Vol. 64, No. 10
Activity of Herpes Simplex Virus
Type 1
Latency-Associated
Transcript
(LAT) Promoter in
Neuron-Derived
Cells: Evidence for
Neuron
Specificity and for
a
Large
LAT
Transcript
JOHN C. ZWAAGSTRA,1 HOMAYON GHIASI,"2 SUSAN M. SLANINA,1 ANTHONY B. NESBURN,"2
S. C. WHEATLEY,3 K. LILLYCROP,3 JOHN WOOD,4 DAVID S. LATCHMAN,3 KAMALESH PATEL,'
AND STEVEN L. WECHSLER'2*
Ophthalmology Research, Cedars-Sinai Medical Center, Halper Building 111, 8700 Beverly Boulevard, LosAngeles, California 90048-18691*;Department ofOphthalmology, UCLA School of Medicine, LosAngeles, California 900242;
andDepartment of Biochemistry, University College and Middlesex School of Medicine,3
andSandozResearch Institute,4 London, WCIE6BNUnited Kingdom
Received 4 August 1989/Accepted 17 July 1990
By using chloramphenicol acetyltransferase (CAT) assays in neuron-derived cell lines, we show here that
promoter activity associated with the herpes simplex virus type 1 latency-associated transcript (LAT) had
neuronalspecificity. Promoteractivityin these transient CATassayscoincided withaDNAregion containing
excellent RNApolymerase II promoterconsensus sequences. Primerextension analysis ina LAT
promoter-CATplasmidconstructplacedthestartoftranscription about28 nucleotidesfrom thefirstT in theconsensus
TATAboxsequence. Neuronalspecificityof thispromoterwassuggested byexamining theeffectofsequences
upstream of the promoter on CAT activity in neuronal versus nonneuronal cells. In nonneuronal cells,
promoter activity was decreased 3- to 12-fold with the addition of upstream sequences. In contrast, in
neuron-derived cells, the addition of upstream sequences did not decrease promoter activity. The LAT
promoterpredicted byourtransientCATassayswaslocatedover660 nucleotidesupstreamfromthe5'endof
thepreviously mapped 2-kilobase (kb) LAT. This unusual location wasexplained by in situ and Northern
(RNA) blot hybridization analysesthat suggested that LATtranscriptionbegannearthepromoterdetectedin
ourCATassays,ratherthannearthe5'endofthe2-kb LAT. In situhybridizationwithneuronsfrom latently
infected rabbits detected small amounts of LAT RNA within 30 nucleotides of the consensus TATA box
sequence. Thissuggested that LATtranscription begannearthisTATA box. Northern blothybridization of
RNA from ganglia of latently infected rabbits revealed a faint8.3-kb band ofthesame sense as LAT. We
conclude that (i) the LAT promoter has neuronal specificity, (ii) the LAT promoter is located over 660
nucleotides upstreamofthe5' endofthepreviously characterized stable 2-kb LAT, (iii) LAT transcription
beginsabout 28 nucleotidesfromthe first ToftheconsensusTATA boxsequenceand extendstonearthefirst
available polyadenylation site approximately 8.3 kb away, and (iv) this 8.3-kb RNA may be an unstable
precursorof themorestable 2- and 1.3-kb LATs.
Following primary infection in humans, herpes simplex
virustype1(HSV-1) establishes latent infections insensory
neurons (14). During HSV-1 latency, detectable viral
tran-scription is limited to an area within the genomic long
repeatsinthevicinityof theimmediateearlygeneICP0(20, 21, 25).Atleasttwoabundantandstablelatency-associated transcripts (LATs) (2 and 1.3 kilobases[kb])thatsharetheir
5' and3' endsarederivedbyalternativesplicing (30).These
LATspartially overlapthe3' endof ICP0andareantisense
(complementary)totheICP0mRNA(20, 25, 29, 30).The 5'
endof the stable 2-kb(and 1.3-kb)LATin the internallong
repeathasbeenmapped towithinafew bases (28-30). This
position corresponds to nucleotide 119462 of the genomic sequence(9, 15).Asecondcopyof the LATgeneispresent in the corresponding location ofthe terminal long repeat.
RecentreportswithsomeLATdeletionmutantssuggestthat
LAT may play arole in reactivation of the virus from the
latent state (2, 7, 24).However, this is notsupported byall
LATmutants (1, 6).
Basedonsequenceanalysis,weinitiallyproposedthat the
LATpromoteris located over660 nucleotidesupstream of
*Correspondingauthor.
themapped5'end of the stable LATs (30, 31),andlater, by using chloramphenicol acetyltransferase (CAT) assays, we
confirmed that in Vero cells, LAT promoter activity
coin-cides with this region (32). This location for the LAT
promoter is further supported by more recent studies in
which viral mutants lacking the predicted promoter region
failed toproduceany LATRNAduringlatentinfections (2,
7, 11, 24).
Since LAT is the only HSV-1 gene that is abundantly
expressed during neuronallatency (20), the LAT promoter
must becontrolled differently fromother HSV-1gene
pro-moters(22, 23). Furthermore,the LAT promoter is likelyto
have neuronalspecificity, since LAT is abundantinlatently
infected neurons (20, 25) but is present at onlylow levels
duringacutetissueculture infection(22).We have therefore
extendedourstudies of the LAT promotertoneuron-derived
cellstoconfirmthelocationofthe LATpromoterandtolook
for neuronal specificity.
We show here thatinneuron-derived celllines, promoter
activity mappedto thesamelocationasit did inVero cells. Furthermore,wefound that in nonneuronalcells,sequences
upstreamof the TATA box decreased promoter activity in
cis, while in neuron-derived cells, inhibition by these
cis-acting sequences was not observed. These upstream
se-5019
JOURNALOFVIROLOGY,Oct. 1990, p.5019-5028
0022-538X/90/105019-10$02.00/0
Copyright ©1990,American Society for Microbiology
on November 10, 2019 by guest
http://jvi.asm.org/
quences may confer neuron specificity on the HSV-1 LAT
promoter invivo. Wealso show here that LAT transcription began near the promoter and appeared to continue for approximately 8.3 kb, the location of the first-occurring consensus polyadenylation site. A likely interpretation of
thisdata isthat this 8.3-kb RNA may beanunstable primary LAT transcript that gives rise to the previously mapped, relatively abundant 2- and 1.3-kb LAT transcripts.
MATERIALS AND METHODS
Cellsandvirus. Plaque-purified herpes simplex virustype 1 (HSV-1), strain McKrae, was grown as previously
de-scribed(20) and was usedforallinfections.Cellsweregrown
asmonolayers in minimal essentialmedium, Dulbecco mod-ified Eagle medium, or F12 (GIBCO Laboratories, Inc.) supplemented with 10% fetal calf serum and antibiotics. Neuroblastomacells(NB41A3;American TypeCulture
Col-lection CCL147)are ofmouseorigin. Theseneuroblastoma cells retain several neuronal markers, including acetylcho-linesterase activity. Inaddition, they arenonpermissive for HSV-1 infection (27). Immortalized neurons weremade by fusing
H18TG2
cells, a5-azaguanine-resistantmouseneuro-blastoma cell line, with neonatal rat dorsal root ganglia
neurons. These cells are nonpermissive for lytic HSV-1
infectionand express LATfollowingHSV-1
infection
(S. C.Wheatley,C.Dent,K.Lillycrop,L. M. Kemp,J. N.Wood,
and D. S.Latchman, submittedforpublication).
Plasmids. Allrestrictionfragmentswere derivedfromthe BamHI B restrictionfragmentfrom HSV-1 strain F(16).The LAT genefromposition -2592to+663relativetothe 5'end of the stable 2-kb LAT was divided into five restriction fragments. FragmentsAtoC and thedetailsof theircloning
in the proper orientation in front of the CAT gene within
plasmid pSVOCAT(4) have beenpreviously described(32). Fragments A+ andA++ weresimilarly cloned and consist
of
fragment
A with additional upstream sequences asde-tailed in the legend to Fig. 2 and in
Fig.
