R E S E A R C H
Open Access
Inflammation contributes to NKX3.1 loss and
augments DNA damage but does not alter
the DNA damage response via increased
SIRT1 expression
Bilge Debelec-Butuner
1,2, Nursah Ertunc
1and Kemal Sami Korkmaz
1*Abstract
The oxidative stress response is a cellular defense mechanism that protects cells from oxidative damage and cancer development. The exact molecular mechanism by which reactive oxygen species (ROS) contribute to DNA damage and increase genome instability in prostate cancer merits further investigation. Here, we aimed to determine the effects of NKX3.1 loss on antioxidant defense in response to acute and chronic inflammation in anin vitromodel. Oxidative stress-induced DNA damage resulted in increased H2AX(S139)phosphorylation (a hallmark of DNA damage), along with the degradation of the androgen receptor (AR), p53 and NKX3.1, upon treatment with conditioned medium (CM) obtained from activated macrophages or H2O2. Furthermore, the expression and stability of SIRT1 were increased by CM treatment but not by H2O2treatment, although the level of ATM
(S1981)
phosphorylation was not changed compared with controls. Moreover, the deregulated antioxidant response resulted in upregulation of the pro-oxidant QSCN6 and the antioxidant GPX2 and downregulation of the antioxidant GPX3 after CM treatment. Consistently, the intracellular ROS level increased after chronic treatment, leading to a dose-dependent increase in the ability of LNCaP cells to tolerate oxidative damage. These data suggest that the inflammatory microenvironment is a major factor contributing to DNA damage and the deregulation of the oxidative stress response, which may be the underlying cause of the increased genetic heterogeneity during prostate tumor progression.
Keywords:ROS, NKX3.1, Inflammatory microenvironment, Prostate tumor, DNA damage
Introduction
Oxidative stress contributes to the initiation, promotion and progression of carcinogenesis. Excessive levels of re-active oxygen species (ROS) are generated by exposure to oxidative stress and cause sustained DNA damage. DNA damage occurs during the initiation step of car-cinogenesis, and most likely results in abnormal gene expression. Because ROS accumulation results in a sub-sequent failure of signal transduction at the promotion step, the cells undergo a loss of genomic fidelity during the progression step [1]. Notably, ROS are generated in excess amounts during chronic inflammation, and ROS-mediated DNA damage alters the genetic composition.
This damage may promote oncogenic transformation, which occurs when genes encoding essential factors in-volved in DNA repair, apoptosis and cell cycle regulation are affected [2]. Others have shown that ROS contribute to carcinogenesis by activating signaling pathways that regulate cellular proliferation, angiogenesis and metastasis [3-5]. ROS serve as secondary messengers for the activa-tion of key transcripactiva-tion factors in response to pro-inflammatory cytokines, and they also regulate the transcription of genes involved in inflammatory responses [6,7]. Additionally, a number of protein kinase pathways, such as the MAPK pathway, are activated by oxidative sig-nals during inflammation and inflammatory diseases. These pathways synergistically contribute to the activation of cytokine release, and combined with the loss of adhe-sion and the release of angiogenic factors, they may even-tually contribute to cellular proliferation, differentiation
* Correspondence:[email protected]
1
Department of Bioengineering, Cancer Biology Laboratory, Faculty of Engineering, Ege University, Bornova, 35100 Izmir, Turkey
Full list of author information is available at the end of the article
and tumor progression [7]. ROS play important roles in multiple signal transduction pathways, such as those me-diated by TNFα[8] and p53. These two pathways can acti-vate each other, as DNA damage caused by TNFα-induced ROS directly induces cell cycle and/or apoptosis regula-tion by p53 [9]. In addiregula-tion to the ability of cells to trigger proliferation in response to sustained ROS production at low levels, excess ROS generation or the accumulation of ROS may induce cell death depending on the ROS con-centration and cell type [4]. Although ROS can induce apoptosis or necrosis depending on the oxidative level [3], the appropriate cellular responses to ROS production are critical events and may protect the cell from sustained oxi-dative damage and support cell survival.
