Nprotein
prepared
fromthis source cansupport the
replication
ofwild-type
or DIgenomicRNA invitro(unpublished results).
Apparently,
thesmallamountofthe
protein remaining
nearthe topofthegradient is insufficienttosupport
genomic
RNAreplication.
DISCUSSIONAt least three soluble forms ofthe N
protein
ofVSV,
distinguishable
by
their rates of sedimentation onglycerol
gradients,
are present in infected cells. One form iscom-plexedwith theNS
protein
ina1:1molarratio,
and theother two forms contain much less NSprotein
or appear to beN-protein
self-complexes.
The resultsfromtheexperiments
on November 10, 2019 by guest
http://jvi.asm.org/
100000o
75000
50000
-25000
A NS
N
3 4 5 6 7
Timepostinfection,hr
0
0
40000
c
CM0- 30000
:- 20000
0L
0)
10000
5 10
Fraction#
B
CY
0.
0 3 hrpi
-_- 4hrpi
_- 5hrpi
0- 6hrp _- 7hrpi
0 5 10
Fraction #
FIG. 4. Relative levels of the N and NS proteins in asoluble
formas afunction of time after infection. Theradioactivityboundto
the N and NSproteinsin each lane of each of the blots inFig.3was
determinedby excisingtheappropriateareasof the membranes and
gammacounting.The resultsarepresentedfor the Nprotein (A)and
theNSprotein (B)from 3 to7hp.i.
prestntedin thiscommunicationsupportthe conclusion that the nucleocapsid protein of VSV is synthesized as a rela-tively slowly sedimenting form capable of bindingtothe NS proteintoformacomplex whichserves asthe substrate for the replication and encapsidation of replicating genomic RNA molecules (Fig. 1, 2, and 6). The more rapidly sedi-menting N-protein complexesareunableto supportgenomic RNAreplication in vitro. Thepulse-chasedata presented in Fig. 1 and 2 show that the two more rapidly sedimenting forms of the N protein are derived from the more slowly sedimenting form. Figure 8 diagrams ways in which the various N-protein complexes might be formed. In both schemes, the N protein in the N-NS protein complex can
bind toreplicating RNA molecules to formanucleocapsid,
15000
10000,
5000
0
B N/NS
3 4 5 6 7
Timepostinfection, hr
FIG. 5. Relative abundancesof totalNS,totalN,and the N-NS
protein complexas afunction of timep.i.The data obtained from the
quantitativeanalysis of viralproteins ininfected cells (Fig. 3)are
plottedastotal N and NS(A)orthe Nproteinin the N-NSprotein complex (B).Forthedata inpanel A,thetotal counts of 1251 bound
to the N or NS protein were summed for each fraction of the
gradientateach timepoint;forpanel B,only thecountsboundtothe
Nproteinin fractions1 to5weretotaled.
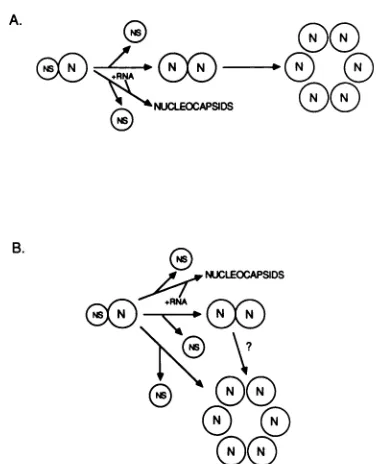
releasing the NS protein (23, 24). In scheme A, instead of bindingtoreplicating nucleocapsids,the N-NSprotein com-plex dissociates, releasingthe NSproteinandresultinginthe formation of N-N protein complexes exhibitingan interme-diatesedimentation rate.The numberof Nproteinsin these complexes is not known, and the diagram is not meant to suggestthatthey are dimers. Theroleof the NS proteinin these complexes is unclear, and so it is not shown here. These intermediately sedimenting N-protein complexes as-sociate to form the most rapidly sedimenting form of N protein found in the pellet fraction of the gradients. The stoichiometry of N proteins in these complexes is not known. Scheme Athereforepredictsa sequential formation of the three forms of N protein. Scheme B, on the other hand, predicts that the formation of each form of the N-protein complexes is independent of the other, but does allow for the possible conversion of the intermediately sedimentingformtothemostrapidly sedimenting formof N complexes. We cannotpresently distinguish between these twomodels.
12000
-10000
-A
U 3hr pi -& 4hr pi -5 hrpi
-06hrpi
0 7hrpi
0)
C 70
0~
0
8000
-6000
-4000
-0
CL 04
M~
0.
