ARTICLE
No Evidence of Persisting Measles Virus in Peripheral
Blood Mononuclear Cells From Children With Autism
Spectrum Disorder
Yasmin D’Souza, MSca, Eric Fombonne, MDb, Brian J. Ward, MDCMa
aDivision of Infectious Diseases, McGill University Health Center, Montreal, Quebec, Canada;bDepartment of Psychiatry, McGill University, Montreal Children’s Hospital, Montreal, Quebec, Canada
Financial Disclosure: In the United Kingdom, Dr Fombonne has provided advice on the epidemiology and clinical aspects of autism to scientists advising parents, to MMR vaccine manufacturers (for a fee), and to several government committees between 1998 and 2001. He has been consulted by ad hoc US committees from the Institute of Medicine and the American Academy of Pediatrics reviewing MMR safety. Since June 2004, Dr Fombonne has been an expert witness for vaccine manufacturers in the US thimerosal litigation. None of his research has ever been funded by industry. Dr Ward served on a number of Canadian and US government advisory committees addressing the issues of vaccine use and safety between 1994 and 2006. He has provided expert testimony for both the US and Quebec vaccine injury compensation programs. Dr Ward has also provided advice and teaching to Canadian government and industry groups in the area of vaccine immunology. He has conducted and participated in several studies of measles vaccine safety sponsored by Canadian and US government funding agencies. He has also conducted a small number of phase I and phase II industry-sponsored clinical trials of nonlicensed vaccines for smaller biotechnology companies. He has conducted a single, company-sponsored, immunologic study of a licensed, acellular pertussis vaccine.
ABSTRACT
OBJECTIVES.Despite epidemiologic evidence to the contrary, claims of an association between measles-mumps-rubella vaccination and the development of autism have persisted. Such claims are based primarily on the identification of measles virus nucleic acids in tissues and body fluids by polymerase chain reaction. We sought to determine whether measles virus nucleic acids persist in children with autism spectrum disorder compared with control children.
PATIENTS AND METHODS. Peripheral blood mononuclear cells were isolated from 54 children with autism spectrum disorder and 34 developmentally normal children, and up to 4 real-time polymerase chain reaction assays and 2 nested polymerase chain reaction assays were performed. These assays targeted the nucleoprotein, fusion, and hemagglutinin genes of measles virus using previously published primer pairs with detection by SYBR green I. Our own real-time assay targeted the fusion gene using novel primers and an internal fluorescent probe. Positive reac-tions were evaluated rigorously, and amplicons were sequenced. Finally, anti-measles antibody titers were measured by enzyme immunoassay.
RESULTS.The real-time assays based on previously published primers gave rise to a large number of positive reactions in both autism spectrum disorder and control samples. Almost all of the positive reactions in these assays were eliminated by evaluation of melting curves and amplicon band size. The amplicons for the remaining positive reactions were cloned and sequenced. No sample from either autism spectrum disorder or control groups was found to contain nucleic acids from any measles virus gene. In the nested polymerase chain reaction and in-house assays, none of the samples yielded positive results. Furthermore, there was no difference in anti-measles antibody titers between the autism and control groups.
INTERPRETATION.There is no evidence of measles virus persistence in the peripheral blood mononuclear cells of children with autism spectrum disorder.
www.pediatrics.org/cgi/doi/10.1542/ peds.2006-1262
doi:10.1542/peds.2006-1262
Key Words
measles, MMR, autism, autistic spectrum disorder, real-time PCR
Abbreviations
MMR—measles-mumps-rubella MV—measles virus ASD—autism spectrum disorder PCR—polymerase chain reaction RT—reverse transcription
PBMC—peripheral blood mononuclear cell IBD—inflammatory bowel disease HBSS—Hank’s buffered salt solution GAPDH— glyceraldehyde-3-phosphate dehydrogenase
cDNA— complementary DNA UNG— uracyl DNA-glycosylase PBS-T—phosphate-buffered saline-Tween
Accepted for publication Jul 11, 2006
Address correspondence to Brian J. Ward, MDCM, Montreal General Hospital, 1650 Cedar Ave, Room D7-153, Montreal, Quebec, Canada H3G 1A4. E-mail: brian.ward@mcgill.ca
C
ONTROVERSY OVER MEASLES-MUMPS-RUBELLA(MMR) vaccine erupted in 1998 when it was suggested that the measles virus (MV) component of the vaccine was responsible for autistic enterocolitis, a new form of au-tism spectrum disorder (ASD) characterized by the pres-ence of ileo-colonic lymphonodular hyperplasia, chronic inflammatory colonic disease, and loss of acquired cog-nitive skills after a period of normal development.1Toaddress this concern, several epidemiologic studies were performed that found no association between MMR vaccine and ASD.2–14This controversy has been the
sub-ject of several authoritative reviews and statements by, among others, the American Academy of Pediatrics and the Institute of Medicine.15–17
Despite the rising tide of epidemiologic evidence against any MMR-ASD link, several polymerase chain reaction (PCR)-based investigations have been published that seem to support the association.18–20 Using nested
reverse-transcription (RT)-PCR targeting the MV hem-agglutinin and fusion genes to study peripheral blood
mononuclear cells (PBMCs), Kawashima et al19reported
the presence ofⱖ1 MV gene in 2 (18%) of 11 samples
from subjects with inflammatory bowel disease (IBD) and 3 (33%) of 9 children with ASD compared with 0 of 8 control samples. By direct sequencing, these authors implicated wild-type virus in 1 of the positive IBD cases and vaccine-strain virus in the remainder. In 2002, both
Uhlmann et al18 and Martin et al20 published studies
claiming the detection of MV nucleic acids in intestinal samples from children with the alleged new variant
ASD. Using real-time TaqMan PCR, Uhlmann et al18
reported the detection of MV fusion and hemagglutinin genes in biopsies from 75 of 91 children with ASD (vs 5 of 70 developmentally normal children). Similarly,
Mar-tin et al20 described the detection of MV fusion and
hemagglutinin genes in biopsies from 62 of 68 ASD children (vs 4 of 39 biopsies from controls). Both groups reported the detection of MV nucleoprotein gene by in situ PCR (42 of 57 and 25 of 28 ASD biopsies in the Uhlmann et al18and Martin et al20studies, respectively,
vs 1 of 5 control samples in both cases). No sequence information was provided in either study, despite the apparent ease with which amplicons were generated and the high copy numbers reported (range: 1–300 000 cop-ies/ng RNA).
These molecular data have been used to implicate MMR vaccination in the development of ASD in at least a subset of affected children. In effect, it has been argued that these molecular data trump the epidemiologic evi-dence at the level of the individual child. Unfortunately, the media attention that accompanied the initial claims and the reporting of the ongoing controversy have led to a loss of confidence in MMR vaccine. MMR coverage in the United Kingdom fell from 92% in 1995–1996 to ⬃80% in 2004 –2005, resulting in several major measles outbreaks andⱖ3 deaths.21,22
Although concerns have been expressed regarding the molecular data generated by proponents of the
MMR-ASD hypothesis23and ethical concerns have been
raised about one of the reports,24no one has attempted
specifically to confirm or refute the molecular data18–20to
resolve the controversy. We sought to replicate the real-time PCR and nested PCR results reported by Uhlmann et al18 and Kawashima et al,19respectively, by applying
their primer pairs to PBMCs isolated from children with ASD and developmentally normal controls. The princi-pal objective of this study was to determine whether or not the amplicons produced in these assays were MV-specific by sequencing. In addition, we developed an in-house, probe-based PCR assay to detect the MV fu-sion gene in these same samples. We found no evidence to support the contention that MV persists in any of the PBMC samples from ASD patients or developmentally normal children. Although many samples yielded posi-tive results in the real-time PCR assays based on the Uhlmann et al18primers, none of the amplified products
were of MV origin by sequencing. No sample was found to be positive using the Kawashima et al19assays or our
probe-based fusion gene real-time RT-PCR assay. Finally, we found no significant difference in anti-MV antibody titers in ASD subjects and age-matched controls.