To reduce the oxidative damage caused by ROS, the antioxidant response system is activated through several constitutive and inducible detoxification mechanisms, including the expression of enzymes such as glutathione peroxidases (GPx), catalase, superoxide dismutases (SODs), peroxiredoxins (PRDXs), glutathione-S-transferases (GSTs), NADP (H) quinone oxidoreductase (NQO1), epoxide hy-drolase, heme oxygenase (HO-1), UDP-glucuronosyl trans-ferases (UGTs), and gamma-glutamylcysteine synthetase. The upregulated expression of these enzymes exhibits a distinct cellular defense to protect cells from oxidative damage and cancer development [7,10].
In addition to the mechanisms that regulate ROS, cell proliferation is controlled by stress-sensing molecules such as Sirtuin 1 (SIRT1), an NAD-dependent deacety-lase that responds to the levels of redox pairs NAD+/ NADH and NADP+/NADPH. Under oxidative conditions, SIRT1 deacetylates a number of transcription factors, in-cluding p53, NBS1 and FOXO, which subsequently con-tribute to cellular metabolic responses, such as cell cycle regulation and DNA damage [7,11,12]. Therefore, SIRT1 has been linked to tumor cell survival by deregulating apoptosis and promoting senescence. Particularly, in pros-tate cancer, the deacetylation of AR by SIRT1 represses androgen-induced AR transcription and contributes to AR-induced tumorigenesis [13].
NKX3.1 is an androgen-regulated gene that encodes a homeobox protein with a tumor suppressor function in prostate cells [14,15]. The AR response is ubiquitous in prostate tumors, and NKX3.1 is upregulated by andro-gens; in contrast, NKX3.1 loss has been reported in prostate tumors [16]. Furthermore, the functional loss of NKX3.1 expression upon cytokine exposure has been re-ported in previous studies of the inflammatory micro-environment [17,18], strengthening its tumor suppressor role in prostate carcinogenesis. The pro-inflammatory cytokines TNFα and IL-1βinduce the C-terminal phos-phorylation of NKX3.1 by casein kinase 2 (CK2), result-ing in a shortened half-life [17,18]. Additionally, loss of NKX3.1 expression in pathogenE. coliinfected prostate
lobes in mice has been shown to be correlated with re-duced AR expression [19]. It was previously reported that the loss of NKX3.1 expression was related not only to the loss of AR transactivating function [17,18] but also to high ROS level upon cytokine exposure, particu-larly TNFα[17]. Concurrently, the loss of p53 expression was also observed in the inflammatory microenvironment, promoted the progression of prostate cancer, perhaps cor-relating with increased oxidative stress. This effect was partially restored by suppressing AKT and MDM2 phos-phorylations, leading to p53 degradation [18].
In this study, we aimed to identify the role of cytokine-induced NKX3.1 loss in the deregulation of the antioxi-dant defense during acute and chronic exposure to both cytokines and ROS. Therefore, the effect of antioxidant treatment on the inflammation- and/or oxidative stress-induced degradation of NKX3.1, AR and p53 was ana-lyzed. Cultures of the prostate cancer cell line LNCaP were exposed to conditioned medium (CM) with adjusted amounts of pro-inflammatory cytokines (TNFα) for 24 h for acute treatment and for 2 weeks for chronic treatment. Cells were also chronically treated with H2O2for 2 weeks
to compare the effects of pro-inflammatory cytokines and ROS exposure.
Materials and methods
Macrophage differentiation and conditioned media (CM) collection
The U937 monocyte cell line was cultured in RPMI 1640 medium including 10% FBS (fetal bovine serum) at 37°C with 5% CO2. To achieve macrophage
differenti-ation and cytokine production, cells (8×105) were seeded into 75-cm2culture flasks 2 h prior to treatment. Next, PMA was added at a final concentration of 16 nM for 16 h, and the adherent clusters (differentiated mono-cytes) were maintained. The cells were washed twice be-fore the addition of 20 ml of fresh medium, and the cells were then allowed to rest for 3 h. Then, lipopolysacchar-ide (LPS) was added at a final concentration of 10 ng/ml to induce cytokine secretion. The cells were incubated for an additional 24 h, and the supernatant (conditioned medium - CM) was collected and filtered (using a 0.22-μm filter) for further use. To ensure that the CM was cell-free, diluted CM was cultured in an empty flask (25 cm2) for one week and analyzed.