2000
I.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.67.288.68.520.2] [image:6.612.322.537.73.438.2]A
A B 1 2 -a4 5 9 10 t1 12 13
B
e
-AB 123 4591011213
19S
q
FIG. 6. Activity of fractionated soluble proteinsprepared at4and 7 hp.i. Soluble proteinswerepreparedfrom VSV-infectedcellsand
separated by glycerol gradientcentrifugation. Each gradientwasfractionated into13 1-mlfractions.Aportionof eachfractionwasmixed with
nucleocapsids fromcells coinfected with VSV and the MS-TDIparticle,and replication wasallowedtoproceed for 90minin thepresence
of [3H]UTP. The micrococcal nuclease-resistant species (encapsidated) were then analyzed by agarose gel electrophoresis. (A) RNA
produced by usingthe 4-h protein sample; (B) RNA produced by usingthe 7-h protein sample. In both panels the numbers refertothegradient
fractions thatwere tested, lane A isacontrolunfractionated reaction,and lane Bisareactionwithnucleocapsids andnosolubleproteins.
Theposition of the MS-TDI 19SRNAgenomeand anti-genome is indicated. We have previously reported that the NS protein in the
soluble proteinfraction of infected cells can exchange with
the NS protein on nucleocapsids (23, 24). Perhaps the NS
protein in the complex with the N proteincanalso exchange with free NS protein, and during this exchangetwoor more Nproteinsmayassociate with each other if they arein high enoughconcentration. The fact that the more rapidly
sedi-menting forms of N proteinaremostabundantatlater times afterinfection, when the totalamountof soluble N protein is high, supportsthis idea.
Ithas been suggested that the NS protein binds tothe N proteinto preventit from self-associating (1, 9, 14, 24). This wouldsuggestthatthebinding siteonthe N protein for NS is thesamesite that is recognized when N bindstoitself,or thatbinding oftwoor moreNproteinstothemselves masks theNS-binding siteoralters thestructureof the N protein in such a way that NS can no longer bind to it to form an equimolar complex. The fact that N-protein complexes exist inastableform in cells in thepresenceofamolarexcessof NS protein indicates that the formation of N-NS protein complexes from N complexes and free NS is unlikely to
occur.
Thefunction(s) of the N-proteinself-complexes is unclear atpresent. We have beenunable todetect activity of these
complexes in supporting the replication and encapsidation of viralgenomes (Fig. 6) (23, 24). Anargument canbe made, therefore, thatoneorboth of the morerapidly sedimenting forms of the Nproteinmayrecognizeand encapsidate leader
RNA in the cell, but not the replicating genome. Several lines of evidenceindirectly supportthis notion. First, Blum-bergetal. (3) havedemonstrated in vitro encapsidation of leader RNAbyusingapreparation of N protein that exhibits
sedimentationproperties similartothose of themostrapidly
sedimenting form of N protein (Fig. 6). Second, leader RNA isencapsidated ininfected cellsatlatetimes after infection with VSV (5), a time when the level of the N-protein complexes is high.Experimentsto testthis hypothesisarein progress.
Itis of interest that differences inisoelectricpoint between soluble N protein and nucleocapsid-associated N protein have beenreported (12). One major form of N proteinwas
detected in the solubleprotein fraction of cells, withtwoor
three additional forms detected in intracellular and viral nucleocapsids. Additionally, a phosphorylated form of N proteinwas detectedonly in virions. These results suggest thatmodifications of theprimarystructureof Nprotein are
occurring in vivo. These modificationsmaybeatleastpartly responsible for the appearance of the various sedimenting
A
U
I
I
B
~u
Li
...
13 1 13FIG. 7. Sedimentation analysisofnucleocapsid-derived N protein. [35S]methionine-labeled N protein was purified fromVSV-infected cellular nucleocapsids by using guanidine hydrochloride and cesium chloride gradient centrifugation. The proteinpreparation was then
subjectedtosedimentationonglycerol gradients containing1 MNaCl,50 mM Tris(pH7.8),and 1 mM EDTA eitherdirectlyafterpurification
(A)orafter incubationat37°Cfor 30minin0.2 MNaCI-50mM Tris-1 mM EDTA(pH7.8)topromotethe formation of disklikeN-protein
complexes (B). Aportionof theproteins in eachgradientfractionwasanalyzed bySDS-PAGE.
4
19S
- amb4ma n
tomb. 4moqm I.- __o
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.146.475.76.199.2] [image:7.612.146.474.537.681.2]A.
B.
00
SsQ -p0 0NU
O) NUCLEOCAPSIDS 0
0 -0
FIG. 8. Possible relationships between the three differentially sedimentingforms ofthe N protein. For details, see thetext. species ofNproteinwhichwedetect. Itwill be of interestto
analyzeeach of the formsof N proteinforaltered biochem-ical properties thatmay correlate with the observed
differ-encein sedimentation, the ability to self-associate, and the ability to function as substrate for the encapsidation of replicating genomic RNA molecules.