METHODS
Study Subjects
Anti-coagulated blood (2–3 mL with ethylenediami-netetraacetic acid) was collected from 54 sequential chil-dren with ASD (autism, 41; pervasive developmental disorder not otherwise specified, 13; mean: 4 years old; 48 boys) referred to the Autism Spectrum Disorders Program and 34 control children (mean: 4 years old; 26 boys) seen in the outpatient pediatric clinic of the Mon-treal Children’s Hospital. Children with ASD were as-sessed by a standardized protocol involving the admin-istration of the Autism Diagnostic Interview-Revised25to
the main caregiver and a direct examination of the child with the Autism Diagnostic Observational Schedule
Ge-neric.26 Both measures were administered by trained
staff. The children were all seen by a clinical expert in autism (E.F.) who formulated the final diagnosis using
Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, criteria.27 Controls with normal development
were selected among children attending an outpatient clinic of the same hospital who were to undergo blood testing for other reasons. The absence of ASD in the controls was verified by parental interview, review of medical charts, and the administration of the Autism Screening Questionnaire.28Parents of children from both
patient population. Furthermore, the observation by Ka-washima et al19that MV nucleic acids could be identified
in the blood of 33% of children with autistic enteroco-litis suggested that the use of PBMCs would be adequate to address the principal hypothesis. The operator (Y.D.) was blinded to study group (ASD or control) while per-forming RNA manipulations and the in-house fusion gene and Uhlmann assays.
PBMC Separation
PBMCs were isolated by Ficoll-Plus (GE Healthcare, Buckinghamshire, United Kingdom) density gradient centrifugation as described previously.29 Briefly, whole
blood was centrifuged (300gfor 10 minutes), and plasma
was stored at ⫺20°C until used for ELISA testing. The
remaining blood cells were resuspended in Hank’s buff-ered salt solution (HBSS, Wisent, St-Bruno, Quebec, Canada) to a total of 10 mL and layered onto 4 mL of Ficoll-Plus. After centrifugation (1020gfor 30 minutes), the PBMC interface was removed and washed twice in
10 mL of HBSS (300gfor 10 minutes). The PBMCs were
counted by hemocytometer before the second wash. The final cell pellet was resuspended in 2 mL of bovine serum albumin (Sigma, Oakville, Ontario, Canada) con-taining 7.5% dimethyl sulfoxide (American Chemicals Ltd, St Laurent, Quebec, Canada) and frozen in liquid nitrogen until it was used for RNA extraction.
Positive and Negative Control Samples
PBMCs from 2 laboratory volunteers were cultured in RPMI 1640 (Wisent) containing 15% fetal bovine serum (Wisent) and supplemented with Hepes (0.01M final concentration: Wisent), glutamine (2 mM final tration: Wisent), gentamicin (0.1 mg/mL final concen-tration: Wisent), and penicillin/streptomycin (100 IU/ 100 g/mL final concentrations, respectively: Wisent). Cells (1⫻106) were stimulated with 1g/mL
phytohe-magglutinin (Sigma) for 1 hour at 37°C, followed by infection with the Edmonston strain of MV (gift from S. Ratnam, Newfoundland Public Health Laboratory, New-foundland, Canada) at a multiplicity of infection of 1. Uninfected PBMCs isolated from the same volunteers served as negative controls. These samples were used to optimize assay conditions.
RNA Extraction and Measurement
PBMCs were thawed and washed in 10 mL of HBSS
(300g for 10 minutes), and subsequent RNA isolation
was conducted with the RNeasy kit according to the manufacturer’s instructions (Qiagen, Valencia, CA), with terminal digestion of residual DNA using the ribo-nuclease-free deoxyribonuclease kit (Qiagen). Total
RNA was eluted in 30 L of distilled water, and its
concentration was determined by fluorescence (Ri-bogreen, Invitrogen, Burlington, Ontario, Canada). In-tegrity of the harvested RNA was assessed on 1%
ribo-nuclease-free agarose gels. It is noteworthy that
Kawashima et al19extracted total RNA from PBMCs by a
similar method, also based on guanidine salts but using a specific RNA-binding resin instead of a silica-based membrane.
Primer Selection
Name and direction, GC content, melting temperature, position, sequence, gene-amplified and expected size of band for the in-house primer-probe set, as well as the Uhlmann and Kawashima primers are shown in Table 1.
The housekeeping gene used by Uhlmann et al18and in
our in-house assay was human glyceraldehyde-3-phos-phate dehydrogenase (GAPDH). This constitutively ex-pressed gene serves as an internal control for RNA ade-quacy, because it is subject to the same errors in RNA extraction and preparation as the gene of interest. We had originally planned to include in-house probe-based assays for both the nucleoprotein and fusion genes. However, during optimization studies, our nucleopro-tein assay proved to have a lower sensitivity limit of 10E4 copies per reaction and was abandoned. To some extent, this decision was dictated by the number of assays that we wished to perform and the limited amount of RNA available.
Probe-Based, In-House, Real-Time Fusion Gene Assay
Oligonucleotide primers and probes were designed and synthesized according to the Qiagen guidelines for cus-tom real-time PCR primer and probe design (https:// customassays.qiagen.com/design/inputsequences.asp).30
The oligonucleotide probe was labeled with fluoro-ami-docarboxymethyldethia coenzyme A, a fluorescent re-porter dye. All of the positions in the primers and probe correspond with the Edmonston, vaccine-strain MV se-quence reported in the National Center for Biotechnol-ogy Information nucleotide sequence database, acces-sion No. AF266288. The in-house primers and probe contain superbases, which are proprietary, modified nu-cleotides that permit highly specific binding to GC-rich DNA templates. The sequences for the GAPDH primers and probe are proprietary information of Qiagen. The cycling conditions for controls, samples, and standards were as follows: reverse transcription: 50°C for 20 min-utes; activation: 95°C for 15 minmin-utes; PCR conditions: 42 cycles of 94°C for 0 seconds; 56°C for 30 seconds; and 76°C for 30 seconds at a ramp rate of 2°C/second (Light-cycler, Roche Applied Science, Laval, Quebec, Canada). Each reaction tube contained: 10L of master mix, 0.2
L of RT, 1L of probe, 1L of combined forward and reverse primers, 4.8L of distilled water, 1L of uracyl
DNA-glycosylase (UNG) and 2 L of RNA (2–332 ng).
RT-PCR kit, Qiagen). The expected amplicon size was 111 base pairs (bp).