Measurement of cytokines in CM
for the courses of CM treatments were optimized as re-ported in our previous study [18]. TNFα was chosen as a measure of the CM concentration, which was adjusted by diluting the CM with normal medium before applica-tion to the LNCaP cells. As a result, the concentraapplica-tions of macrophage-secreted cytokines were adjusted and maintained at picogram levels. In our studies, the effect-ive concentration of TNFα was 400 times less than the concentration of recombinant TNFα (rTNFα) (sigma, UK) reported in other studies [17-19].
Cell culture and treatments
LNCaP cells were obtained from the American Type Cul-ture Collection (ATCC, Manassas, VA, USA) and were propagated as recommended using RPMI 1640 medium supplemented with 10% FBS, L-glutamine (2 mM), peni-cillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C with 5% CO2. For the acute exposures, the CM (62, 125,
and 250 pg/ml of TNFα) treatments were performed for 24 h; for the chronic exposures, the CM treatments con-tinued for 2 weeks, and lower doses (50 and 100 pg/ml of TNFα) were used. TNFαconcentrations were adjusted by diluting the CM using RPMI 1640 medium as described previously [18]. A chronic oxidative condition was also in-duced by treating the cells with 50, 100 or 200μM H2O2
for 2 weeks for comparison of the effects of cytokine ex-posure and oxidative stress.
Transfections
TheNKX3.1open reading frame was amplified (using the primers F: GGATCCATGCTCAGGGTTCCGGAGCCG and R: GAATTCGGTTGTCACCTGAGCTGGCATTA) and cloned into the pcDNA4/HisMax-TOPO vector (Invi-trogen, USA) according to the manufacturer’s instructions to obtain HM-NKX3.1 and the HM-vector constructs. Then, transfections were performed using the Fugene HD reagent (Roche, Germany) for 24 h. The cells were incu-bated for an additional 18 to 42 h, as appropriate.
The siNKX3.1 and siAR transfections were performed as recommended by the supplier (Dharmacon). Briefly, 4×105 cells were seeded into 6-cm plates, and the medium was changed (w/o antibiotics). A transfection mix was prepared by adding 6μl of Dharmafect II (tube 1) and 200 pmol of siNKX3.1, siAR or scrambled siRNA (tube 2) into 94 μl of transfection medium (w/o antibi-otics and serum). After incubation for 5 min at RT, the tubes were mixed and incubated for 15 min at RT and then added onto the cells dropwise. The transfected cells were incubated for an additional 24 h before harvesting.
Antibodies
The following antibodies were purchased from the man-ufacturers: AR (Millipore, USA), p53, pH2AX(S139) and pATM(S1981) (Abcam, UK), GAPDH (Ambion, UK), β
-actin and SIRT1 (Sigma, UK), Caspase-3 (R&D, UK), and
β-tubulin (ABM, UK). The NKX3.1 custom antibody was a gift from Prof. Dr. F. Saatcioglu (University of Oslo). The HRP-conjugated anti-mouse and anti-rabbit (Amersham, UK) and the AlexaFluor 488- and 594-conjugated second-ary antibodies (Invitrogen, USA) were purchased and used as recommended by the manufacturers.
DCFH assay
LNCaP cells (8×103) were seeded into 96-well plates, and the transfections were carried out on the following day. Two days later, the cells were incubated with DCFH-DA (2′ 7′- dichlorodihydrofluorescein diacetate, Molecular Probes, 10μM) for 30 min at 37°C. Next, the treatments were performed following gentle washes using phenol red-free medium. Finally, the fluorescence intensity was measured every 20 min for up to 3 h using a Fluoroscan fluorometer (Thermo Science, USA).
Protein extraction and western blotting
For protein extraction, LNCaP cells were lysed using a modified RIPA buffer (10 mM Tris-Cl (pH: 8.0), 1% Triton X-100, 0.1% SDS, 0.1% Na deoxycholate, 1 mM EDTA, 1 mM EGTA, 140 mM NaCl) containing protease and phosphatase inhibitors. Then, the concentrations were de-termined using the BCA assay (Sigma, UK). SDS-PAGE and western blots were performed under standard condi-tions with 50 μg of protein lysate per lane. The proteins were separated on 10-12% gels and transferred to PVDF membranes (Amersham, UK) using a wet transfer blotter. The PVDF membrane was blocked with 5% dry milk in TBS-T (Tris-Buffered-Saline solution containing 0.1% Tween 20). The primary and secondary antibody incuba-tions were performed in TBS-T containing 0.5% dry milk or 5% BSA at RT for 1 h or at 4°C o/n. The membranes were developed using the ECL prime reagent (Amersham, UK) for 5 min and were photographed using Kodak X-Ray films in a dark room.