3. For someexperiments, similar constructs were made by using a
dif-ferent promoterless CATplasmid,
p1O6CAT
(3),in place of pSVOCAT.CAT assays andquantitation procedures. CAT assays and
quantitation procedures have been previously described
(32). Briefly, cell monolayers atapproximately 60% conflu-ency, on 60-mm plates, were transfected with CAT
con-structs by thecalcium phosphate precipitation method (5).
After46 h, the cells were harvested and cellextracts were
prepared. Within an experiment, equal cell numbers were
used and, if necessary, corrections were made for the
amount ofproteinpresentin the extracts. Acetylatedforms
of
['4C]chloramphenicol
were detected by thin-layer chro-matography and subsequent autoradiography. The amountofacetylated andunacetylated chloramphenicol was
quanti-tated by excising the spots from the thin-layer plates and
counting in a liquid scintillation counter. In some
experi-ments, samples ofcell extracts were analyzed by DNA dot blothybridizations with aCAT-specific probe to determine the relative amount of CAT DNA that had entered the cells
during the transfection. Within a cell line, no differences
were seenbetween the transfection efficiencies of fragments
A+ +, A+, or A. Thus, differences between the CAT
activitiesof these plasmids withinacell line were not a result ofdifferences in transfection efficiency. The differences in
transfection efficiencies between cell lines was partially compensated for by using 10 ,ug of each plasmid in
immor-talized neurons, BHK cells, and L cells; 5 ,ug of each
plasmidin neuroblastoma cells and CV-1cells;and 2.5,ugof eachplasmid in Verocells.
Primer extension. As modified from a procedure of P. Krause and J. Ostrove (personal communication), 10 ,ug of RNAfrom transfected Vero cells(isolated bythe
guanidin-ium-cesium chloride method [8])was suspendedin 10
RI
of 20 mMTris-hydrochloride, pH 7.6-100 mM NaCl-0.1 mM EDTA. A 10-ng portion of 32P-end labeled CAT primer (approximately 5 x105
cpm) was added. The sequence of theprimer from 5' to 3' wasGATGCCATTGGGATATAT CAACGGT. This sequence is complementary to the CAT mRNA sequence located between nucleotides 127 and 151 downstream from the first T of the LAT TATA box in theCAT constructusedfor the transfection. This location was
confirmed by partial sequencing of the plasmid and
corre-sponds to CAT nucleotides 28 to 52 relative to the ATG codon at which CAT translation initiates. The difference between these numbers is the result ofaHindIll linker, a
multiple cloning region, and noncoding CAT sequences between the end of the LAT sequences and the startofthe structural CAT sequences in this plasmid. The reaction mixture was heat denatured for 3 minat90°C, hybridizedat
55°C for 10 min, and slowly (approximately 1 h) cooled to
30°C. Primer extension of the hybridized primer was done with 20 U of cloned Moloney murineleukemia virus reverse transcriptase(Bethesda ResearchLaboratories, Inc.) at 37°C for 1 haccordingtotheinstructions of the manufacturerby
using 2 mM deoxynucleoside triphosphate, 100 ng of bovine
serumalbumin per Pd, and 20U of RNasin(PromegaBiotec)
per 20-pdl reaction. The reaction was stopped by adding
EDTA to 10 mM. The extendedprimer wasprecipitated with 2 volumes of 95% ethanol, washed with 70% ethanol,
sus-pended in formamide-dye loading buffer, heat denatured at
95°C for 3 min, quick chilled on ice, and run on a 10%
acrylamidesequencing gel containing 7 Murea.Thegelwas
thenprocessed for autoradiography. In one experiment the same oligonucleotide used in the primer extension reaction was used to prime a sequence reaction from the A-CAT plasmid by using a Sequenase DNA sequence kit from United States Biochemical Corporation. Products from the sequence reaction were run next to the primer-extended product on a10% sequencing gel containing 7 M urea.
Rabbits.New Zealand White male rabbits(approximately
2 kg each) were used for all animal experiments. These animals develop a primary and recurrent herpetic disease (13) which mimics HSV-1 keratitis in man.
Latent ganglionic HSV-1 infections. Latent ganglionic
HSV-1 infections were done as previously described (20). Briefly, rabbits were bilaterally infected without corneal scarification by placing approximately 1 x 105 to 2 x
105
PFU of virus into the conjunctival cul-de-sac, closing the eye, and rubbing gently for 30 s. Rabbits surviving after 4 weeks were considered latently infected (12).
Rabbittrigeminal ganglia. Rabbit trigeminal ganglia were taken from sacrificed rabbits and immediately placed in liquid nitrogen for RNA extractions for Northern (RNA) blots or into the preservative periodate-lysine-paraformalde-hyde (26)for sectioning prior to in situ hybridizations.
In situ hybridizations. Fixing, embedding, and cutting sections of trigeminal ganglia were done as described previ-ously (19). Hybridizations to identify RNA were done aswe previously described (20, 30) by using 32P-labeled synthetic oligonucleotides as probes. Slides were exposed to photo-graphic emulsion for 2 to 3 days. Pretreatment ofslides with RNase, but not DNase, eliminated hybridization. Three to five sections of ganglia from each of four latently infected
on November 10, 2019 by guest
http://jvi.asm.org/
HSV-1 LAT PROMOTER IN NEURONAL CELLS 5021
rabbits and two uninfected rabbits were examined with each probe. No hybridization with any of the probes resulted in an accumulation of grains over any neurons from uninfected rabbits. Positive probes showed an accumulation ofgrains
over the nuclei of some neurons from latently infected
rabbits compared with the amount of background grains on
the slide and compared with uninfected neurons (see Fig. 5). Positive probes detected positive neurons from at leasttwo
latently infected rabbits, as judged by detection of at least one positive neuron on at least two different slides from a
givenrabbit.
Northernblot hybridizations. Total RNA was isolated from
uninfected orlatently infected rabbit trigeminal ganglia fro-zeninliquidnitrogen orfrom uninfected or acutely infected (multiplicity ofinfection of 20; 18 h postinfection) CV-1 cells as previously described (20). Northern blot hybridizations were done as we previously described (20, 30).
Hybridization probes. Random-primed labeling with 32p
wasdone on linearized plasmids following the instructions of the manufacturer (Amersham Corp.). Synthetic
oligonucle-otides (20-mers) were synthesized by using beta-cyanoethyl
phosphoramidite chemistry on a Pharmacia Gene
Assem-bler.The sequencesof the 20-mers were based on sequences
for HSV-1 strains 17 syn+ (15) and F (31). End labeling of
oligonucleotides with
[y-32P]ATP
was done as described previously (8).RESULTS
CAT activityinneuron-derivedcells.Byusing CAT assays, we previously showed that in Vero cells, LAT promoter
activity coincides with RNA polymerase II promoter con-sensus sequences (32)that arelocatedover 660nucleotides upstreamfromthe 5' endofthe stable 2-kb LAT (30, 31). To
determinewhether LAT promoteractivity in neuron-derived cells mapped to the same unusual upstream region, we
assessed the ability of portions ofthe LAT gene to
consti-tutively functionas apromoterinimmortalizedneuronsand in neuroblastomacells. DNArestrictionfragments from the LAT gene were individually cloned into the plasmid
pSVOCAT (4) in front of the CAT structural gene. The proper orientation of each fragment was confirmed by
re-striction enzymeanalysis.
Cells were transfected with individual CAT constructs, and 46 h later CATactivity was measured. Representative
results are shown in Fig. 1. Thelocation ofeach fragment relative tothe 5' endofthe stable 2-kb(and 1.3-kb) LAT is
shownatthe bottom of thefigure.Numbers above each lane indicate the percent conversion of
[14C]chloramphenicol.