Theexperiments reported hereleadtotheconclusion that the N protein is ableto supportthe replication and encapsi-dationofreplicatingDI RNAgenomesonlyif it iscomplexed
withtheNSproteinina1:1molar ratio. However, Pattonet
al. (21), usinganinvitrosystemprogrammedwith Nprotein mRNA and DInucleocapsids, have reported thatN protein alone can support this process. An explanation for this
discrepancy can beoffered. In both systems, the N protein that supports RNAreplication sediments slowlyonglycerol gradients (11, 21, 23, 24) (Fig. 6). Inthe reportbyPatton et
al. (21), N protein that was allowed to age in vitro before
being tested for the ability to support RNA replication demonstrated a much decreased activity. Subsequent glyc-erol gradient analysis revealed that aging was associated
with an increased sedimentation rate of the N protein (11).
Ourpulse-chasedata (Fig. 1 and2)demonstratethatagingof theN protein occurs in vivo, resulting in the conversion of
slowly sedimenting active N protein (as an N-NS complex)
to more rapidly sedimenting forms unable to support DI
genome replication and encapsidation. These experiments
suggest one oftwo things: either the N protein is modified
immediately after (or during)its synthesis (either covalently
or by binding to the NS protein), and this modification is
slowly reversed to allow N to interact with itselfor even
nonspecifically with cellular RNAs, or the N protein is
synthesizedinanunmodifiedform whichcanassociatewith
NS but not with itself, and a modification to the protein occurswhichfavorsN-N interactions,leadingtothe appear-ance of the more rapidly sedimenting forms of N protein.
Whichever the case, it appears that to have replicating
activity, the N protein mustbe in aform whichis
non-self-complexedand in cellsthe NSproteincanbindtoNtokeep
itinthisactiveform. Inthe coupledtranscription-replication
system of Patton et
al.
(21), N protein that supported DI genomeRNAreplicationwasnon-self-complexed,
and in the experiments reportedhere and elsewhere(23, 24),the activeform of N was shown to be an N-NS
protein complex,
andnot N-N complexes. These experiments point to a crucial
role for the NS proteinin maintaining a pool ofN protein in the cell in a form that can support the encapsidation of replicatinggenomic RNAmolecules, that
is,
as anequimolar
complex ofthese two proteins. Complexes between N and NS which exhibit stoichiometries other than 1:1 have been described (9, 11; R. W. Peluso, unpublished results), but their relevance toviralgenomic RNAreplication and encap-sidation is uncertain.
ACKNOWLEDGMENTS
This research was supported by Public Health Service grant Al 22116 from the National Institute ofAllergy and Infectious Diseases. The excellent assistance of George Rosenberg is acknowledged.I thank Anne Deatly and Frank LaFerla for helpful comments and suggestions on the manuscript.
LITERATURE CITED
1. Arnheiter, H., N. L. Davis, G. Wertz, M. Schubert, and R. A. Lazzarini. 1985. Role ofthenucleocapsid protein in regulating vesicular stomatitis virus RNA synthesis. Cell 41:259-267. 2. Banerjee, A.K. 1987.Transcription and replication of
rhabdovi-ruses. Microbiol. Rev 51:66-87.
3. Blumberg, B. M., C. Giorgi, and D. Kolakofsky. 1983. Nprotein of vesicular stomatitis virus selectively encapsidates leader RNA in vitro. Cell 32:559-567.
4. Blumberg, B. M., C. Giorgi, K. Rose, and D. Kolakofsky. 1984. Preparation and analysis of the nucleocapsid proteins of vesic-ular stomatitis virus and Sendai virus, and analysis of theSendai virus leader-NP gene region. J. Gen. Virol. 65:769-779. 5. Blumberg, B. M., and D. Kolakofsky. 1981. Intracellular
vesic-ular stomatitis virus leader RNAs are found in nucleocapsid structures. J. Virol. 40:568-576.
6. Cartwright, B. 1973. The distribution of virus proteins in BHK 21 cells infected with vesicular stomatitis virus (Indiana C). J. Gen. Virol. 21:407-411.
7. Chatis, P. A., and T. G. Morrison. 1982. Characterization of the soluble glycoprotein released from VSV-infected cells. J. Virol. 45:80-90.
8. Clinton, G. M., B. W. Burge, and A. S. Huang. 1978. Effects of phosphorylation and pH on the association of NS protein with vesicular stomatitis virus cores. J. Virol. 27:340-346.
9. Davis, N. L., H. Arnheiter, and G. W. Wertz. 1986. Vesicular stomatitis virus N and NS proteins form multiple complexes. J. Virol. 59:751-754.