Real-time cell proliferation assay
The Xcelligence proliferation assay platform was used for real-time measurements. Briefly, the LNCaP cells (8×103) were transfected with an HM vector and HM-NKX3.1 (24 h), seeded into 96-well plates (E-plates, Roche GmbH, Germany) and cultured for 24 h. The treatments were per-formed as described, and the proliferation rate and mor-phological changes were monitored. Impedance values were collected every 10 min for 48 h.
cDNA synthesis
synthesis kit (Invitrogen, USA) as recommended by the manufacturer.
Real time PCR
To study the expression of specific genes, quantitative RT-PCR was performed using a SYBR Green PCR kit and the LC480 PCR system (Roche, Germany). The rela-tive abundance of each transcript was calculated using the comparative cycle threshold (CT) method with GAPDH as an invariant control. The following primers were used: GPX2_F: CAGTCTCAAGTATGTCCGT, GPX2_R: AGGC TCAATGTTGATGGT; GPX3_F: CTTGCACCATTCGG TCT, GPX3_R: CGGACATACTTGAGGGTAG; PRDX6_F: TAGTGTGATGGTCCTTCCAAC, PRDX6_R: AGCGGA GGTATTTCTTGC; QSCN6_F: GAGGCTACGTGCACT ACT, QSCN6_R: CTGCAAGGCGAGCATTGA; ENOX2_ F: CTGAACGTGAAGCACTG, ENOX2_R: ATCAAGAC GGTGCAAGTAG; SOD1_F: TGTACCAGTGCAGGTCC, SOD1_R: GCCAATGATGCAATGGTC; SOD2_F: TGTCC AAGGCTCAGGTT, SOD2_R: CTGAAGGTAGTAAGCG TGC; NKX3.1_F: TCTATCAGCATCTGACAGGTGAA, NKX3.1_R: AGCAGGGTTTGTTATGCATGTAG; SIRT1_ F: TGCGGGAATCCAAAGGATAATTCAGTGTC, SIRT1_ R: CTTCATCTTTGTCATACTTCATGGCTCTATG; and GAPDH_F: CATTGCCCTCAACGACCACTTT, GAPDH_ R: GGTGGTCCAGGGGTCTTACTCC.
Statistics
Student’s t test was applied to determine the statistical significance between pairs where necessary.
Results and discussion
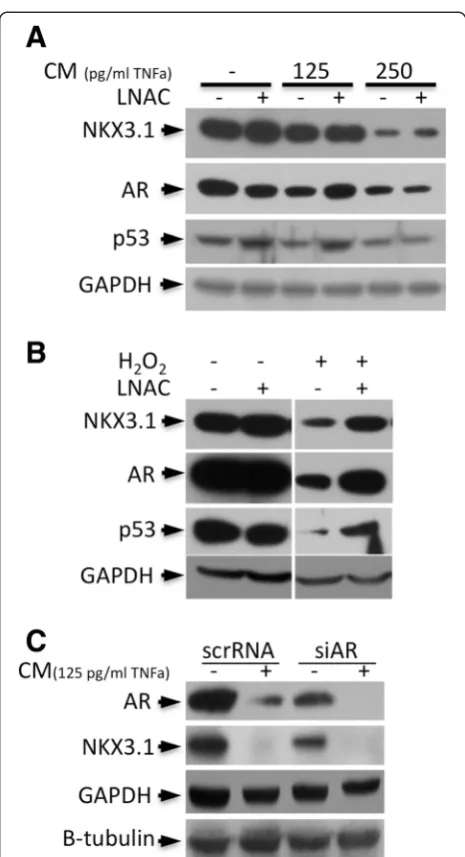
NKX3.1, AR, and p53 degradation is restored by LNAC treatment
To determine whether inflammatory cytokine or oxida-tive exposure is the major factor in ROS-dependent re-duction of AR, NKX3.1 and p53 protein levels, LNCaP cells were treated with CM or H2O2with or without the
antioxidant, N-Acetyl-L-cysteine (LNAC). The CM treat-ments resulted in the degradation of AR, p53 and NKX3.1 in a dose-dependent manner, which is consistent with our previous studies [18]. Interestingly, LNAC treatment par-tially restored AR, but not NKX3.1, at low (125 pg/ml of TNFα) concentrations of CM (Figure 1A). Because the antioxidant treatment could not significantly change the expression level of NKX3.1 and AR under inflammatory conditions (CM treatment), cytokine exposure was sug-gested as the major cause of NKX3.1 and AR depletion. Nevertheless, AR and NKX3.1 degradation in the presence of H2O2 were almost completely restored back to basal
levels by LNAC (Figure 1B), suggesting that there is clear crosstalk between ROS and cytokine signaling. Also, the p53 level was slightly increased by LNAC in the control cells, and it was completely restored back to the basal
expression level at lower doses of CM (Figure 1A). Because LNAC also enhanced the p53 level after H2O2treatment
(Figure 1B), these results implied that p53 degradation under inflammatory conditions was dependent upon the increased ROS levels. Furthermore, we performed AR si-lencing together with CM treatment in LNCaP cells, and the results showed that the reduced NKX3.1 level was a consequence of the synergetic effects of repression of AR transactivation due to AR depletion and CM-mediated proteasomal degradation (Figure 1C).