Fragment A (-940 to -662) had CAT activity in immor-talizedneurons (Fig. la, lane A) and in neuroblastoma cells
(Fig. lb,laneA)thatwascomparabletothatofpSV2CAT (a
strongpositivecontrolcontaining CAT under the control of the simian virus 40 early promoter) (Fig. la and b, lane
pSV2) in the same cells. This was indicative of
significant
promoter
activity
byfragment
Ain these cells.Fragments B andC, which encompass theregionfrom-662 to+663, hadno CAT activity in either neuron-derived cell line. These results were identical to those we previously reported in Vero cells (32). This mapped the in vitropromoteractivity
associated with the LAT gene to the same location (i.e.,
within fragment A) in two neuron-derived cell lines as in Verocells. As shown in theexpandedviewatthe bottom of
Fig.
lb,
fragment A contains several consensus sequencesnormally associated withanRNApolymerase II promoter,
including aTATAbox, a potentialCAATbox, and several
a
rmtnortahzed Neurons
b
NEtArobtlasI(m
Iekls
139 <05 <05 12.4 %Conversion
* Acetylated
** * * ~~Unacetylated
A B C p)SV2
1 <05 <0.5 9.7 %.,Conversion
* Acertylated
* * * Unacelylaled
A B C pSV2
.94(:1 (iP92 151 +43
HarIII Pviitl t1FfllT Hvin * 2kbstabhe LAT SpiSpi CAMT .TATA
TAATGAIAT1688 1l 6ii
FIG. 1. CATactivity in neuron-derived cells ofdifferent DNA
fragments derived from the LAT gene. Different portions ofthe
LAT gene (32) cloned in front ofthe gene for CAT (in plasmid
pSVOCAT [4])wereindividually transfected into immortalized
neu-rons(a) or neuroblastoma cells(b), and CATactivitywasmeasured
asdescribed inMaterials and Methods(lanesA, B, andC). Lanes
pSV2areplasmid pSV2CAT (4), containing the simian virus40early
promoter, as apositivecontrol.Transfectionsweredonewith 10 ,ug
of eachplasmid in immortalizedneuronsand 5j±g of eachplasmid in
neuroblastoma cells. Acetylated forms of [14C]chloramphenicol
weredetectedbythin-layer chromatography andsubsequent
auto-radiography. TheLATgenefragments aredesignatedAtoC (32)
and arerepresentedschematicallyatthe bottom ofpanelb(alsosee
Fig. 3). Nucleotidepositionsarerelativetothe 5'end ofthestable
2-kbLAT(nucleotide+1) (28, 30), whichcorrespondstonucleotide
119462 ofthe HSV-1 genome (9, 15). Numbers above each lane
indicatethe percentconversion ofunacetylated
[14C]chloramphen-icoltoacetylated forms.
Spl sites (30, 31). This region also contains a sequence
having significant homology to a Vmw65 binding site
(TAATGARAT) (32).
Sequencesupstream offragmentAdecrease LAT promoter
activity in nonneuronal cells. To look forpotential cis regu-lation by sequences upstream of the LAT promoter, two
additionalCAT constructs were made.FragmentA+(-1271
to -662) consisted offragmentAplus 331 upstream
nucle-otides. Fragment A+ + (-2592 to -662) consisted of
frag-mentAplus 1,652 upstreamnucleotides.
Fragments
A+ +, A+, andAhaveacommon3'end.FragmentsA+ and A+ +were cloned into plasmidpSVOCAT in thesame manneras fragment A.
The effect of these upstream sequences on promoter
activityinneuronal and nonneuronal cell lineswasexamined
VOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.333.545.75.381.2]5022 ZWAAGSTRA ET AL.
(83) fl31 "03 f8 89T 1,. 3: 5 : OKr2 90 1
859 93 7 12 2 I3 3 5 : 65 8 3062t7.4
.e
4 1 _i4' . {
-i;,.
ofrvL
acetvi1ated
Nont
a--etya DNwA _a-e
FIG. 2. Theeffect ofupstream sequences onCATactivity drivenbythe LAT promoterinneuron-and nonneuron-derivedcells. DNA
fragments containingthe LAT promoterregion and differentamountsof upstream sequenceswerecloned intopSVOCATatthesamelocation
usedfor the fragments in Fig.1.FragmentsA, A+,and A++havea common3' end(nucleotide-662relativetothe 5'endof the stable 2-kb
LAT). This is26nucleotides downstream from the firstTintheTATAboxsequence.FragmentAis 278 nucleotideslong. Fragment A+ is
609nucleotideslong. FragmentA++is 1,930 nucleotideslong. ThesefragmentsareshownschematicallyinFig.3.Plasmidswereindividually
transfected into neuron-derived cells (neuroblastoma or immortalized neurons) or nonneuron-derived cells (BHK, Vero, CV-1, or L).
Representative resultsareshown.Immortalizedneurons(lanes4to6),BHKcells(lanes7to9),andLcells(lanes15to17)weretransfected
with10 tg of eachplasmid. Neuroblastoma cells(lanes1 to3)and CV-1 cells(lanes12and13)weretransfectedwith 5 ,ugofeachplasmid,
andVerocells(lanes10 and11)weretransfected with2.5 ,ugof eachplasmid. Numbers above the lanes indicate thepercentconversion of
[14C]chloramphenicol. Numbers inparentheses indicatetheactivity relativetofragmentAfor each cellline.
byCAT assays. Representativeresultsareshown inFig. 2. The percentconversionof['4C]chloramphenicolis indicated
above each lane in Fig. 2. The numbers in parentheses
indicatethe percentactivity of the otherplasmids relativeto A(100%) within each cell line. In this particular experiment, the addition of the upstream sequences contained in frag-ments A++ and A+ had little effecton the activity of the LAT promoterin neuron-derived cell lines (83 to 131% of
fragment A activity; lanes 1 to 6). In other experiments, these upstream sequencesincreasedpromoteractivityupto
threefold (summarizedinFig. 3). In contrast, in BHK cells
(lanes7 to9),theupstream sequencescontained in fragment
A++and A+reducedfragmentA's promoteractivityabout
threefold. The effect of upstream sequences was further
examined in (nonneuronal) Vero and CV-1 cells, using fragment A+ +. The upstream sequences caused a reduction inCATactivity of about 6- to 12-fold in these cells (lanes 10 and 12).
Sincebothof the neuron-derived cell lines we used were
ofmouseorigin, itwasimportant to compare these results to
those in nonneuronal cells of mouse origin. In L cells (a
commonly used mouse cell line) there was a greater than
threefold reduction in LAT promoter activity by upstream sequences(Fig.2,lanes 15 and 16).Thiswassimilar to BHK
cells(Fig. 2,lanes 7to9) and confirmedthatthis
phenome-non wasrelatedtotheneuronal andnotthespeciesorigin of
the cells.
To partially correct for transfection efficiency between
differentcelllines,different amountsof plasmid were used in
different cell lines (within a cell line, equal amounts of all
plasmidswereused). We used 2.5 ,ug of plasmid per plate for Vero cells, 5 ,ug of plasmid per plate for CV-1 and
neuro-blastomacells, and 10 ,ug of plasmid per plate for
immortal-ized neurons, L cells, and BHK cells. Within a given cell
line,
these concentrations gave similar CAT activity withfragmentAand with the positive control plasmid pSV2CAT
(Fig. laand b, lanes A and pSV2 for neuronal cells; Fig. 2, lanes 16 and 17 for L cells; not shown for other nonneuronal
cells). Thus, in CAT assays, fragment A (containing the
minimalLATpromoter) had promoter activity similar to that
ofpSV2CAT and relative topSV2CAT appeared to be an
equally effectivepromoterin all celltypes.