Primers and Cycling Conditions for Uhlmann PCR Assays
Nucleoprotein 1, fusion 1, and hemagglutinin 1 primer sequences (Integrated DNA Technologies, Coralville, IA) were as described by Uhlmann et al18(Table 1). Cycling
conditions were as follows: RT: 50°C for 20 minutes; activation: 95°C for 15 minutes; and PCR conditions: 94°C for 15 seconds, 56°C for 30 seconds, and 72°C for 30 seconds, and a fourth segment of 5 seconds was added to each cycle depending on the targeted gene (nucleoprotein: 78°C; hemagglutinin: 79°C; fusion: 74°C) to minimize the amplification of nonspecific prod-ucts (Lightcycler, Roche Applied Science). Each reaction tube contained 10L of master mix, 0.2L of RT, 2L (0.5M) each of forward and reverse primers, 3.8L of distilled water (or 2.8L of distilled water and 1L of UNG for the Uhlmann fusion and hemagglutinin assays), and 2L of RNA. In all but 1 case,⬎5 ng of sample RNA were used (range: 5–332 ng) per reaction. All of the sample reactions were performed in duplicate. An MV-spiked aliquot was run in parallel for each sample (QuantiTect SYBR green RT-PCR kit, Qiagen). Expected amplicon sizes were 150 bp for each of the Uhlmann MV assays and 226 bp for the Uhlmann GAPDH primers. The Uhlmann et al18study reported the use of 2 primer sets
for each fusion, nucleoprotein, and hemagglutinin assay (ie, 6 primer sets total). However, it is unclear which and how many of their samples were positive using which primer sets. The F1/F2 and N1/N2 primer sets partially overlap one another; we, therefore, arbitrarily chose the
first sets for inclusion in our study (F1 and N1). The H2 primer set performed poorly during optimization in our hands and was omitted from further study.
Primers and Cycling Conditions for Kawashima Nested PCR Assays
Primer sequences (Integrated DNA Technologies) were
as described by Kawashima et al19(Table 1). We
modi-fied their protocol slightly by omitting the initial inde-pendent complementary DNA (cDNA) synthesis step. In addition, instead of conventional PCR, we used real-time PCR with detection by SYBR green I for both PCR rounds. Cycling conditions for the first round of nested RT-PCR were as follows: RT: 50°C for 20 minutes; acti-vation: 95°C for 15 minutes; PCR conditions: 94°C for 15 seconds, 56°C for 30 seconds, and 72°C for 30 seconds; melting conditions: 95°C for 0 seconds, 65°C for 30 seconds, and 95°C for 0 seconds at a ramp rate of 0.1°C/ second (Lightcycler, Roche Applied Science). Each reac-tion tube contained 10L of master mix, 0.2L of RT, 2L (0.5M) of forward and reverse outer primers, 4.2
L of distilled water, 0.5L of UNG, and 2L of RNA.
In all but 1 ASD and 1 control case, ⬎3 ng of sample
RNA were used (range: 3–200 ng) per reaction. All sam-ple reactions were performed in duplicate (QuantiTect SYBR green RT-PCR kit, Qiagen). Expected amplicon sizes were 834 bp for the fusion assay and 595 bp for the hemagglutinin assay. Cycling conditions for the second round were as follows: activation: 50°C for 15 minutes; PCR conditions: 94°C for 15 seconds, 56°C for 30 sec-onds, and 72°C for 30 seconds; and melting conditions: 95°C for 0 seconds, 65°C for 30 seconds, and 95°C for 0
TABLE 1 Primer and Probe Characteristics for the Real-Time and Nested PCR Assays Oligonucleotide GC
content, %
Melting Temperature,
°C
5⬘ positiona
3⬘ positiona
Sequenceb Gene Amplified Amplicon
Size, bp
In-house F forward 50 64.1 6895 6914 TCGGACCAGATATTGAGGAG Fusion 111
In-house F reverse 43 64.8 7005 6985 GCAACATATTAAAGCGgGGAT
In-house F probe 53 68.1 6968 6984 TTGGAGgGTTGATAGgG
Uhlmann N1 forward 60 59.6 1325 1344 TCAGTAGAGCGGTTGGACCC Nucleoprotein 150
Uhlmann N1 reverse 60 60.3 1475 1456 GGCCCGGTTTCTCTGTAGCT
Uhlmann F1 forward 50 56.5 5593 5612 TGACTCGTTCCAGCCATCAA Fusion 150
Uhlmann F1 reverse 43 55.1 5843 5823 TGGGTCATTGCATTAAGTGCA
Uhlmann H1 forward 55 57.2 7448 7467 TTCATCGGGCAGCCATCTAC Hemagglutinin 150
Uhlmann H1 reverse 65 59.5 7598 7579 CTCTGAGGTGTCCTCAGGCC
Uhlmann GAPDH1 56 53.9 NA NA GAAGGTGAAGGTCGGAGT GAPDH 226
Uhlmann GAPDH2 45 50.8 NA NA GAAGATGGTGATGGGATTTC
Kawashima PtF7 forward 54 61.6 4777 4798 ACTCCACAACCGAACCGCACAA Fusion (outer) 834
Kawashima PtF14 reverse 48 54.6 5611 5591 GATGGCTGGAACGAGTCATAA
Kawashima PtF11 forward 52 56.4 5304 5324 GACATCAGTATCCCACAGCCT Fusion (inner) 181
Kawashima PtF13 reverse 56 62.4 5591 5461 TGGCAGAGACGTTCACCTTGAGACC
Kawashima H3 forward 44 55.4 8106 8130 CAGTCAGTAATGATCTCAGCAACTG Hemagglutinin (outer) 595
Kawashima H6 reverse 44 56.4 8677 8701 CTTGAATCTCGGTATCCACTCCAAT
Kawashima H5 forward 52 59.1 8369 8392 TCCCGACAACACGAACAGATGAC Hemagglutinin (inner) 332
Kawashima H6 reverse 44 56.4 8677 8701 CTTGAATCTCGGTATCCACTCCAAT
seconds at a ramp rate of 0.1°C/second. Each reaction tube contained 10L of master mix, 2L (0.5 M) of
forward and reverse inner primers, 4 L of distilled
water, and 2L of DNA from the first round (QuantiTect SYBR green PCR kit, Qiagen). Expected amplicon sizes were 181 bp for the fusion assay and 332 bp for the hemagglutinin assay.
External Standard Curves
The standards were synthesized in vitro from DNA seg-ments amplified by PCR from plasmids containing full-length, Edmonston vaccine-strain MV nucleoprotein, fu-sion, and hemagglutinin genes kindly provided by Dr Alex Valsamakis (the Johns Hopkins Hospital, Balti-more, MD). Briefly, DNA transcripts containing a T3 polymerase site were generated for each targeted se-quence. Using real-time PCR amplification (Lightcycler FastStart DNA Master SYBR green I kit, Roche Applied Science), the in-house forward primer and all 3 of the Uhlmann forward primers were modified to contain the T3 polymerase recognition sequence AATTAACCCTCA-CTAAAGGGACT. The resulting PCR products were pooled and purified on 3% agarose gels. Bands of the correct size were excised, and the products were ex-tracted using the QIAquick gel extraction kit according to the manufacturer’s instructions (Qiagen). The ex-tracted DNA products were then in vitro transcribed using the Megascript kit (Ambion, Austin, TX) to gener-ate the corresponding RNA standard templgener-ates. Tran-scription reactions were conducted for 14 to 16 hours at
37°C. Any residual DNA was removed using 2 L of
Turbo deoxyribonuclease (Ambion) for 30 minutes at 37°C, followed by a further 2-minute incubation at room
temperature with 4 L of deoxyribonuclease
inactiva-tion reagent (Ambion). The supernatant was removed and washed using the MegaClear kit according to the manufacturer’s instructions (Ambion). Final standard template RNA concentrations were determined by fluo-rescence (Ribogreen, Invitrogen) using the Lightcycler (Roche Applied Science). An aliquot of each of the final purified RNA templates was separated by electrophoresis on 1% ribonuclease-free agarose gels to verify purity and size. Standard curves were generated for each assay using 10-fold serially diluted RNA standards to quantify MV gene copy numbers detected over a range of 0 to 107
copies. Carrier transfer RNA (Roche Applied Science) was added to dilutions E4 to E0 at a concentration of 10
ng/L to prevent the standard RNA from adhering to
nonspecific sites. Results of all of the assays are reported in both qualitative (eg, positive/negative based on the
presence of any fluorescence peak at ⬍41 cycles) and
quantitative terms (eg, estimated copy number per mi-croliter based on extrapolation from the standard curve).
