Demyelinating Encephalomyelitis-Like Illness
abstract
X-linked Charcot-Marie-Tooth disease (CMTX1) is a clinically heteroge-neous hereditary motor and sensory neuropathy with X-linked trans-mission. Common clinical manifestations of CMTX1 disease, as in other forms of Charcot-Marie-Tooth (CMT) disease, are distal muscle wasting and weakness, hyporeflexia, distal sensory disturbance, and foot deformities. Mutations in the connexin-32 gene (gap junction protein
b1 [GJB1]) are responsible for CMTX1 disease. In this report, we describe a patient with CMTX1 disease presenting with recurrent attacks of transient and episodic acute demyelinating encephalomy-elitis (ADEM)–like symptoms without previous signs of lower extrem-ity weakness or foot deformities; the patient, as well as his asymptomatic mother, exhibited a novel GJB1 mutation (p.Met1Ile). Differential diagnosis of recurrent and transient ADEM-like illness, if unexplained, should include the possibility of CMTX1 disease. Pediat-rics2014;134:e270–e273
AUTHORS:Gun-Ha Kim, MD,aKyoung Min Kim, MD,bSang-il
Suh, MD, PhD,bChang-Seok Ki, MD, PhD,cand Baik-Lin Eun,
MD, PhDa
aDepartments of Pediatrics, andbRadiology, Korea University College of Medicine, Seoul, Korea; andcDepartment of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
KEY WORDS
Charcot-Marie-Tooth disease, connexin 32, encephalomyelitis, acute disseminated, peripheral nervous system diseases
ABBREVIATIONS
ADEM—acute demyelinating encephalomyelitis CMT—Charcot-Marie-Tooth (disease) CMTX—X-linked Charcot-Marie-Tooth (disease) CNS—central nervous system
EMG—electromyelogram GJB1—Gap junction proteinb1 NCS—nerve conduction study/studies
Dr Eun conceptualized this report and revised the manuscript; Dr G.-H. Kim reviewed the references and drafted the initial manuscript; Dr Ki performed genetic diagnosis, guided us through the interpretation of genetic analysis, and revised the manuscript critically; Dr K.M. Kim conceptualized the report, collected thefigures, and critically reviewed the manuscript; Dr Suh commented on the imaging studies, revised thefigures, and critically reviewed the manuscript; and all authors approved thefinal manuscript as submitted.
www.pediatrics.org/cgi/doi/10.1542/peds.2012-3243
doi:10.1542/peds.2012-3243
Accepted for publication Dec 4, 2013
Address correspondence to Baik-Lin Eun, MD, PhD, Department of Pediatrics, Korea University Guro Hospital, 97 Gurodong-gil, Guro-gu, Seoul 152-703, Korea. E-mail: bleun@korea.ac.kr
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2014 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE:The authors have indicated they have nofinancial relationships relevant to this article to disclose.
FUNDING:No external funding.
Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heteroge-neous hereditary motor and sensory neuropathy characterized by distal muscle wasting and weakness, sen-sory disturbance, hyporeflexia, and pes cavus foot deformity. It is a common disorder with a population frequency of∼1:2500.1CMT1A is the most common
CMT disease type, accounting for 40% to 50% of all cases, and is caused by the overexpression of the peripheral my-elin protein 22 gene (PMP22). X-linked Charcot-Marie-Tooth (CMTX) disease (CMTX1) is the second most common type of CMT disease (7% to 12% of all patients) and is associated with muta-tions in the gap junction proteinb1 gene (GJB1), which encodes connexin-32.2 In
this report, we describe the case of a patient presenting with recurrent attacks of transient and episodic acute demyelinating encephalomyelitis (ADEM)– like illness without apparent signs of
lower extremity weakness or foot de-formities, in whom we identified a novel GJB1mutation.
PATIENT PRESENTATION
In November 2007, a 14-year-old boy presented with right-side motor weak-ness and aphasia. MRI (3.0T, Magnetom Skyra; Siemens, Erlangen, Germany) re-vealed abnormally increased T2 signal and diffusion restriction in the splenium of the corpus callosum and centrum semiovale (Fig 1 A, B, C, and D). These lesions spared the subcortical Ufibers, which showed no signal enhancement. Brain magnetic resonance angiography, EEG, and cere-brospinalfluid examination revealed normalfindings, including a normal lac-tate level (1.2 mmol/L) and absence of oligoclonal bands. Laboratory tests, in-cluding a complete blood count, electro-lytes, renal function test, urinalysis, and coagulation parameters, were negative.