Figure 1Antioxidant LNAC treatment restores AR, NKX3.1 and p53 expression levels.In LNCaP cells, the expression levels that were reduced by treatment withA. CM (including 125 and 250 pg/ml of TNFα) andB. H2O2(250μM) for 24 h were partially restored back to
NKX3.1 is required for antioxidant gene expression to limit oxidative damage
NKX3.1 is an important androgen-regulated transcrip-tion factor in prostate and testis tissues. Here, CM (250 pg/ml of TNFα) treatments were performed for 3, 6 and 24 h. The results indicate that NKX3.1 loss was cor-related with antioxidant responsive gene expression in LNCAP cells. We found that the expression of the pro-oxidants Quiescin Q6 (QSCN6) and Ecto-NOX disulfide thiol exchanger 2 (ENOX2) was increased by 2.7 and 1.3-fold relative to the controls, respectively. The anti-oxidant glutathione peroxidase-2 (GPX2) was upregu-lated 6.2-fold, and glutathione peroxidase-3 (GPX3) and peroxiredoxin-6 (PRDX6) were downregulated 12.5- and 2.4-fold, respectively. Further, the antioxidant superoxide dismutase-1 (SOD1) was downregulated 1.5-fold, whereas superoxide dismutase-2 (SOD2) was upregulated 2.3-fold after 24 h of CM treatment (Figure 2A). Because an ap-proximately 30-fold reduction of NKX3.1 was observed upon CM exposure, these data support the finding that the loss of NKX3.1 could be related not only to cytokine
exposure [17] but also to the loss of the androgen receptor.
To investigate whether the loss of the oxidative stress response in prostate cancer cells was related to the loss of NKX3.1 expression or androgen signaling during in-flammation, NKX3.1 was depleted by transfection with siRNA for 48 h, and the mRNA levels of specific genes were quantified. The analyzed genes were classified into two groups. The first group consisted of the genes whose expression was dose-dependently affected by treatment with CM. This group included GPX2 and QSCN6, which were upregulated by 5.6-fold and 1.9-fold, respectively, and PX3, PRDX6 and SOD1, which were downregulated by 2.4-fold, 2-fold and 2.5-fold, respectively (Figure 2B and C). The second group consisted of the genes whose expression was influenced by NKX3.1 depletion but not by CM treatment (Figure 2B and C). This group included ENOX2, which was upregulated by 3.7-fold compared to the control, and SOD2, which was downregulated by 2.5-fold. NKX3.1 depletion was confirmed by a 4.8-fold decrease in the mRNA level (Figure 2). In addition, the
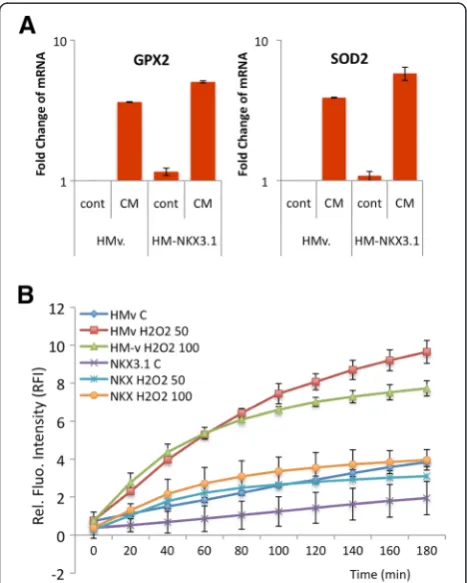
native expression of NKX3.1 in LNCaP cells was supple-mented with ectopic NKX3.1 expression, and its role in oxidative regulation was investigated. Ectopic NKX3.1 ex-pression significantly increased the exex-pression of GPX2 (3.7 to 5.1) and SOD2 (3.8 to 5.9) upon CM exposure (Figure 3A). These data suggested that the restored NKX3.1 expression back to its normal levels enhanced the antioxidant response in the inflammatory microenviron-ment. Moreover, the intracellular ROS level was examined using the DCFH assay following the treatment of NKX3.1-overexpressing LNCaP cells with H2O2(50 and 100 μM).