Contamination Control
The known susceptibility of PCR to contamination and false-positive results31 was limited by the partial
substi-tution of deoxythimidine triphosphate (dTTP) and de-oxyuridine triphosphate (dUTP) in all of the amplifica-tions and pretreatment of samples with heat-labile uracil-DNA glycosylase ([UNG] Roche Applied Science). UNG catalyzes the hydrolysis of the N-glycosylic bond between the uracil and sugar, leaving an apyrimidinic site in uracil-containing single- or double-stranded DNA. This approach can markedly reduce carry-over contam-ination in PCR by removing uracil incorporated into amplicons, thereby blocking reamplification. Although Uhlmann et al18used 0.01 U of UNG per reaction, it has
subsequently been shown that even 0.5 U per 25L PCR
mix can sometimes fail to degradeⱕ250 copies of
con-taminating DNA.32 We, therefore, incubated each test
sample with between 0.5 and 1 U of UNG per 20L of
PCR mix for 10 minutes at 20°C before PCR for the in-house fusion, Uhlmann (fusion and hemagglutinin), and Kawashima (fusion and hemagglutinin) assays. UNG was inactivated during the RT step (50°C for 20 minutes). Also, DNA Zap (Ambion) was applied to the flow hood, capillary holder, all of the pipettes, tip boxes, and tweezers before each PCR work session.
We closely monitored positive reactions in our non-template and uninfected PBMC controls for any sign of contamination. In addition, all of the samples that flagged positive were routinely rerun in triplicate or quadruplicate to confirm the result before proceeding with further analysis (ie, gel electrophoresis, melt curve inspection, and, ultimately, sequencing). As noted be-low, we modified the Kawashima assays during optimi-zation studies to eliminate the independent first-strand cDNA synthesis. This change both increased efficiency and reduced the potential for contamination. In addi-tion, we monitored positive reactions by melting curve analysis after the first and second rounds of nested PCR to screen for contamination.
Protocol for Samples That Flagged Positive Using Uhlmann Assays
using the Zero Blunt TOPO PCR Cloning Kit for Se-quencing (Invitrogen). Transformants were detected ac-cording to the manufacturer’s protocol. Plasmids were prepared using the PureLink HiPure plasmid DNA puri-fication kit (Invitrogen). Each resulting plasmid
prepa-ration was digested with 1 unit EcoRI (New England
Biolabs, Ipswich, MA) at 37°C for 1 hour then separated on 1% agarose gels at 110 V for 1 hour. Digests contain-ing a band of 4000 bp (expected plasmid vector size) were sent for sequencing using T3 primer (McGill Uni-versity Genome Center, Montreal, Quebec, Canada).
Mega3 software33was used for computer-based analyses
of nucleotide sequences. The Basic Local Alignment Search Tool (BLAST; National Center for Biotechnology
Information)34 was used to find regions of similarity
between amplified sequences and all sequences depos-ited in databases. The program compares these nucleo-tide sequences and calculates the statistical significance of matches.
Protocol for Samples That Flagged Positive Using Kawashima Assays
Positive samples were first analyzed by melting curve analysis after both the first and second round of nested PCR. Next, 5L of first- and second-round PCR product were separated by electrophoresis on 1% agarose gels for 2 hours at 80 to 90 V. In the event that a second-round product had the expected band size, this sample product was pooled and separated on a 1% agarose gel for 3 hours at 55 V, then excised and gel extracted using the QIAquick gel extraction kit (Qiagen). Samples were sent for direct di-deoxy sequencing using the primers that generated the products (McGill University Genome
Cen-ter). Mega3 software33 was used for computer-based
analyses of nucleotide sequences. BLAST34was used to
search the nucleotide database for sequence similarities.
Anti-Measles Immunoglobulin G ELISA
Singh and colleagues35–37have reported higher anti-MV
antibodies in ASD children (vs normal children), arguing that this difference supports the presence of a persistent virus. Briefly, 96-well microtiter plates were coated with
1g/mL of MV antigen in carbonate-bicarbonate buffer
(pH 9.6) overnight at 4°C. Plates were washed 3 times with phosphate-buffered saline-Tween 0.05% (PBS-T) buffer (pH 7.4) and blocked with phosphate-buffered saline-5% nonfat milk at 37°C for 1 hour (300L/well). The blocking buffer was removed and standard (NIBSC [National Institute for Biological Standards and Control, Potters Bar, United Kingdom] 5 IU/mL: twofold serial dilutions) or test plasma (diluted 1:200) were added (100L/well) and incubated at 37°C for 2 hours. After washing 3 times with PBS-T, assays were completed
with goat anti-human-immunoglobulin G-alkaline
phosphatase (100L/well at 1:5000: Sigma) at 37°C for
30 minutes, further PBS-T washes, and 100 L/well of
TM Blue solution (1 mg/mLp-nitrophenylphosphate in
50 mM sodium bicarbonate buffer [pH 9.6] containing 1 mM magnesium chloride; Serologicals Corp, Norcross, GA). The color reaction was stopped with 50L/well of 0.5 mol/L sulfuric acid (Sigma) and was read at 405 nm (Microplate Reader model 3550: Bio-Rad, Richmond, CA). Antibody concentrations in test plasma were esti-mated by extrapolation from the standards included on each plate and are reported as international units.
Ethical Approval
Approval for this study was obtained from the Montreal Children’s Hospital Research Ethics Board. Fully in-formed, written consent was obtained from parents or legal guardians of all of the patients, including controls.
RESULTS
Subject Characteristics
Fifty-one of 54 children from the ASD group and 31 of 34 from the control group had received MMR vaccine. From the ASD group, 2 children had not been vacci-nated, and 1 child’s status was unknown. From the control group, vaccination status was unknown for 3
children. The majority of children with ASD (n ⫽ 43;
84%) and the developmentally normal children (n⫽28;
90%) had received 2 doses. The Quebec immunization schedule recommends a 2-dose MMR schedule at ages 12 and 18 months.14Only 1 (control) child had received
3 doses. Gastrointestinal symptoms, including constipa-tion, cramping, diarrhea, and abnormal stools, were re-ported in 43 (79.6%) of 54 children from the ASD group and 11 (32.3%) of 34 children from the control group. Regression was reported in 20 (37%) of 54 subjects in the ASD group. The time between vaccination and blood analysis was similar between the 2 groups. The mean time (⫾SD) between vaccination and blood sampling for
the ASD group was 30.4 (⫾15.9) months and 26.6
(⫾15.3) months for the control group (2-tailedttest,t⫽
1.056, not significant).