To ensure against the unlikely possibility of a
plasmid-specific artifact, some experiments were also done with fragments cloned into a different CAT-containing plasmid
(p106CAT [3]). Similar results were obtained (not shown).
1RL UL IRL IRSUS w
GenomeI1RL|~
Ger r
Nuctkdso 116,000 117,000 11,000 119,000 120,000 121,000 12Z000
Po6skin
-3000 -2000 -1000 +1 +1000 +2000
Poskionrebbve
toS' o 2kbLAT
2kbstableLAT 2kbloL3T
CATactiviy
inreuronal derh,scells
TATAbox 1.3 kb LAT A++ _ 8 (83-190%)
A+ (89-306%)
A _(100%) B
C CATactivityin A++ (S30%)
ron4Wuronal A+ i_ (1035%)
ceil A _ (100%)
Be
cc
FIG. 3. LAT genefragmentsusedinCATconstructs.The upper
portion of the figureshowsthe HSV-1genomicorganization inmap
units (mu). The expanded region indicates nucleotide positions
relativetotheentire HSV-1DNAsequence(15)and relative tothe
5'end (+1)of thestable 2-and 1.3-kb LATs. The locationsofthe TATAbox sequences and of the 2- and1.3-kbLATs areshownfor
reference. The LAT genefragments are shown as horizontal
rec-tanglesand arelabeledA++, A+,andA toC, as in Fig. 1 and 2.
Solid rectangles indicate CAT (promoter) activity comparable to
that of pSV2CAT in the same cells. Dotted rectangles indicate
reducedactivity. Openrectanglesindicatenoactivity. Numbers in
parenthesesshow the range ofactivity ofA++andA+relativeto
Ainover5independent experiments.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.158.467.72.218.2] [image:4.612.324.554.409.599.2]HSV-1 LAT PROMOTER IN NEURONAL CELLS 5023
a.
b.
LATsense ge
23
4 3:
200
- 23cx
-23
* oo
j82
.6 JA7
(P,asrnd,,
3 4 5
c-FIG. 4. Primer extension mapping of the transcriptional start site of the LAT promoter-CAT plasmid. Primer extension was done as
described in Materials and Methods by using a 32P-end-labeled 25-nucleotide primer complementary to CAT mRNA, and the gel was
processedforautoradiography. All lanes represent the result of primer extension with RNA from transfected Vero cells. (a) Lane 1,p106CAT (promoterless CAT); lane 2, plasmid A(p1O6CATwithfragment A); lane 3,untransfected control. Marker lane is32P-end-labeledHinfl-cut
ijx174 DNA. The size of the primer extension product is approximately 123 nucleotides, corresponding to a transcriptional start site
approximately28nucleotidesfrom thefirst T of the TATA box sequence. (b) The same oligonucleotide used for primer extension experiments
was used as a primer in a set of sequencing reactions (see Materials and Methods). This generated the sequence shown in lanes 1 to 4, which
is complementaryto the RNAproduced by this plasmid with the LAT TATA box region as a promoter. The predicted sequence of theregion
fromthe TATA box to the beginning of theHindllllinker used to clone the LAT sequences into the plasmid is shown to the left of the gel.
Lane5 shows theresultof aprimerextension experiment similar to that in panel a. The extended primer comigrates with the first T in the
HindlIl linker. Thisis 28 nucleotides from the first T in the TATA box.
Theunlikely possibilitythatthe decrease in promoter
activ-ity seen with A++ and A+ could be due to a decrease in
transfection efficiency (compared with A)of these plasmids innonneuronal compared with neuronal cells was addressed asfollows.In someexperiments,aportionof the cell extract used for the CAT assay was monitored for transfection
efficiency by DNA dot blots by using a CAT-specific
32P-labeled DNA probe. As expected, although transfection
efficiencies differedbetween celllines,withinagiven cell no
differences in transfection efficiency that could account for the decreased promoteractivity of A++ or A+ compared with A were detected (not shown).
In vitro LAT transcriptional start site. To determine the
approximatelocationofthestartoftranscriptionin the CAT constructs, primer extension experiments were done as
described in Materials and Methods. RNA was prepared from transfectedoruntransfected cells andhybridizedwitha
32P-end-labeled25-nucleotide-longsynthetic oligonucleotide primer. The primer was complementary to CAT mRNA sequences located 127to 151 nucleotides downstreamfrom
the first T of the LAT TATA box (see Materials and
Methods). The primer was extended with reverse
tran-scriptase,denatured,andsizedon apolyacrylamidegel (Fig. 4a). Lane 1shows the result of
primer
extension withRNA preparedfrom cells transfected withp1O6CAT(promoterlessCAT,withoutfragment A).Lane3showsaprimerextension
reactionwith RNA from cells thatwere nottransfected. No
product is seen in either control lane. Lane 2 shows the result ofaprimer extensionreaction with RNA from cells transfected with
p1O6CAT
containing fragment A. We esti-mated the size of themajor extendedproduct asabout 123 nucleotides. Smaller faint bands are probably the result of premature termination of the primer extension reaction.Since the primerwas complementary to nucleotides 127to
151 relative to the first T of the TATA box, a size of 123
nucleotides placed the start of transcription at about 28
nucleotides from the first T of the TATA box (151 minus 123). This would be HSV-1 nucleotide 118802(9) or nucleo-tide-660 relativetothe 5'end of the stable LATs. The LAT sequence in this region is "...GCCGATCGCGG...," with theunderlined Tbeing theapproximatetranscriptional start site.
To further confirm thetranscriptional startsite, the result ofa similarprimerextension reactionwas sizedrelative to
the sequenceof the CATplasmid on asequencing gel (Fig.
4b). The CATplasmid sequencewasobtainedbyusing the
same oligonucleotide primer to prime a sequence reaction
from the A-CATplasmid (seeMaterials andMethods). The
extended primer comigrated with the underlined T in the sequence "...CGGCGITCGAA..." onthis gel. The
corre-sponding LAT sense strand sequence is "...GCCGCAAG CTT.." This is the samelocation relativetothe TATA box
(28 nucleotides fromthefirstT)asestimated fromFig.4a. In this case, this base isanA, notthe T foundatthis location in the LAT sequence. This A is the second base in the
HindIlI linker usedtoclone the LAT Afragmentin this CAT
construct.
Our finding that in vitro transcription started approxi-mately28nucleotides downstream from theLAT TATAbox
strongly supportsthe notion that CAT
transcription
in these plasmids is under the control of the LAT sequences infragment A. Furthermore it demonstrates that the LAT sequences neartheTATA boxare
capable
offunctioning
as a typical promoter in vitro. This supports the notion thatfragmentAcontains the LAT promoter and makes it
likely
thatduringneuronallatencyin
vivo,
LATtranscription
maybegin at or nearthe sameposition (i.e., -660).
In situ hybridization near the LAT promoter. The CAT
VOL.64, 1990
I
A--A
A
A-
G--
3--s
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.168.457.77.270.2].,:~Aor
~
w.,i't.,'o 4wowaft.
4w
.4
I
.1
t / <w ss\s,* 4'
SK,..
p~~~~~P
It,
ol . C 4
S.