Upon H2O2treatment, a significant reduction in the ROS
level was observed not only in control cells with basal NKX3.1 expression but also in cells with ectopic NKX3.1 expression (Figure 3B). The data demonstrate that LNCaP cells require NKX3.1 expression for regulation of the anti-oxidant response during inflammation in the prostate.
SIRT1 leads to oxidative stress resistance in the inflammatory microenvironment
The accumulation of damage to DNA, proteins and lipids is characterized by an increase in intracellular
oxidative stress levels due to a progressive decrease of ROS scavenging [18,20]. Several lines of evidence indi-cate that the loss of oxidative tolerance is age dependent and associated with DNA damage as well as metabolic deregulation [21]. The SIRT1-mediated increase in oxi-dative stress tolerance and the concurrent activation of the p53-mediated DNA damage response were corre-lated with an extended lifespan in mouse models. A de-crease in p53 activity reduces the essential role of p53 in tumor prevention in older animals [22]. Thus, a dra-matic increase in the frequency of cancer provides a likely explanation for the correlation between tumori-genesis and the accumulation of DNA mutations [23,24] that might be related to a decreased stress tolerance and increased genetic heterogeneity during multiple inflam-matory exposures over a lifetime.
p53 is found to be completely lost or mutated at a high frequency in advanced prostate cancer [25], but many other tumor suppressors also contribute to the regulation of cell proliferation and apoptosis. An in-crease in the stability of p53 by hyperacetylation via sev-eral acetylases activates p53 to trigger apoptosis and cell cycle arrest. Conversely, the deacetylation of p53 induced by sustained SIRT1 expression enhances the destruction of p53 through ubiquitin-mediated proteasomal degrad-ation [26]. This work suggests that the activdegrad-ation of SIRT1 (via decreased metabolic events) unexpectedly increases the genetic heterogeneity in the inflammatory micro-environment, where the DNA damage response may not be activated. Because high expression of SIRT1 is a com-mon and relevant pathologic event in prostate cancer [27], we examined DNA damage, the subsequent acti-vation of SIRT1 and the DNA damage response. We observed that oxidative stress strongly increased the
γ-H2AX(S139) levels (a hallmark of DNA damage) upon treatment with CM and H2O2, concurrent to the increase
in SIRT1 expression/stability upon CM treatment, al-though the γ-H2AX(S139) levels were not increased upon treatment with H2O2. However, the antioxidant (LNAC)
restored SIRT1 expression to its basal level in cells ex-posed to high concentrations of CM (Figure 4A). Because the CM-induced DNA damage cannot be restored by LNAC, the γ-H2AX(S139) level remained high upon CM treatment and was partially restored upon H2O2
treat-ment. CM treatment results in a significant amount of DNA damage, suggesting that the level of DNA damage is high in the inflammatory microenvironment but not in more highly oxidative conditions, in which the putative re-sponsive pathways remain to be elucidated. On the other hand, because the relative SIRT1 mRNA level (Figure 4B) correlated with the SIRT1 protein level, we hypothesized that SIRT1 is upregulated under inflammatory conditions and that additional control mechanisms affect protein sta-bility. These data suggest that SIRT1 is an important
Figure 3Overexpression of NKX3.1 leads to the suppression of oxidative stress. A. The CM (250 pg/ml of TNFαfor 24 h)-mediated increase in GPX2 and SOD2 expression is upregulated upon NKX3.1 overexpression in LNCaP cells.B. The H2O2-induced intracellular ROS
metabolic regulator that senses the metabolic rate and oxi-dative level under oxioxi-dative stress tolerance conditions.