RNA Integrity
Extracted RNA was intact in all of the samples as shown by the presence of discrete 28S and 18S bands on 1% ribonuclease-free agarose gels. The mean amounts of
RNA (⫾SD) for the ASD and control groups were 47.4
(⫾38.2) ng/L and 18.70 (⫾30.4) ng/L, respectively. The difference in extracted RNA amounts is most likely attributable to lower PBMC numbers in the control
group. The mean cell counts (⫾SD) for the ASD and
control groups were 3.2 ⫻ 106 (⫾2.5) and 1.5 ⫻ 106
Optimization of Standard Curves and Assays
Reagent optimization was relatively straightforward, be-cause the SYBR green and probe kits have been preop-timized for use with the Lightcycler. Nonetheless, we verified the band sizes of all of the standards and positive controls on gels. All of the amplified products were of the appropriate size. Based on amplification of the spe-cific RNA templates produced for each real-time PCR assay, the detection limits were 36.5, 35.5, 24.0, and 17.5 copies/L for the Uhlmann nucleoprotein, fusion, and hemagglutinin gene assays and our in-house fusion gene assay, respectively. Slopes for the corresponding standard curves of the optimized assays ranged from ⫺3.505 to⫺3.201 withrvalues between 0.99 and 1.00, indicating high efficiency of the PCR reactions. All of the assays yielded strong positive results using the positive control RNA harvested from infected PBMCs, and am-plicons from the positive controls generated sequences that were 100% identical to the deposited Edmonston MV sequence (accession AF266288). Although there was considerable nonspecific amplification from the negative control RNA (ie, uninfected PBMCs) in all 3 of the Uhlmann primer-based assays, the melting peaks of these amplification products were generally distinct from those from the positive control RNA. Independent first-strand cDNA synthesis was omitted from the Ka-washima protocol, because MV RNA from the positive control was not consistently detected during optimiza-tion studies. Instead, we combined first-strand cDNA synthesis and the first round of PCR to exploit the highly sensitive RTs contained in the SYBR green kit. In addi-tion, in our hands, the Kawashima fusion primers did not produce visible amplification products of the correct size from the positive control RNA after the first round of PCR. However a product was detected by the Lightcycler
with an appropriate melting curve. Both the fusion and hemagglutinin Kawashima assays yielded strong bands on gels after second-round PCR using the positive con-trol RNA.
Uhlmann Primer-Based Assays
Almost all of the PBMC samples yielded positive signals in all of the assays based on the Uhlmann primer pairs (see Fig 1). Because of the high number of positive results using the Uhlmann primers, PCR was not rou-tinely repeated. Using the Uhlmann nucleoprotein prim-ers, 37 (100%) of 37 of the ASD samples and 18 (100%) of 18 of the samples from developmentally normal con-trols were positive. The Uhlmann fusion gene primers yielded 93% positive results in the ASD samples (39 of 42) and 100% of controls (17 of 17). The Uhlmann hemagglutinin assay yielded 100% positive results in the ASD samples (40 of 40) and 100% of controls (13 of 13). On inspection of the melting curves, however, it was clear that only a proportion of PCR products had melting peaks that overlapped with the melting peak of the positive control. For example, only 17 (46%) of 37 of the positive ASD results in the nucleoprotein gene assay had the appropriate melting peak, whereas 6 (33%) of 18 of controls had the appropriate melting peak. Melting peaks were not always reproduced in duplicate samples. Nevertheless, these assays generated copy numbers. The
mean copy numbers (⫾SD) using the Uhlmann
nucleo-protein assay were 5.66E4 (⫾4.41E4) for the ASD group
and 2.03E4 (⫾1.28E4) for the control group. For the
Uhlmann fusion assay, 8 (20.5%) of 39 of the positive ASD results and 3 (17.6%) of 17 controls had the ap-propriate melting peak (mean assigned copy number [⫾SD]: 1.47E3 [⫾1.17E3] for the ASD group and 5.66E2 [⫾5.66E2] for the control group). Finally, 11 (27.5%) of
FIGURE 1
40 of the positive ASD results and 1 of 13 (7.7%) of the controls had the appropriate melting peak for the Uhl-mann hemagglutinin assay (mean assigned copy
num-ber [⫾SD]: 3.81E4 [⫾5.10E4] for the ASD group and
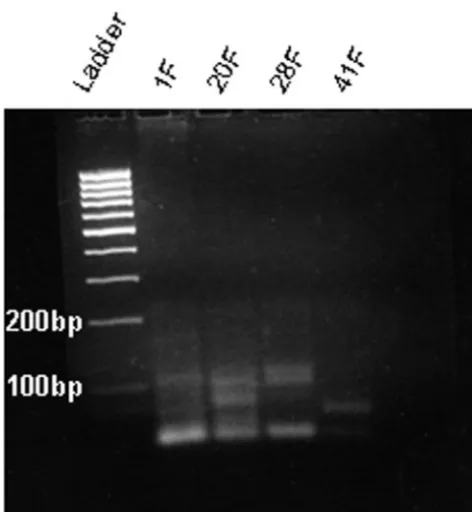
2.16E3 [⫾7.25E3] for the control group). Almost all of the remaining positive results were eliminated by anal-ysis of amplicon size on 3% agarose gels. The Uhlmann assays gave rise to PCR products of ⬃100 bp or less in most cases, demonstrating their lack of specificity (Fig 2). However, 2 of the nucleoprotein gene assay amplicons, 3 of the fusion gene assay amplicons, and 4 of the hem-agglutinin assay amplicons (all from ASD subjects) were of the appropriate size (150 bp; Fig 3). On direct se-quencing, none of these amplification products gener-ated interpretable sequence data. Therefore, we at-tempted to clone these amplicons for sequencing. Seven of the 9 appropriately sized amplicons were successfully cloned and sequenced, but none of these revealed MV RNA. BLAST searches instead revealed a number of different mammalian genes, suggesting nonspecific am-plification of PBMC RNA.34It is unclear why 2 of the 9
positive samples could not be cloned; cloning can be particularly difficult with nonspecific amplification prod-ucts.
Results for the Kawashima Nested PCR Assays
Sufficient sample RNA was available from 23 ASD sub-jects and 16 control subsub-jects to complete nested PCR
testing using the Kawashima primer pairs. No samples were positive using the Kawashima hemagglutinin as-say. One ASD sample generated a band of the correct size (181 bp) using the Kawashima fusion primers but was negative by melting curve analysis after first-round PCR, leading us to suspect contamination. This sample was negative when rerun in quadruplicate.
Probe-Based Fusion Gene Assay
The in-house fusion gene probe-based assay generated positive results for all of the positive control samples and internal standards but did not yield any positive results in test samples from either ASD or control PBMCs.
Anti-Measles Antibody Titers
The mean titer (⫾SD) for the ASD group was 4.3 (⫾3.9)
IU/mL vs 4.6 (⫾4.0) IU/mL for the control group
(2-tailed t test, P ⫽ 0.722, not significant). Three outlier samples were removed from this analysis (1 sample from the ASD group at 120.3 IU and 2 samples from the control group at 266.6 and 86.7 IU). Although
unvacci-nated children (n⫽2) and children of unknown status
(n ⫽ 4) were included in the antibody analysis, their
inclusion did not skew the mean titers (data not shown). Anti-measles antibodies were detectable in all of the subjects, regardless of vaccination history.