, ¢S t\
_ J~~~~~~wT-1.
le. _ , is_
4
_lk
,.,'
A 4 .h
Ia
--- ..#
A"~*
*~~~~~~~
--"4
. --~~~~~~~~~~~~~~~~~~~~~~~~~~
,~~~~~~
:r
--,
G4 l S* t #,*,
~~ ~
.i~
~
~
~
i~
s,
< X' i*,~~~~W
-4'~~~~~~~~~4
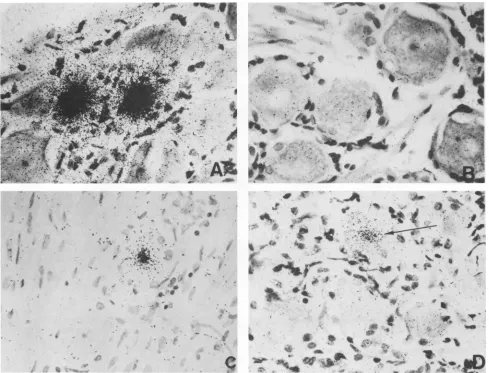
FIG. 5. Insituhybridization of trigeminal ganglia fromlatentlyinfected rabbits. Trigeminalgangliafrom latently infectedoruninfected
rabbitswereremoved, sectioned, and processed for in situ hybridization as described inMaterialsand Methods. Allprobes weresynthetic
oligonucleotides(20-mers) end labeled with[32P]ATP.(A)Section fromalatentlyinfected rabbit hybridizedto aprobecorrespondingto(the
complement of) nucleotides +1292 to +1311from within the stable 2-kb LAT. (B) Uninfectedrabbitwith the same probe as inpanelA.(C)
Latently infected rabbitwith aprobe fromtheregionbetween the TATA box and the5' end of the stable 2-kb LAT (probe 8 from Fig. 6).
(D) Latently infected rabbit with a different probe from the region between the TATA box and the5'end of the stable 2-kb LAT (probe 11
from Fig.6). The arrow points to a faintly positive cell.
assaysand the primer extension results reported above both
stronglysuggestthat the LAT promoter is located over 660 nucleotides upstream of the start of the stable LATs. To look for small amounts of LAT RNA between the LAT TATA
box and the5'end ofthestableLATs,wecarefully analyzed
a series of in situ hybridizations to sections from trigeminal
gangliaofrabbits latently infected with HSV-1.
Figure5shows representative in situ hybridization results in which 32P-end-labeled synthetic oligonucleotides were used as probes. These 20-mers were based on the DNA sequenceof this region (15, 31) and were constructed so that RNAdetected with these probes would be the same sense as LAT RNA. To eliminate possible false positives, computer
analysis was done to ensure that no 20-mer would have
homology to any other portion of the LAT region (30). Several 20-mers were also rejected because they hybridized
to neurons from uninfected rabbits. Panel A shows the
strong hybridization typical of a probe corresponding to a
region within the stable 2-kb LAT to a section from the
trigeminal gangliaof a rabbitlatently infected withHSV-1. Panel B shows lack ofhybridization with thesameprobeto
a section from an uninfected rabbit. Panels C and D show
fainthybridizationtolatentlyinfected sectionsusing probes
betweenthe LATpromoterand the5' end of the stable2-kb
LAT.The numberof grainsin these positive cellsisgreatly reducedcompared with panel A, indicating muchless LAT RNA in the positive cell. In addition, the number ofcells showingfainthybridizationwasonlyabout 10% of that seen
withprobes hybridizing to the 2-kb LAT.
The faint hybridization seenwiththe probes in panels C andD(Fig. 5)was atthe limitof detectability in our latently infected rabbit system. Under normal circumstances, in which one expects to see numerous, strong hybridization signals, these probes might have been scored as negative. However, it was clear that these probes were not simply "negative."Inthisanalysis we therefore consideredaprobe
positiveifatleastone neuron wasfaintly positive (asinFig.
SC orD) on atleasttwo sections from each of at leasttwo
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.64.553.71.444.2]HSV-1 LAT PROMOTER IN NEURONAL CELLS 5025
-6100 // -900 400 -700 -00 -500 -400 -300 -200 -100 +1 100
II/ 'I
TATA
Probes 1 2 3
BatnHI EooRv LAT- RNA M
5'
4 5 6 78910
H ES
Start 1.3& 2 kbLAT
11 2b>
3,
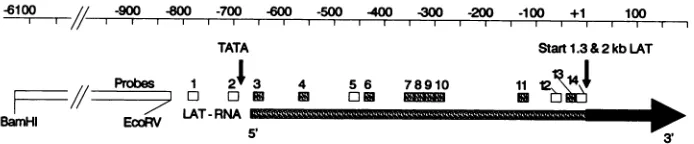
FIG. 6. Detection of low-abundance RNA between the TATA box sequence and the 5' end of the stable 2-kb LAT by in situhybridization. Results from the experiment partially shown in Fig. 5are shown schematically. In situ hybridization with each32P-endlabeled20-mer was
performedon 3 to5 sections of trigeminal ganglia from each of four rabbits latently infected with HSV-1. The 20-mers were based on the
sequenceofthis region and were of complementary sense to LAT RNA. The approximate locations of the probes relative to the 5' end of thestable 2-kb LAT (+1) are shown at the top.Exactlocations are: 1, -789 to -770; 2, -707 to -688; 3, -658 to-639;4, -569 to -550;
5,-470 to -451; 6, -440 to-421; 7, -359 to -340; 8, -338 to -319; 9, -319 to -300; 10, -299 to -280; 11, -140 to -121; 12, -70 to-51;
13,-39 to -20; and 14, -20 to -1. Shaded boxes correspond to low levels ofhybridizationsimilar to Fig.SCand D,detected in at least two ofthefourlatently infected rabbits (one or more neurons in at least two sections per rabbit). Open boxes indicate that no hybridization was
detected.The dark rectangle indicates the start of intense hybridization (as shown in Fig. 5A)at thestart of the stable 2- and 1.3-kb LATs
(30). Thelargeopen rectangle (note break) represents our previous results (29, 30), showing no hybridization to a BamHI-EcoRV restriction
fragment(-6141 to -824). The direction of LATtranscription is from left to right, as indicated by the large arrow.
latently infected rabbits. Each probe was hybridized to 3 to 5 sections of trigeminal ganglia from each of four rabbits
latentlyinfected with HSV-1. On this basis, at least 9 of the 12probes tested between the TATA box and the 5' end of thestable 2-kb LAT hybridized faintly to sections of
trigem-inal gangliafrom latently infected rabbits (Fig. 6). The lack ofdetected hybridization with the remaining three probes wasprobably due to technical limitations related to detecting
extremely small amounts of RNA. None of these probes
hybridizedtosections from uninfected rabbits (not shown).
Similar results were also obtained with sections of human
trigeminalganglia (not shown).
The start of transcription appeared to be close to the
TATA box consensus sequence. Probe 1 (-791 to -772),
approximately 85 nucleotides upstream ofthe TATA box, andprobe 2 (-707 to -688), immediately to the left of the TATAbox(at -688 to -684), werenegative (Fig. 6, probes 1 and 2). In addition, we have previously shown that a
restriction fragment probe from -6140 to -824
(BamHI-EcoRV) doesnothybridizetolatently infected ganglia from
rabbitsorhumans (29, 30)(Fig. 6). Probe 3 (-658 to -639), thefirstprobe totheright ofthe TATA box, waspositive.
Thisplacedthestartoftranscriptionbetweenprobes 2and 3
orwithin 30nucleotides ofthefirstTof the TATA box. This
isin excellent agreement with our invitroprimer extension
data,whichplaced thestart ofLATtranscription 28 nucle-otides from the TATAbox. These results strongly suggest thatthisTATAboxsequenceisthefunctionalTATAboxof the LAT promoter. The greatly reducedamountof detect-able RNA between the promoter and the stdetect-able LATs suggests that the RNA from this region was relatively unstable.
Northern blothybridization ofRNAfromlatently infected rabbits.Northern blothybridizationof RNA fromtrigeminal ganglia oflatently infected rabbits did not reveal any small
bandsin the 500-to700-base range that mightrepresent the
region between the TATA box and the 5' end of the 2-kb
LAT. However, by using probe 8 from Fig. 6, which is located between the TATA box and the 5' end of the 2-kb LAT, a veryfaint band with an estimated size of 8 to 9 kb
was detected (Fig. 7, lane 2; designated 8.3 kb). For
com-parison,
an equal amount of the same RNApreparation
hybridizedwithaprobethat was specificfor the 2-kb LAT
regionis shown in lane 1. Thisprobewasthesame size and
specificactivity astheprobe in lane 2.