Furthermore, because the SIRT1 level was increased in the cells treated with CM, we elucidated the DNA dam-age response activation by examining the p-ATM(S1981) level. Interestingly, we found that ATM(S1981) phosphor-ylation was not significantly increased upon CM treat-ment. LNAC only induced p-ATM(S1981)in control cells and not after CM exposure, suggesting that the reduced
oxidative stress levels might trigger a cell cycle progression and DNA damage response, presumably via SIRT1-mediated p53 and NBS1 activations respectively. However, this hypothesis requires further investigation. We also ex-amined whether apoptosis was induced in response to DNA damage, and found that the ATM-mediated re-sponse upon CM exposure did not generate caspase-3 cleavage in LNCaP cells, either with or without LNAC treatment (Figure 4B). These data demonstrate that
Figure 4Inflammation results in sustained oxidative damage to DNA. A. The levels of the DNA damage markerγ-H2AX(S139)and the metabolic regulator SIRT1 are remarkably affected by treatment with CM and H2O2in the absence of LNAC.B. SIRT1 mRNA expression in LNCaP
cells is altered by treatment with CM (including 125 and 250 pg/ml of TNFα) and 250μM H2O2with and without 10 mM LNAC.C. Caspase 3 and
p-ATM(S1981)levels remain lower in the treated cells in comparison to controls, indicating that the cells failed to activate apoptosis and the DNA damage response upon oxidative DNA damage. LPS (10 ng/ml) treatment was used as positive control for caspase-3 cleavage and activation.
D. LNCaP cells were exposed to CM (including 50 and 100 pg/ml of TNFα) and H2O2(50, 100 and 200μM) for 2 weeks. Chronic exposure to CM,
but not H2O2, results in the loss of NKX3.1.E. Similar to the acute treatments (24 h), chronic CM exposure (100 pg/ml of TNFα) also results in
increases in QSCN6 and GPX2 expression but a decrease in GPX3 expression in LNCaP cells. Red bars represent upregulation, green bars represent downregulation.F. Chronic CM exposure, but not treatment with H2O2, results in an increase in the intracellular ROS level; chronic exposure to
apoptosis was not activated, whereas the cells continued to proliferate with damaged DNA, resulting in genetic het-erogeneity. However, this proliferation was suppressed by the metabolic activity sensor and NAD+-dependent deace-tylase SIRT1, which slows down the cell cycle through the deacetylation of functional p53 to allow time for DNA damage repair. This process requires the presence of func-tional NKX3.1 in prostate cells [28,29]. This mechanism might result in oxidative stress tolerance and is commonly observed in cancer progression.