DISCUSSION
The main objective of this study was to compare the real-time PCR primers and nested PCR primers used by
Uhlmann et al18 and Kawashima et al19 with our own
real-time PCR primers to determine whether or not MV nucleic acids are present in the PBMCs of children with ASD and age-matched, developmentally normal con-trols. The majority of samples generated positive results in the Uhlmann assays. However, melting curve analyses revealed that only a proportion of these PCR products had the correct melting temperature (ie, identical to the positive MV control). Furthermore, only 2 products gen-erated by the Uhlmann nucleoprotein assay, 3 by the Uhlmann fusion assay, and 4 by the Uhlmann hemag-glutinin assay had the correct amplicon size when sep-arated by gel electrophoresis. We successfully obtained sequences for 7 of these 9 products, but none were MV RNA. No specimen was positive using the Kawashima primer-based assays or our own probe-based fusion gene assay. We also found no significant difference in an-ti-MV antibody titers in ASD subjects and age-matched controls. These data do not support the purported asso-ciation between MMR vaccination and ASD, including the hypothesis that MMR induces regression in a subset of ASD children. These data are consistent with the recently published findings of Afzal et al,38who found no
evidence of persistent MV in leukocyte preparations from MMR-vaccinated children with ASD using TaqMan
FIGURE 2
and nested PCR. Finally, our data reinforce the epidemi-ologic evidence against such an association.15,39
In the course of this work, we experienced many of the vulnerabilities of using PCR technologies to support a claim of association between an organism and an event. Without meticulous attention to these vulnera-bilities, errors in interpretation can occur, as we believe have occurred in the MMR-ASD controversy. First, real-time PCR software will provide copy number informa-tion for almost any amplificainforma-tion product whether or not the product is specific. As a result, this technology can provide what seems to be a highly objective measure of the presence of an organism when such an inference is not justified. Uhlmann et al18claimed that copy number
in their positive samples was generally low (ie, typically
between 1 and 3⫻105MV gene copies/ng total RNA)20
but did not carry their analyses any further. Using the Uhlmann primer pairs, we also obtained copy number
estimates between 3.6 and 3.5⫻103MV gene copies/ng
total RNA (100 and 10 000 copies/L) but demonstrated that these amplicons were not of MV origin by rigorous application of a protocol for melt-curve analysis, ampli-con size determination by electrophoresis, and, ulti-mately, sequencing. There is no evidence that Uhlmann et al18 used a similar protocol for dealing with positive
results. A detailed review of the Uhlmann reagents and protocol provides several plausible explanations for their reported results. First, as noted above, Uhlmann et al18
likely used suboptimal concentrations of UNG in their assays, reducing the efficacy of this contamination con-trol strategy. Second, a ClustalW40 search for sequence
similarity between the human sequences we amplified and the Uhlmann primers and probes revealed several clustered regions of homology, ranging from 3 to 9 nu-cleotides (Fig 4, which is published as supporting infor-mation on www.pediatrics.org/content/full/118/4/1664). It is plausible that these regions of homology permitted
primers, probes, or both to bind, leading to false-positive results. Our data reinforce the principle that great cau-tion must be exercised in interpreting copy number data in the absence of further verification and validation of the amplification products.
Several limitations of our own study deserve men-tion. Although real-time PCR is regarded by many as the gold standard for the detection of microorganisms in human disease, this technique has significant disadvan-tages as well. When the hypothesized target nucleic acid is likely to be present in small quantities, the key issue in real-time PCR is guarding against almost inevitable con-tamination results. Amplification of rare nucleic acids requires rigorous adherence to a set of standard operat-ing procedures, includoperat-ing separate work stations for RNA and DNA handling, decontamination of work ma-terials before every PCR setup, and the optimal use of reagents such as UNG. A single PCR run has the capacity to generate sufficient material to contaminate large areas and all of the subsequent amplifications of the same target sequence. Despite our best efforts, we, like oth-ers,41 experienced several episodes of contamination in
the course of this work. For example, whenever the nontemplate and negative PBMC controls flagged posi-tive, all of the data from that PCR run were discarded and, if sufficient sample RNA was available, the run was repeated. Had we not anticipated such occurrences, we could easily have been misled. If there is reason to question an experimental PCR result, it is always wisest
to repeat the experiment.41 In this context,
uninten-tional contamination is a plausible explanation for Ka-washima et al’s19observation that MV nucleic acids are
present in a variety of conditions, including IBD,19
auto-immune hepatitis,42and epilepsy.43Moreover, both
Uhl-mann et al18 and Martin et al20have reported the
pres-ence of MV nucleic acids in a proportion of control subjects.
FIGURE 3
Second, we took some liberties with the PCR strate-gies used by Uhlmann et al18and Kawashima et al.19For
example, Uhlmann et al18performed TaqMan RT-PCR,
which is a probe-based, fluorescent reporting chemistry, using the EZ TaqMan kit and the ABI 7700 Sequence detector from Applied Biosystems. We chose to use SYBR green I, a nonspecific detection chemistry, because we were interested in assessing the performance of the Uhlmann primers without the use of probes. The SYBR green approach allows the user to carry out initial, ex-ploratory screens of different primer sets before using a probe-based protocol. Interestingly, there is a report sug-gesting that SYBR green I detection is more precise and produces a more linear decay plot than TaqMan detec-tion.44 The use of an internal probe, as in TaqMan, is
supposed to provide a higher level of specificity. In the-ory, nonspecific amplification because of mispriming or primer dimers should not generate a signal in TaqMan assays, because such amplification is ignored by the flu-orescence detector. However, absence of detection is not the same as absence of artifacts. Nonspecific artifact am-plification can certainly affect amam-plification efficiency and subsequent quantification. Because of the theoreti-cal specificity of the TaqMan approach, artifacts that interfere with amplification efficiency typically cannot be detected. As a result, many authorities believe that intercalating dyes should be used to optimize primers and reaction conditions before any quantification exper-iments to ensure the absence of amplification artifacts.45
Our data suggest that the Uhlmann primers are probably not sufficiently efficient to detect MV genes, because they performed suboptimally during our SYBR green I screening. The only other major difference between the TaqMan and the SYBR green approaches is the choice of
RT: TaqMan RT requires MnOAc2to function as a
probe-cleaving nuclease and a DNA polymerase, whereas the SYBR green kit contains a combination of Omniscript and Sensiscript RTs optimized to detect a wide range of RNA template amounts. Another difference is the fact
that Uhlmann et al18 used their nucleoprotein gene
primers to detect the MV nucleoprotein gene by in situ PCR, whereas we used the same primers to detect MV nucleoprotein gene by real-time RT-PCR. Although these primers were not used in precisely the same way, the fact that they resulted in nonspecific amplification in our hands should certainly raise concerns about the in situ data.
A final and obvious difference between our study and those of Uhlmann et al18and Martin et al20is the fact that
we targeted PBMCs instead of gut biopsy material. As noted above, we did not feel that it was ethically justi-fiable to subject the children in this study to endoscopy and biopsy despite the fact that almost 80% of the chil-dren with ASD had gastrointestinal complaints (vs 32% of our control population). Although there is good evi-dence that natural MV accumulates mutations when it
persists in the central nervous system of subjects with subacute sclerosing panencephalitis,46–49there is no
rea-son to postulate that the mutation pattern in a virus persisting in the gut would be qualitatively different from that of a virus persisting in the PBMCs. Although such tissue-specific changes in mutation pattern are the-oretically possible, it seems highly unlikely that the lack of amplification of MV genes in our study using 6 dif-ferent primer pairs and several difdif-ferent PCR strategies can be explained in this way.