Enterovirus, herpes virus, varicella-zoster virus, and Japanese encephalitis virus were not detected in the cerebrospi-nalfluid samples tested by polymer-ase chain reaction. Arylsulfatpolymer-ase A and very long chain fatty acid levels were normal. The patient had neither past illness nor a family history of any in-heritable neurologic illnesses. Symp-toms were completely absent on day 2 without any treatment. Follow-up MRI performed after 1 month showed marked improvement of the abnormal T2 signal hyperintensity in the corpus callosum and centrum semiovale (not shown). However, 4 years later, the pa-tient returned after the onset of a second attack of episodic right-side hemiparesis and dysarthria. During episodes, deep tendon reflexes were normal. Brain MRI revealed approximately the same dis-tribution of hyperintense foci as those found during thefirst attack (Fig 1 E and F). We searched the literature to look for
FIGURE 1
Episode 1: A and B, Axialfluid-attenuated inversion recovery images (repetition time/echo time/inversion time = 9000/119/2200 milliseconds) through the splenium of the corpus callosum and the centrum semiovale show the bilaterally symmetric regions of abnormal T2 hyperintensity. C and D, Diffusion-weighted images (DWIs; b = 1000) show high signal intensity in the same regions. The lesions showed low signal intensity on an apparent diffusion coefficient map (not shown). Episode 2: E and F, DWIs show the recurrent diffusion-restricted lesions, manifesting as high signal intensity in regions similar to those observed during episode 1. Episode 3: G and H, DWIs at the levels of the splenium of the corpus callosum and the centrum semiovale show symmetric diffusion-restricted lesions in the white matter of both cerebral hemispheres. White matter lesions that had been seen during the second attack are preserved.
CASE REPORT
PEDIATRICS Volume 134, Number 1, July 2014 e271
at Viet Nam:AAP Sponsored on August 28, 2020
www.aappublications.org/news
analysis after finding similar case re-ports of CMTX disease. An electromyo-gram (EMG) was normal, and a nerve conduction study (NCS) of the legs re-vealed slightly delayed conduction ve-locities, but was assumed to be not conclusive (Table 1). Hemiparesis and dysarthria were shown, but completely disappeared within a few hours. These episodes recurred, sometimes 2 or 3 times in a single day. Although the ra-diologicfindings were somewhat incom-patible, we suspected a demyelinating illness such as ADEM, and the patient was treated with immunoglobulin (0.4 g/kg per day for 5 days) and intra-venous methylprednisolone (30 mg/kg per day for 3 days). The patient’s symp-toms suddenly disappeared on day 4. However, 2 months later, a third attack presented with episodic left-side hemi-paresis lasting a few hours. Between events, deep tendon reflexes were slightly diminished at the ankles, but no obvi-ous weakness was observed in the lower extremities. Brain MRI revealed
findings similar to the previous attacks (Fig 1 G and H). EMG and NCS were re-peated on both arms and legs, and EMG and NCS revealed sensorimotor poly-neuropathy of the demyelinating type and decreased motor and sensory nerve conduction velocities (Table 1) with normal muscle unit potentials. We treated the patient with intravenous methylprednisolone for 3 days and then tapered to oral steroid (1 mg/kg per day,
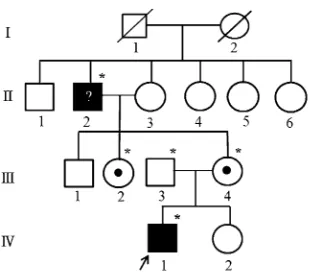
abnormal T2 signal intensity were ob-served. EMG and NCS were followed up 1 month later and showed no significant changes (Table 1). When the results for genetic analysis were available, se-quencing of the GJB1 gene revealed a novel G to T transversion at nucleotide position 3(c.3G.T), which has been predicted to result in an amino acid change from methionine to isoleucine at codon 1 (p.Met1Ile). The patient’s mother was also confirmed as having the same GJB1mutation and similar results were shown on her NCS and EMG (Table 1). We performed polymerase chain reaction and restriction fragment length poly-morphism analysis for the proband, the maternal grandparents, both parents, and the maternal aunt and found that the proband, the maternal grandfather, mother, and maternal aunt have the sameGJB1mutation (Fig 2). However, it is unclear whether his grandfather has a neuropathy because his limping gait may have been due to a history of car accident and there was no opportunity to confirm the mutation by using NCS. Un-fortunately, we failed to obtain the patient’s uncle’s blood sample because he resided outside the country, but he did not report any symptoms by the time of this writing.