Lanes 1 and 2 (Fig. 7) show a typical exposure for the
detectionandanalysisof the stable LATs.
(The
1.3-kbLATisnotseenin lane 1 because this probe is entirely within the intron of the 1.3-kb LAT.) The band designated 8.3 kb is almostindistinguishable in lane 2 and would notnormally be considered meaningful. However, by overexposingthe au-toradiogram, the 8.3-kb band was detected above the back-ground (lane 3). The detection of this band in RNA from latently infected rabbits wasarare event.Thisband was not detectedby other laboratories (2, 11) nor was it detected in the majority of our RNA preparations (not shown). Since this RNAwasbarely detectable withthe much more
sensi-tive in situ hybridization assay, the inability of Northern
blots to detect this RNAconsistentlywas notunexpected. The simplest explanation for the 8.3-kb band was that it represented anRNA initiating near the LAT promoter and
(because of its size) extending through and past the 2-kb LAT. Examination of the published HSV-1 sequence (15) revealed that the first consensus polyadenylation signal (AATAAA)wasapproximately8.3 kb downstreamfrom the LATpromoter,just 34 nucleotidespriortothe ICP4 polya-denylation signal on the opposite DNA strand (see Fig. 8). Wedetectedasimilar8.3-kb bandduringacutetissue culture infection and wereabletomapittothe regionbetween the TATA box and the potential polyadenylation site byusing oligonucleotide probes flanking these regions (results not shown). A similar result with RNA from acutely infected
tissue culture cells was recently reported (2), while this
manuscript was inpreparation. Thus, the faint 8.3-kb band wedetectedinlatently infectedneuronsmaybe anunstable,
possibly polyadenylated, primary LAT transcript from which the 2- and 1.3-kb LATs arederived.
DISCUSSION
When we first proposed that the LAT promoter was
locatedover660 nucleotides upstream of the2-kbLAT, we
also raised thepossibilitythat LATtranscription mightstart nearthis promoterregion(30, 31).This was basedonthe fact
that ingangliafromlatentlyinfectedrabbits,weoccasionally detected whatappearedtobeveryfaint in situhybridization withprobes upstream of the 2-kbLAT(30).Since then other labs have reported in situ hybridization in this region in
ganglia from latently infected mice (2, 10). However, these
experiments were doneby using large, nonoverlapping
re-striction fragments as
probes
and the start oftranscriptioncouldnotbe
precisely
determined. In this report, theuseof shortoligonucleotide (20-mers) probes
enabledus to deter-mine thatduringneuronallatency
inrabbits, transcription
ofVOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.136.483.74.146.2]-8.3 kb
*28S
[image:8.612.319.552.69.247.2]*18S
FIG. 7. Northern blot hybridization of RNA from ganglia of latentlyinfected rabbits. RNAwasisolated from trigeminal ganglia oflatently infected rabbits, and Northern blot hybridizationswere
done as described in Materials and Methods by using
32P-end-labeled 20-mers asprobes. Each lane containsapproximately 20 Fg of totalRNA. Lane 1 showshybridizationwithanoligonucleotide
correspondingto (the complement of) nucleotides +261 to +280.
This region is within the 1.3-kb LAT intron and therefore hybridizes
to the 2-kb LAT but not the 1.3-kb LAT (30). Lane 2 shows hybridization with oligonucleotide number 8 from Fig. 6,which is
midway between the TATA box sequence and the 5' end ofthe
stable 2-kb LAT. These probes were labeled tothe same specific activity, and thesame amountofradioactivity wasused toprobe lanes 1 and 2. Lanes 1 and 2representanautoradiogramexposureof
12 h andarefrom the samegel. Lane 3 isan 8-day overexposed autoradiogram of lane 2. The point marked 2 kb indicates the
location of the major 2-kb LAT. The point 8.3 kb marks the large bandvisible in lane 2and 3. Theapparentsize of this band is 8to9 kb, based on the mobilities of the 18S and 28S ribosomal RNAs (indicated by asterisks) and the 2- and 1.3-kb LATs. The 8.3-kb designation is consistent with thisapparentsize and correspondsto
the distance from the TATA box sequence tothe first consensus
polyadenylationsequence.None of theseprobeshybridizedtoRNA fromuninfected rabbits (30).
the faintLAT RNA beganimmediately downstream fromthe
putativepromoter,within 30nucleotides of the first T in the
TATAbox sequence. This is the first report in which the
start of"upstream" LAT transcription in latently infected
neuronshas been fine mapped. This result stronglysupports
thenotion that this TATA boxsequenceispartof the LAT
promoter.
We also reported here the detection by Northern blot
analysis of a minor 8.3-kb LAT transcript in ganglia of
latently infected rabbits. This is the first visualization by
Northernblot of this 8.3-kb LAT duringneuronal latency. A
similarlarge LATband has been detected byNorthern blots
of RNAfromacutelyinfectedcells and mapped to
approx-imately the region between the LAT promoter and the nearest potential polyadenylation sequence approximately
8.3 kbdownstream (not shown) (2). Basedon thesequence
of strain 17 syn+ (9, 15), the distance from the TATAbox
(nucleotide 118774)tothestartof thepolyadenylation signal (nucleotide 127143) is8,369nucleotides. Thisdistance could varyby several hundred bases in different strainsbecause of
differences in the repeated regions near the junction. The
location of this faint, large LAT is consistent with our
TRL UL
IRLIRS
UTRSNucleoUdepositon 118,00 125,000 132:000
CPO
TATA
&3kbLAT (unstable)
ICP4 PolyA
-I? ?
) \~~'7
2kb LAT(gable)
i? 2kb LATremoved ?
1.3kb LAT(stable)
6.3 kb LAT ?(unstable)?
bobgicslyactiveLAT?
FIG. 8. Proposed structure of the LAT gene. The HSV-1
ge-nomic organization in map units (mu) is shown atthe top. The
expanded region indicates the nucleotide positions relativeto the
entire HSV-1 DNA sequence. The approximate locations of the
ICPO and ICP4genesareshown forreference.Transcriptionofthe
LAT gene in the internal long repeat begins approximately 28
nucleotides from the first T of the TATA box (118774) approxi-matelyatnucleotide 118802 and extendstonearthepolyadenylation signal locatedatnucleotide 127143.The derivationofthe stable
2-and 1.3-kb LATsfromthe unstable 8.3-kb transcriptis likelybut
remainstobedemonstrated.The seriesof smallarrowsatthe end of
the8.3-kb LAT indicates that terminationmaybeinefficient, since
inoccasional Northern analyses of RNA from acutelyinfectedcells, using probes that correspond to the 5' end ofLAT, wedetected
LAT bands withapparentsizes of 12to 14 kb(not shown). If the
2-kb LAT is an intron, the 6.3-kb LAT shown maybe apossible result.
earliest results that detected weak in situ hybridization
signals with probes derived from BamHI SP, which is
downstream from the 2-kb LAT(20).
We originally proposed that the LAT promoter was
lo-catedover660 nucleotides upstream fromthe 5' end ofthe
2-kb LAT(30, 31),basedonsequenceanalysis showingthat
this area contained an excellent TATA box consensus
se-quence as well as other RNA polymerase II consensus sequences. Subsequently, wepublishedthe first report that
this upstream region was capable of promoter activity in
transientCATassaysinVerocells(32). This report
demon-stratesthat thisregionisalsocapableof promoteractivityin
transient assays in neuron-derived cells, as would be
ex-pectedfor the LAT promoter. Furthermore, wefound that
this promoteractivity hadneuron specificity. These results
again support the notion that this regioncontains the LAT
promoter.Theauthenticityof thisputativeLAT promoteris
furthersupported by recentreports (2, 7, 11, 24) indicating
that virus mutantswith deletions encompassing the TATA
boxregiondonotexpress anyLATduringneuronallatency.