In the case of the Kawashima assays, we attempted to replicate their results using the same RNA source (PB-MCs), a similar method for RNA extraction (guanidine salts with an ethanol wash) and a potentially more sen-sitive PCR method that combines the strengths of real-time and nested PCR. In addition, by eliminating the first-strand cDNA synthesis step, we were able to elim-inate a step where contamination could potentially oc-cur. Despite the similarities between the 2 protocols, we were still unable to identify MV nucleic acids from 23 ASD samples and 16 control samples. These observations stand in sharp contrast to the 33% yield from PBMCs isolated from ASD children reported by Kawashima et al.19 There is no reason to believe that the minor
tech-nical differences between our approaches explain these discrepant results.
In light of our findings, it is worth noting that some of the authors who contributed to the Kawashima et al19,
Uhlmann et al18, and Martin et al20 articles had earlier
reported the detection of MV proteins and nucleic acids in the bowel tissues of subjects with IBD using a variety of techniques.50–52This claim also provoked considerable
media attention and undermined public confidence in the safety of measles-containing vaccine.53In a series of
carefully designed experiments, Iizuka and colleagues54,55
demonstrated that the monoclonal antibody used in many of the early studies supporting the MMR-IBD link (MAS 182r) cross-reacted with an unidentified human protein. A novel monoclonal antibody raised against the positive clone (4F12) was found to react to Crohn dis-ease, ulcerative colitis, and non-IBD colitis control tis-sues, providing evidence of its nonspecificity. Further-more by sequencing, Iizuka et al55demonstrated that the
RT-PCR amplicons generated using primers targeting the MV nucleoprotein gene were actually derived from a human rather than a viral gene. The host-derived pro-tein has not yet been identified but may play a role in intestinal inflammation, because neither MAS 182r nor 4F12 antibodies seem to react with control tissues. Our observations raise the possibility that detection of ⱖ1 host-derived genes may again have been mistakenly interpreted as persisting MV, this time in children with ASD.
for prolonged periods of time in some individuals (eg, subacute sclerosing panencephalitis and HIV infected). However, a large burden of proof is required to move from the biological plausibility of an association to es-tablishing that such an association truly exists and is causal. To date, the epidemiologic burden of evidence against such an association in the case of MMR and
autism is overwhelming.15,39We now provide evidence
that the PCR data published by Uhlmann et al,18Martin
et al,20 and Kawashima et al19 in support of the more
limited claim of an association between MMR and a subset of children with ASD is also unlikely to be true.
Our data, together with the epidemiologic evidence, demonstrate that arguments against vaccinating chil-dren with MMR because of fear of ASD are not de-fensible on scientific grounds. The risk of death and disability from MV infection has been unequivocally demonstrated. The hypothesized link between MMR and ASD is spurious and undermines the success of measles control programs.56
Editor’s note: Please read the commentary by Dr Samuel Katz in this issue of Pediatrics.
ACKNOWLEDGMENTS
The technical work for this study was funded by the Crohn’s and Colitis Foundation of Canada. Clinical data collection was supported by a grant to Dr Fombonne from the Fonds de Recherche en Sante´ du Que´bec.
We thank Rita Zakarian for blood collection and com-pilation of patient information, Nathalie Martel and An-gela Brewer for technical assistance, and Christine Turenne and Marcel Behr for their help with evaluating sequence data and reviewing an earlier version of this article.
REFERENCES
1. Wakefield AJ, Murch SH, Anthony A, et al. Ileal-lymphoid-nodular hyperplasia, non-specific colitis, and pervasive devel-opmental disorder in children.Lancet.1998;351:637– 641 2. Dales L, Hammer SJ, Smith NJ. Time trends in autism and
MMR immunization coverage in California.JAMA.2001;285: 1183–1185
3. Kaye JA, del Mar Melero-Montes M, Jick H. Mumps, measles, and rubella vaccine and the incidence of autism recorded by general practitioners: a time trend analysis. BMJ.2001;322: 460 – 463
4. Chen W, Landau S, Sham P, Fombonne E. No evidence for links between autism, MMR and measles virus.Psychol Med.
2004;34:543–553
5. DeStefano F, Bhasin TK, Thompson WW, Yeargin-Allsopp M, Boyle C. Age at first mumps-rubella vaccination in children with autism and school-matched control subjects: a popula-tion-based study in metropolitan Atlanta.Pediatrics.2004;113: 259 –266
6. Farrington CP, Miller E, Taylor B. MMR and autism: Further evidence against a causal association.Vaccine.2001;19:3632– 3635
7. Fombonne E, Chakrabarti S. No evidence for a new variant of
measles-mumps-rubella-induced autism. Pediatrics. 2001; 108(4). Available at: www.pediatrics.org/cgi/content/full/108/ 4/e58
8. Madsen KM, Hviid A, Vestergaard M, et al. A population-based study of measles, mumps, and rubella vaccination and autism.
N Engl J Med.2002;347:1477–1482
9. Makela A, Nuorti JP, Peltola H. Neurologic disorders after measles-mumps-rubella vaccination. Pediatrics. 2002;110: 957–963
10. Takahashi H, Suzumura S, Shirakizawa F, et al. An epidemio-logical study on Japanese autism concerning routine childhood immunization history.Jpn J Infect Dis.2003;56:114 –117 11. Taylor B, Miller E, Farrington CP, et al. Autism and measles,
mumps, and rubella vaccine: no epidemiological evidence for a causal association.Lancet.1999;353:2026 –2029
12. Taylor B, Miller E, Lingam R, et al. Measles, mumps, and rubella vaccination and bowel problems or developmental re-gression in children with autism: Population study.BMJ.2002; 324:393–396
13. Smeeth L, Cook C, Fombonne E, et al. MMR vaccination and pervasive developmental disorders: a case-control study. Lan-cet.2004;364:963–969
14. Fombonne E, Zakarian R, Bennett A, et al. Pervasive develop-mental disorders in Montreal, Quebec: prevalence and links with immunizations. Pediatrics 2006;118(1). Available at: www.pediatrics.org/cgi/content/full/118/1/e139
15. Halsey NA, Hyman SL. Conference Writing Panel. Measles-mumps-rubella vaccine and autistic spectrum disorder: report from the New Challenges in Childhood Immunizations Con-ference convened in Oak Brook, Illinois, June 12–13, 2000.
Pediatrics. 2001;107(5). Available at: www.pediatrics.org/cgi/ content/full/107/5/e84
16. Institute of Medicine of the National Academies.Immunization Safety Review: Vaccines and Autism. Washington, DC: The Na-tional Academies Press; 2004.
17. Demicheli V, Jefferson T, Rivetti A, et al. Vaccines for measles, mumps and rubella in children. Cochrane Database Syst Rev.
2005;(19):CD004407
18. Uhlmann V, Martin CM, Sheils O, et al. Potential viral patho-genic mechanism for new variant inflammatory bowel disease.
Mol Pathol.2002;55:84 –90
19. Kawashima H, Mori T, Kashiwagi Y, et al. Detection and se-quencing of measles virus from peripheral mononuclear cells from patients with inflammatory bowel disease and autism.Dig Dis Sci.2000;45:723–729
20. Martin CM, Uhlmann V, Killalea A, et al. Detection of measles virus in children with ileo-colonic lymphoid nodular hyperpla-sia, enterocolitis and developmental disorder.Mol Psychiatry.