DISCUSSION
Our patient did not complain of any
dif-ficulty in walking except during acute episodes, exhibited no foot deformity, and
had no family history of CMT disease. His initial presentation with episodic hemi-paresis led us to diagnose his condition as ADEM-like illness during hisfirst at-tack. While searching the literature for any possible differential diagnosis mainly based on MRIfindings, we subsequently noticed peripheral neuropathy after re-peating electrophysiological testing dur-ing the third attack. Genetic testdur-ing later confirmed theGJB1mutation.
To date,.400 mutations inGJB1 have been reported to cause CMTX1 disease (the Inherited Peripheral Neuropathies Mutation Database is available at http:// www.molgen.ua.ac.be/CMTMutations/ Mutations/MutByGene.cfm). We identified
TABLE 1 Electrophysiological Findings
Conduction Velocity, m/s
Median Motor Nerve Ulnar Motor Nerve Tibial Motor Nerve Peroneal Motor Nerve Median Sensory Nerve Ulnar Sensory Nerve Peroneal Sensory Nerve Sural Sensory Nerve
Second attack — — 41 42 — — 37 35
Third attack 40 39 37 36 32 33 34 32
Follow-up after 1 month
39 38 38 33 32 36 34 32
Patient’s mother 41 54 38 32 35 37 — —
—, not tested.
FIGURE 2
a novel mutation with a G to T change at nucleotide 3, resulting in the substitution of methionine to isoleucine (p.Met1Ile). Because this is the start codon, we suspect that one of the following can result from this change:
complete deletion of the protein because GJB1 mutations are loss-of-function mutations in almost all patients3(some exceptions exist4); an extension by activating an up-stream translation initiation, which is possible for connexin-32 because 10 bases upstream of the initiation codon there is another ATG sequence; an N-terminal deletion by activating downstream translation initiation, which is unlikely for connexin-32 be-cause there is no other methionine until p.34 (first transmembrane [TM] domain).In our case, peripheral polyneuropathy was mild. A similar pattern (mild phe-notype) was also noted in another case report with a codon 1 mutation ofGJB1 (p.Met1Arg).5Because mostGJB1
muta-tions are loss-of-function mutamuta-tions as mentioned previously, this mild pheno-type might be a typical manifestation at an early stage of CMTX1 disease and we should follow a patient’s progress. The second result, an extension by activating
an upstream translation initiation, could be another explanation for mild pheno-type, which needs to be elucidated in future research.
There are several reported cases of CMTX1 disease with transient central nervous system (CNS) involvement. The attacks were provoked by illness, expo-sure to high altitudes,6and
hyperventi-lation7but can be unprovoked as in our
case. Most of these showed signs of pe-ripheral polyneuropathy such as weak-ness, muscle wasting in the legs, or pes cavus.6–10However, a case of CMTX
dis-ease who manifested transient CNS symptoms without any signs of periph-eral neuropathy was reported,11 as in
our patient. Five months after the tran-sient CNS symptoms, hefinally developed signs and symptoms of neuropathy in the form of absent ankle reflexes. The case also suggests that transient CNS symptoms in CMTX1 disease can occur without axonal degeneration in the pe-ripheral nerves, masquerading as ADEM-like illness. In addition, the pathogenesis of CNS symptoms does not seem to in-volve axonal degeneration because the changes in brain MRI were reversible over a short time period. Many re-searchers now think that a decreasing number of functioning gap junctions
between oligodendrocytes and astro-cytes in situations of metabolic stress may affect the CNS phenotype, making both cell types vulnerable to impairment of intercellular exchange of ions and small molecules.12This possibility could
explain the restricted diffusion noted on MRI of patients with CMTX1 disease during their ADEM-like illness. However, there are still no clear mechanisms linking the impairment of intercellular transport of molecules to the diminished integrity of myelin and the axon.