To ensure that our transient CAT assays reflected
tran-scription initiatingfroma position consistent with itsbeing
directed by the TATA box consensus sequences, primer
extension experiments were done with RNA from cells
transfected with theA-promoter region(nucleotides -940to
-662). We found thattranscription began about 28
nucleo-tides from the first T of the TATA box sequence. This
confirmed that in vitro this region can function as a
pro-moter, further supporting the notion of this region as the LATpromoter.Thestartoftranscriptionthatwedetermined
by primer extension was in agreement with our in situ
12h 12 h
[image:8.612.113.242.73.264.2]1
2
...
2kb
8day exposure .3
....
on November 10, 2019 by guest
http://jvi.asm.org/
HSV-1 LAT PROMOTER IN NEURONAL CELLS 5027
hybridization data in latently infectedneuronswhich placed
thestartoftranscriptionwithin30nucleotides of theTATA
box. Thisposition is also inagreementwiththatdetermined
for thestartof LATtranscription duringacutetissue culture
infection withan RNAprotection assay (2).
Theimmortalized neuroncell line and the neuroblastoma
cell line used for transient assays in this report are both nonpermissive for lytic HSV-1 infection. Furthermore, fol-lowingHSV-1 infection,LATisexpressed inthe
immortal-ized neuron cell line (Wheatley et al., submitted). These
characteristics mimic neurons during in vivo latency and make these cellsparticularlyrelevantfortheseexperiments. By usingtheseneuron-derived celllines, wefoundthat the presence of sequences upstream of the LAT promoter had
differential effectsonpromoteractivityin neuron-compared
to nonneuron-derived cells. In Vero, CV-1, BHK, and L
cells,upstream sequencesdecreasedpromoteractivity by
3-to 12-fold.This apparentdownregulationwas notobserved
inimmortalized neurons or neuroblastomacells. Infact, in
some
experiments
inneuron-derivedcells, addition ofthese upstream sequencesenhancedpromoteractivityasmuchas300%relative to the Afragment (Fig. 3). Itis possible that
down
regulation
innonneuronal cellswascausedbynonneu-ronal trans-acting cellularfactors interacting withthesecis
upstream sequences. This notion is supported by
prelimi-nary observations that cotransfection of nonneuronal cells
with exogenous upstream sequences appeared to compete
outdownregulation by theupstream sequencesinfragment
A+. Inaddition, transfection with largeamountsofthe A+
orA++ CATconstructs eliminated downregulation, again
suggesting
acompetitive
effect(datanotshown).In summary, we have (i) used transient CAT assays in neuron-derived cells to map LAT-associated promoter
ac-tivity
to alocationover660nucleotides upstreamfromthe 5'end ofthe stable 2-kb LAT,
(ii)
shownby primer extensionthat in CAT constructs,transcription beganabout 28
nucle-otides from the firstT ofa consensus TATA box sequence
locatedatnucleotide 118774(-688), (iii) shownbya combi-nation ofNorthern blots and in situhybridization of latently infected neurons that transcription of a large 8.3-kb LAT
began
within30nucleotidesof the firstTof thisTATAbox sequenceataboutnucleotide118802(-660),and(iv) shownthat the LAT promoterhad neuronal
specificity.
Although
wehavenotproventhat the 2- and 1.3-kbLATsare derived from the 8.3-kb LAT, this appears to be the
simplest
and the mostlikely hypothesis,
since mutantslacking only
the immediateregion
around the LAT promoterdid not
produce
eitherthe 8.3 kb LATorthe 2- and 1.3-kb LATs(2, 11).Thelikely
derivation ofthe 2- and 1.3-kb LATsfrom a
primary
8.3-kb LAT raises thequestion
of theprocessing
eventsinvolved.The1.3-kbLAT canbe derived from the 2-kb LAT by a simple splice (30). Likewise, the2-kb LAT could be derived
by
asimple splice
from the 8.3-kb LAT.However,
if the 2-kb LAT is derivedby
asingle
splice,
either the 2-kb LATorthe 5' endofthe8.3-kb LATwould have to be an intron. We havenotbeen able to find
anyexample ofa5' intron in the literature.
Therefore,
it islikely
thateither(i)
the 5' endof the 2-kb LAT containsan asyetundetected smallexonfromthe 5'endofthe8.3-kbLAT or
(ii)
the 2-kb LAT is an intron. In the latter case, thebiologically
active LATmight
be anapproximately
6.3-kbpolyadenylated
RNAresulting
from the removal ofa 2-kbintron fromthe 8.3-kb
primary transcript (Fig.
8).Although
we cannot point to a
specific
6.3-kb LATband,
we havefoundthat Northernblotsfrom
acutely
infected cellsusually
detect
regions
ofLAT-specific
RNAs between the 2- and8.3-kb LATs(data not shown). Some of this material could representsuch a6.3-kb RNA. If the 2-kb LAT is an intron, the coding region for a potential LAT protein would be somewhere in the putative 6.3-kb LAT. This might explain why a LAT protein coded by the 2-kb LAT has not been detected(28, 31).
It is also possible that the 2- and 1.3-kb LATs are not derivedby splicing but ratherarestableregions (perhaps due
tosecondary structure)thatareleft intact afterdegradation
of the less stable 8.3-kb primary transcript. In this case,
predictions ofthebiologically active regionof the LAT gene would be more difficult.
One intriguing feature of the 8.3-kb transcript is that it spanstheHSV-1junction. Thus, duringacuteinfection,the copy of the LAT gene in the internal repeat produces a
primary transcript of 8.3 kb that extends approximately 800 bases into the short repeat. Since the HSV-1 genome is
circularizedshortlyafteracuteinfection andalsoappearsto
be circularizedduring latency (17, 18) the LAT geneinthe
terminal repeat would make an identical 8.3-kb transcript.
However, ifatanytimetheHSV-1genomewereinalinear form,the copyofthe LAT genein theterminalrepeatwould produce a transcript that is truncated at about 7.5 kb by
running off the end of the HSV-1 genome. Recent results with some (2, 7,24) but not all (1, 6) LAT mutants suggest that LAT may be involved in HSV-1 reactivation. It is therefore tempting to speculate that reactivation may be
associatedwithashortperiodoflinearization ofthe genome and thatsomedifference betweenthe complete8.3-kb
tran-scriptand atruncated runofftranscript might play arolein the switch from latency to reactivation. Alternatively, one could also speculate that LAT may play some role in circularization(orlinearization) ofthe genome.
The resultspresentedhere suggest that the LAT promoter iscontrolled, in part, by upstream sequences that resultin
neuronalspecificity. Exactlywhat these sequences are, what cellular and viralfactors they interact with, and how they
regulateLATexpression remain to be determined.
ACKNOWLEDGMENTS
Thisworkwaspartially supported bytheDiscoveryFund forEye
Research,the FactorFamily Foundation,andPublic Health Service
grantsEY07566andEY05939.J.C.Z. isaFactorFamily
Founda-tion Scholar. K.P.isanIris andB. Gerald Cantor Scholar.
Wethank Anita AveryandRichard Lit for technical assistance
and JohnOng andDon Brownforsuggestionsandcriticalreadingof
themanuscript.
LITERATURE CITED
1. Block, T. M., J. G. Spivack, I. Steiner, S. Deshmane, M. T.
McIntosh, R. P. Lirette, and N. W. Fraser. 1990. A herpes
simplex virustype 1latency-associated transcript mutant
reac-tivates with normal kinetics from latent infection. J. Virol.
64:3417-3426.
2. Dobson,A.T.,F.Sederati,G.Devi-Rao,W. M.Flanagan,M.J.
Farrell, J. G.Stevens,E. K. Wagner,and L. T.Feldman.1989.