2002;7(suppl 2):S47–S48
21. Fitzpatrick M. MMR: Risk, choice, chance.Br Med Bull.2004; 69:143–153
22. Department of Health. Statistical Bulletin: NHS Immunisation Statistics 2004 – 05. London, United Kingdom: Department of Health; 2005
23. Afzal MA, Minor PD. Vaccines, Crohn’s disease and autism.Mol Psychiatry.2002;7(suppl 2):S49 –S50
24. Horton R. A statement by the editors of the Lancet. Lancet.
1998;363:820 – 821
25. Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for care-givers of individuals with possible pervasive developmental disorders.J Autism Dev Disord.1994;24:659 – 685
26. Lord C, Risi S, Lambrecht L, et al. The Autism Diagnostic Observation Schedule-Generic: A standard measure of social and communication deficits associated with the spectrum of autism.J Autism Dev Disord.2000;30:205–223
Man-ual of Mental Disorders - DSM IV. Washington, DC: American Psychiatric Association; 1994
28. Kazak Berument SK, Rutter M, Lord C, et al. Autism screening questionnaire: Diagnostic validity.Br J Psychiatry. 1999;175: 444 – 451
29. Bertley FMN, Ibrahim S, Hoskins M, et al. Early exposure to measles vaccine antigens primes for a balanced humoral and cellular response upon revaccination.Vaccine.2004;23:444 – 449 30. Qiagen. QuantiTect Gene Expression Assays. Available at www1.
qiagen.com/Products/Pcr/QuantiTect/GeneExpressionAssays. aspx. Accessed July 28, 2005
31. Pierce KE, Wangh LJ. Effectiveness and limitations of uracil-DNA glycosylases in sensitive real-time PCR assays. Biotech-niques.2004;36:44 – 48
32. Kleiboeker SB. Quantitative assessment of the effect of uracil-DNA glycosylase on amplicon uracil-DNA degradation and RNA am-plification in reverse transcription-PCR.Virol J.2005;2:29 33. Kumar S, Tamura K, Nei M. MEGA3: Integrated software for
molecular evolutionary genetics analysis and sequence align-ment.Brief Bioinform.2004;5:150 –163
34. Altschul SF, Madden TL, Scha¨ffer AA, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search pro-grams.Nucleic Acids Res.1997;25:3389 –3402
35. Singh VK, Lin SX, Yang VC. Serological association of measles virus and human herpesvirus-6 with brain autoantibodies in autism.Clin Immunol Immunopathol.1998;89:105–108 36. Singh VK, Lin SX, Newell E, et al. Abnormal
measles-mumps-rubella antibodies and CNS autoimmunity in children with autism.J Biomed Sci.2002;9:359 –364
37. Singh VK, Jensen RL. Elevated levels of measles antibodies in children with autism.Pediatr Neurol.2003;28:292–294 38. Afzal MA, Ozoemena LC, O’Hare A, et al. Absence of
detect-able measles virus genome sequence in blood of autistic chil-dren who have had their MMR vaccination during the routine childhood immunization schedule of UK.J Med Virol.2006;78: 623– 630
39. Wilson K, Mills E, Ross C, et al. Association of autistic spectrum disorder and the measles, mumps, and rubella vaccine: a sys-tematic review of the current epidemiological evidence.Arch Pediatr Adolesc Med.2003;157:628 – 634
40. Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22: 4673– 4680
41. Ghosh S, Armitage E, Wilson D, et al. Detection of persistent measles virus infection in Crohn’s disease: current status of experimental work.Gut.2001;48:748 –752
42. Kawashima H, Mori T, Takekuma K, et al. Polymerase chain reaction detection of the hemagglutinin gene from an attenu-ated measles vaccine strain in the peripheral mononuclear cells
of children with autoimmune hepatitis.Arch Virol.1996;141: 877– 884
43. Kawashima H, Miyajima T, Mori T, et al. A case of intractable epilepsy positive for the detection of measles virus genome in the cerebrospinal fluid and peripheral mononuclear cells using reverse transcriptase-polymerase chain reaction. Brain Dev.
1996;18:220 –223
44. Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: Comparison of endpoint and real-time methods.Anal Biochem.2000;285:194 –204 45. Bustin SA, Nolan T. Pitfalls of quantitative real-time
reverse-transcription polymerase reaction.J Biomol Tech.2004;15:155– 166
46. Patterson JB, Cornu TI, Redwine J, et al. Evidence that the hypermutated M protein of a subacute sclerosing panencepha-litis measles virus actively contributes to the chronic progres-sive CNS disease.Virology.2001;291:215–225
47. Billeter MA, Cattaneo R, Spielhofer P, et al. Generation and properties of measles virus mutations typically associated with subacute sclerosing panencephalitis.Ann N Y Acad Sci. 1994; 724:367–377
48. Cattaneo R, Schmid A, Eschle D, et al. Biased hypermutation and other genetic changes in defective measles viruses in hu-man brain infections.Cell.1988;55:255–265
49. Cattaneo R, Schmid A, Rebmann G, et al. Accumulated mea-sles virus mutations in a case of subacute sclerosing panencephalitis: interrupted matrix protein reading frame and transcription alteration.Virology.1986;154:97–107
50. Wakefield AJ, Pittilo RM, Sim R, et al. Evidence of persistent measles virus infection in Crohn’s disease.J Med Virol.1993; 39:345–353
51. Lewin J, Dhillon AP, Sim R, et al. Persistent measles virus infection of the intestine: confirmation by immunogold elec-tron microscopy.Gut.1995;36:564 –569
52. Daszak P, Purcell M, Lewin J, et al. Detection and comparative analysis of persistent measles virus infection in Crohn’s disease by immunogold electron microscopy.J Clin Pathol. 1997;50: 299 –304
53. Feeney M, Clegg A, Winwood P, et al. A case-control study of measles vaccination and inflammatory bowel disease.Lancet.
1997;350:764 –766
54. Iizuka M, Masamune O. Measles vaccination and inflamma-tory bowel disease.Lancet.1997;350:1775
55. Iizuka M, Chiba M, Yukawa M, et al. Immunohistochemical analysis of the distribution of measles related antigen in the intestinal mucosa in inflammatory bowel disease.Gut.2000; 46:163–169
56. Meissner HC, Strebel PM, Orenstein WA. Measles vaccines and the potential for worldwide eradication of measles.Pediatrics.
DOI: 10.1542/peds.2006-1262
2006;118;1664
Pediatrics
Yasmin D'Souza, Eric Fombonne and Brian J. Ward
From Children With Autism Spectrum Disorder
No Evidence of Persisting Measles Virus in Peripheral Blood Mononuclear Cells
Services
Updated Information &
http://pediatrics.aappublications.org/content/118/4/1664
including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/118/4/1664#BIBL
This article cites 49 articles, 11 of which you can access for free at:
Subspecialty Collections
b
http://www.aappublications.org/cgi/collection/infectious_diseases_su
Infectious Disease following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
DOI: 10.1542/peds.2006-1262
2006;118;1664
Pediatrics
Yasmin D'Souza, Eric Fombonne and Brian J. Ward
From Children With Autism Spectrum Disorder
No Evidence of Persisting Measles Virus in Peripheral Blood Mononuclear Cells
http://pediatrics.aappublications.org/content/118/4/1664
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.