CONCLUSIONS
We describe a novelGJB1mutation in a patient with CMTX1 disease mas-querading as ADEM-like illness who did not have symptoms of peripheral poly-neuropathy or a known family history. This finding is particularly important for pediatricians who are not familiar with CMTX1 disease with a CNS pheno-type. Differential diagnosis of recurrent and transient ADEM-like illness, if un-explained, should include X-linked peri-pheral polyneuropathy (CMTX1).
ACKNOWLEDGMENTS
We thank Dr Kleopas A. Kleopa of The Cyprus Institute of Neurology and Genetics for useful comments on the manuscript.
REFERENCES
1. Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. 1974;6(2):98–118
2. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease.Lancet Neurol. 2009;8(7):654–667 3. Shy ME, Siskind C, Swan ER, et al. CMT1X phenotypes represent loss of GJB1 gene function.Neurology. 2007;68(11):849–855 4. Kleopa KA, Abrams CK, Scherer SS. How do
mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease?Brain Res. 2012;1487:198–205 5. Brozková D, Mazanec R, Haberlová J,
Sakmaryová I,Subrt I, Seeman P. Six new gap junction beta 1 gene mutations and their phenotypic expression in Czech patients with
Charcot-Marie-Tooth disease.Genet Test Mol Biomark. 2010;14(1):3–7
6. Paulson HL, Garbern JY, Hoban TF, et al. Transient central nervous system white mat-ter abnormality in X-linked Charcot-Marie-Tooth disease.Ann Neurol. 2002;52(4):429–434 7. Srinivasan J, Leventer RJ, Kornberg AJ, Dahl HH, Ryan MM. Central nervous system signs in X-linked Charcot-Marie-Tooth dis-ease after hyperventilation.Pediatr Neurol. 2008;38(4):293–295
8. Hanemann CO, Bergmann C, Senderek J, Zerres K, Sperfeld A-D. Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel connexin 32 mutation.Arch Neurol. 2003;60(4):605–609
9. Taylor RA, Simon EM, Marks HG, Scherer SS. The CNS phenotype of X-linked Charcot-Marie-Tooth disease: more than a peripheral prob-lem.Neurology. 2003;61(11):1475–1478 10. Schelhaas HJ, Van Engelen BG, Gabreëls-Festen
AA, et al. Transient cerebral white matter lesions in a patient with connexin 32 missense mutation.Neurology. 2002;59(12):2007–2008 11. Anand G, Maheshwari N, Roberts D, et al.
X-linked hereditary motor sensory neurop-athy (type 1) presenting with a stroke-like episode.Dev Med Child Neurol. 2010;52(7): 677–679
12. Kleopa KA. The role of gap junctions in Charcot-Marie-Tooth disease. J Neurosci. 2011;31(49):17753–17760
CASE REPORT
PEDIATRICS Volume 134, Number 1, July 2014 e273
at Viet Nam:AAP Sponsored on August 28, 2020
www.aappublications.org/news
DOI: 10.1542/peds.2012-3243 originally published online June 23, 2014;
2014;134;e270
Pediatrics
Services
Updated Information &
http://pediatrics.aappublications.org/content/134/1/e270
including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/134/1/e270#BIBL
This article cites 11 articles, 3 of which you can access for free at:
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
DOI: 10.1542/peds.2012-3243 originally published online June 23, 2014;
2014;134;e270
Pediatrics
Gun-Ha Kim, Kyoung Min Kim, Sang-il Suh, Chang-Seok Ki and Baik-Lin Eun
Encephalomyelitis-Like Illness
Charcot-Marie-Tooth Disease Masquerading as Acute Demyelinating
http://pediatrics.aappublications.org/content/134/1/e270
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.
the American Academy of Pediatrics, 345 Park Avenue, Itasca, Illinois, 60143. Copyright © 2014 has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
at Viet Nam:AAP Sponsored on August 28, 2020
www.aappublications.org/news