Identification of the latency-associated transcriptpromoterby
expressionof rabbitbeta-globinmRNAinmousesensorynerve
ganglia latently infected with a recombinant herpes simplex
virus. J. Virol. 63:3844-3851.
3. Gilman, M. Z., R. N. Wilson, and R. A. Weinberg. 1986.
Multiple protein-bindingsites in the5'-flankingregion regulate
c-fosexpression. Mol. Cell Biol. 6:4305-4316.
4. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982.
Recombinant genomes which expresschloramphenicol
acetyl-transferase inmammaliancells. Mol.CellBiol. 2:1044-1051.
5. Graham,F.L.,andA.J.Van Der Eb.1973. Anewtechniquefor
the assayofinfectivityof humanadenovirus5 DNA. Virology VOL. 64,1990
on November 10, 2019 by guest
http://jvi.asm.org/
52:456-467.
6. Ho, D. Y., and E. S. Mocarski. 1989. Herpes simplex virus latent RNA (LAT) is not required for latent infection in the mouse. Proc.Natl. Acad. Sci. USA 86:7596-7600.
7. Leib, D. A., C. L. Bogard, M. Kosz-Vnenchak, K. A. Hicks, D. M. Coen, D. M. Knipe, and P. A. Schaffer. 1989. Adeletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced
frequency. J. Virol. 63:2893-2900.
8. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular
cloning: alaboratory manual.Cold Spring Harbor Laboratory,
ColdSpringHarbor, N.Y.
9. McGeoch, D. J., M. B. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J.Perry, J. E. Scott, and P. Taylor. 1988.The complete DNA sequence of the long uniqueregion in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574.
10. Mitchell,W. J., R. P.Lirette,and N. W. Fraser. 1990.Mapping
of low abundance latency-associated RNA in the trigeminal
gangliaofmice latently infectedwithherpes simplexvirustype
1.J. Gen. Virol. 71:125-132.
11. Mitchell, W. J.,I. Steiner, M. S. Brown, A. R. MacLean, J. H. Subak-Sharpe, and N. W. Fraser. 1990. A herpessimplex virus type 1 variant,deleted in the promoter region ofthe
latency-associated transcripts does notproduce any detectable minor
RNAspecies during latency inthe mousetrigeminalganglion.J.
Gen. Virol.71:953-957.
12. Nesburn, A. B., M. L. Cook,andJ. G. Stevens. 1972. Isolation
of herpes simplex virus: isolation from rabbit trigeminalganglia
betweenepisodes ofrecurrentocular infection. Arch.
Ophthal-mol.88:412-417.
13. Nesburn, A. B., J. M. Elliott, and H. M. Leibowitz. 1967.
Spontaneousrecurrenceof experimental herpes simplex
kerati-tis inrabbits. Arch. Ophthalmol. Vis. Sci. 78:523-529.
14. Paine, T. F., Jr. 1964. Latent herpessimplexinfectioninman. Bacteriol. Rev. 28:472-479.
15. Perry, L. J., and D. J. McGeoch. 1988. The DNAsequences of thelong repeat region and adjoining parts of the long unique
region in thegenome ofherpes simplex virus type 1. J. Gen.
Virol.69:2831-2846.
16. Post, L. E., A. J. Conley, E. S.Mocarski, and B. Roizman. 1980.
Cloning of reiteratedandnonreiterated herpes simplex virus1
sequences as BamHI fragments. Proc. Natl. Acad. Sci. USA 77:4201-4205.
17. Rock, D. L., and N. W. Fraser. 1983. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature(London) 302:523-525.
18. Rock, D. L., and N. W. Fraser. 1985. Latentherpes simplex
virustype 1 DNAcontains twocopies ofthevirionDNAjoint
region.J. Virol. 55:849-852.
19. Rock, D. L., W. A. Hagemoser, F. A. Osorio, and D. E. Reed.
1986.Detectionof bovineherpesvirustype 1 RNAintrigeminal
ganglia of latently infected rabbits by in situ hybridization. J.
Gen. Virol. 67:2515-2520.
20. Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L.Lewis, J. R. Lokensgard, and S. L. Wechsler. 1987. Detection of latency
related viral RNAs in trigeminal ganglia of rabbits latently
infected with herpes simplex virus type 1. J. Virol. 61:3820-3826.
21. Spivack, J. G., and N. W. Fraser. 1987. Detection of herpes
simplexvirustype 1transcriptsduringlatentinfectioninmice. J.Virol. 61:3841-3847.
22. Spivack, J. G., and N. W. Fraser. 1988. Expressionofherpes
simplexvirus type1(HSV-1) latency-associated transcripts and
transcriptsaffected bythedeletioninavirulentmutantHFEM:
evidence for anew class of HSV-1 genes. J. Virol.
62:3281-3287.
23. Spivack, J. G., and N. W. Fraser. 1988. Expression of herpes
simplexvirustype 1latency-associated transcriptsin the
trigem-inalganglia of mice duringacuteinfection and reactivation of
latentinfection.J. Virol. 62:1479-1485.
24. Steiner, I., J. G. Spivack, R. P. Lirette, S. M. Brown, A. R. MacLean,J. H.Subak-Sharpe, and N. W. Fraser. 1989. Herpes
simplexvirustype 1latencyassociatedtranscripts are evidently
notessentialfor latent infection. EMBO J. 8:505-511.
25. Stevens, J. G., E. K. Wagner, G. B.Devi-Rao,M. L.Cook, and L. T. Feldman. 1987. RNA complementary to aherpesvirus
alpha gene mRNA is prominant in latentlyinfected neurons.
Science 235:1056-1059.
26. Stroop, W. G., D. L. Rock, and N. W. Fraser. 1984.Localization
of herpessimplex virus inthetrigeminalandolfactorysystems
of the mouse central nervous systemduring acute andlatent
infectionsbyin situhybridization. Lab.Invest.51:27-38.
27. Vahlne, A., and E. Lyke.1978.Herpessimplex virus infection of
in vitro cultured neuronal cells (mouse neuroblastoma C1300
cells).J.Gen. Virol. 39:321-332.
28. Wagner, E. K., G. Devi-Rao, L. T. Feldman, A. T. Dobson, Y. Zhang, W. M. Flanagan, and J. G. Stevens. 1988. Physical
characterizationof theherpes simplexviruslatency-associated
transcriptin neurons. J.Virol. 62:1194-1202.
29. Wechsler, S. L., A. B. Nesburn, R. J. Watson, S. Slanina, andH.
Ghiasi. 1988.Finemappingof themajor latency-relatedRNA of
herpessimplexvirus type 1inhumans. J.Gen. Virol.
69:3101-3106.
30. Wechsler, S.L., A. B. Nesburn, R. Watson, S. M. Slanina, and H. Ghiasi. 1988. Fine mapping of the latency-related gene of
herpes simplex virustype 1: alternative splicing produces
dis-tinct latency-related RNAscontainingopen reading frames. J.
Virol. 62:4051-4058.
31. Wechsler, S. L., A. B. Nesburn, J. C. Zwaagstra, and H. Ghiasi.
1989. Sequence ofthe latencyrelated geneof herpes simplex
virustype 1. Virology168:168-172.
32. Zwaagstra, J. C., H. Ghiasi, A. B. Nesburn, and S. L. Wechsler.
1989. In vitro promoter activity associated with the latency
associated transcriptgene of herpes simplex virus type 1. J.
Gen. Virol.70:2163-2169.

![FIG..efragments609transfectedRepresentativewithusedandLAT).[14C]chloramphenicol. 2. The effect of upstream sequences on CAT activity driven by the LAT promoter in neuron- and nonneuron-derived cells](https://thumb-us.123doks.com/thumbv2/123dok_us/1319065.85499/4.612.324.554.409.599/efragments-transfectedrepresentativewithusedandlat-chloramphenicol-upstream-sequences-activity-promoter-nonneuron.webp)