10. Emerson, S. U., and Y.-H. Yu. 1975. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 15:1348-1356.
11. Howard, M., N. L. Davis, J. Patton, and G. W. Wertz. 1986. Roles of vesicular stomatitis virus N and NS proteins in viral RNA replication, p. 134-140. In B. Mahy and D. Kolakofsky (ed.), The biology of negative strand viruses. Elsevier Science Publishing, Inc., New York.
12. Hsu, C.-H., and D. W. Kingsbury. 1980. Vesicular stomatitis virus morphogenesis is accompanied by covalent protein mod-ifications, p. 613-622. In B. Fields, R. Jaenisch, and C. F. Fox (ed.), Animal virus genetics. Academic Press, Inc., New York. 13. Hsu, C.-H., D. W. Kingsbury, and K. G. Murti. 1979. Assembly of vesicular stomatitis virus nucleocapsids in vivo: a kinetic analysis. J. Virol. 32:304-313.
14. Hudson, L. D., C. Condra, and R. A. Lazzarini. 1986. Cloning and expression of a viral phosphoprotein: structure suggests vesicular stomatitis virus NS may function by mimicking an RNA template. J. Gen. Virol. 67:1571-1579.
15. Irving, R. A., and H. P. Ghosh. 1982. Shedding of vesicular stomatitis virus soluble glycoprotein by removal of carboxy-terminal peptide. J. Virol. 42:322-325.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.81.269.72.304.2]16. Kang, C. Y., and L. Prevec. 1970. Proteins of vesicular
stoma-titis virus. II. Immunological comparisons of viral antigens. J. Virol.6:20-27.
17. Kang, C. Y., and L. Prevec. 1971. Proteins of vesicular
stoma-titis virus. III. Intracellular and extracellular appearance of
virus-specific proteins. Virology46:678-690.
18. Kingsford, L., and S. U. Emerson. 1980. Transcriptional activi-ties of different phosphorylated species of NS protein purified from vesicular stomatitis virions and cytoplasm of infected cells.J. Virol. 35:1097-1105.
19. Knipe, D. M., D. Baltimore, and H. F. Lodish. 1977. Separate pathways of maturation of the major structural proteins of vesicular stomatitis virus. J.Virol.21:1128-1139.
20. Little,S.P., and A. S. Huang. 1977. Synthesisanddistribution of vesicular stomatitis virus polypeptides in the absence of
progenyproduction. Virology81:37-47.
21. Patton, J. T., N. L. Davis, and G. W. Wertz. 1984. N Protein alone satisfies the requirement for protein synthesis duringRNA replicationof vesicular stomatitis virus. J. Virol. 49:303-309. 22. Peluso, R. W., and S. A. Moyer.1983. Initiation and replication
of vesicular stomatitis virusgenomeRNA inacell-freesystem.
Proc. Natl. Acad. Sci. USA 80:3198-3202.
23. Peluso, R. W., andS.A.Moyer.1984.Vesicular stomatitis virus proteins required forthe in vitro replicationof defective
inter-fering particlegenomeRNA,p.153-160.InD. H. L.Bishop and R. W. Compans (ed.),Nonsegmented negative strand viruses. AcademicPress, Inc., New York.
24. Peluso, R. W., and S. A. Moyer.1988.Viralproteins required for the in vitroreplication of vesicular stomatitis virus defective interfering particlegenomeRNA.Virology 162:369-376.
25. Peluso, R. W., and G. H. Rosenberg. 1987.Quantitative electro-transfer ofproteinsfromsodium dodecyl sulfate polyacrylamide gelsontopositively charged nylonmembranes. Anal. Biochem.
162:389-398.
26. Rubio, C., C.Kolakofsky, V. M.Hill,and D. Summers. 1980. Replicationandassemblyof VSVnucleocapsids: protein
asso-ciated with RNPs and the effects ofcycloheximideon replica-tion.Virology105:123-135.
27. Soria,M., S. P. Little, and A. S. Huang. 1974. Characterization of vesicular stomatitis virusnucleocapsids. I. Complementary
40S RNA molecules innucleocapsids. Virology61:270-280. 28. Wagner, R.R., M. P. Kiley, R. M. Snyder, and C. A.
Schnait-man. 1972. Cytoplasmic compartmentalization of the protein
and RNAspeciesof vesicular stomatitis virus. J. Virol. 9:672-683.
29. Wagner, R.R., R. M.Snyder,and S. Yamazaki. 1970. Proteins of vesicular stomatitis virus: kinetics and cellular sites of synthesis. J. Virol. 5:548-558.


![FIG. 6.fractionsTheproducedofnucleocapsidsseparated [3H]UTP. Activity of fractionated soluble proteins prepared at 4 and 7 h p.i](https://thumb-us.123doks.com/thumbv2/123dok_us/1336553.87401/7.612.146.474.537.681/fig-fractionstheproducedofnucleocapsidsseparated-utp-activity-fractionated-soluble-proteins-prepared.webp)