Loss of NKX3.1 under conditions of chronic inflammation leads to an abrogated antioxidant response
Chronic inflammation with sustained oxidative stress is well known to promote carcinogenesis [2]. To mimic chronic inflammation in vitro, we fed LNCaP cells with
CM (50 and 100 pg/ml of TNFα) or normal media or H2O2 (50, 100 and 200 μM) for a 2-week period. First,
we analyzed the proteasomal degradation of NKX3.1, which correlated with exposure to increasing concentra-tions of CM (Figure 4C) but not with H2O2treatments,
confirming that cytokines are the major cause of NKX3.1 degradation. Secondly, we investigated the expression of antioxidant genes and detected the upregulation of the pro-oxidant QSCN6 (3.8-fold), marginal upregulation of the antioxidant GPX2 (increase 1.3-fold) and downregula-tion of GPX3 (2.5-fold) after CM (100 pg/ml of TNFα) treatment (Figure 4D). These alterations may be due to the chronic exposure to inflammatory cytokines, which might be sufficient to maintain cell survival. Surprisingly, treatment of cells with H2O2did not result in significant
changes in the expression of antioxidant response factors (data not shown). We also measured the intracellular ROS levels after the chronic treatments to gain insight into the changes in the oxidative conditions within the cells. Chronic CM exposure dose dependently correlated with an increase in the ROS level, but chronic H2O2exposure
maintained the ROS level close to the basal concentration (Figure 4E). These data suggest that inflammatory cyto-kine release is the major factor underlying the deregulated antioxidant response and sustained oxidative damage in prostate cells. Additionally, we found that NKX3.1 loss caused by CM exposure inversely correlated with an in-creased ROS level. When the NKX3.1 level remained stable after H2O2 exposure, there was no change in the
ROS level. Thus, these data suggest that the increased
Figure 5ROS-mediated changes are partially restored upon NKX3.1 expression or LNAC treatment. A. LNAC treatment and
B. NKX3.1 overexpression remarkably suppresses cellular proliferation when LNCaP cells are treated with CM (including 62 and 125 pg/ml of TNFα). LNAC (10 mM) or NKX3.1 overexpression restores the CM-mediated suppression in cell proliferation. HM-NKX3.1 was transfected into the cells 24 h before CM treatment. HM: Hismax control vector, NKX: HM-NKX3.1 transfection. The Xcelligence real-time cell proliferation assay system was used to evaluate cell proliferation. The blue arrows indicate when the treatments were performed.
ROS concentration after chronic CM exposure might be a consequence of NKX3.1 loss.
NKX3.1 suppresses the proliferation enhanced by the inflammatory microenvironment
To investigate the influence of NKX3.1 loss on cell sur-vival, we examined cellular proliferation. In a real-time set-ting for 18 h, we observed that the rate of LNCaP cell proliferation increased after (62 and 125 pg/ml of TNFα including) CM treatment. The proliferation rate was sup-pressed with either LNAC treatment (Figure 5A) or when NKX3.1 was expressed ectopically (Figure 5B). These data demonstrate that NKX3.1 deregulates cell proliferation in-duced by the inflammatory microenvironment by function-ing similarly to LNAC in the response to oxidative stress.
Conclusions
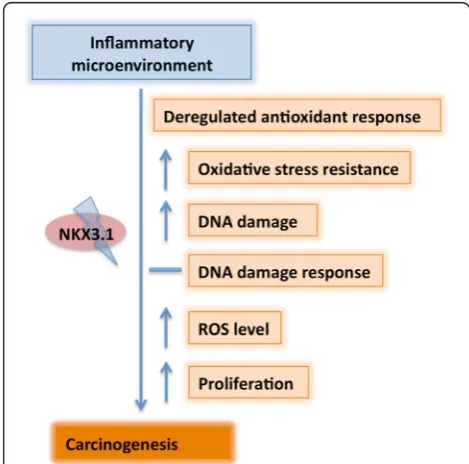
Overall, the inflammatory microenvironment is the major factor leading to sustained DNA damage in LNCaP cells, and the loss of NKX3.1 is an important contributing event. Therefore, the use of antioxidants might have a limited, but important ability to suppress the associated tumori-genic alterations, particularly in prostatic inflammation related cancer development. Upon chronic exposure of LNCaP cells to CM to mimic inflammation-like condi-tions, the loss of NKX3.1 leads to the deregulation of oxi-dative stress scavengers and contributes to increased DNA damage, eventually the prostate tumor progression (Figure 6). The inflammatory microenvironment model proposed here represents a novel approach for investigat-ing the molecular and cellular alterations in cellsin vitro, closely resembling animal studies [19].
Competing interests
The authors declare that they have no competing interests.
Authors’contributions
BDB designed experiments and carried out molecular studies, NE performed treatments and westerns. KSK and BDB drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgment
This research was supported by a grant for project 113S044 (COST action EU-ROS) from TUBITAK to KSK.
Author details
1Department of Bioengineering, Cancer Biology Laboratory, Faculty of Engineering,
Ege University, Bornova, 35100 Izmir, Turkey.2Department of Pharmaceutical
Biotechnology, Faculty of Pharmacy, Ege University, Bornova, Izmir 35100, Turkey.
Received: 4 April 2014 Accepted: 29 January 2015
References
1. Guyton KZ, Kensler TW. Oxidative mechanisms in carcinogenesis. Br Med Bull. 1993;49:523–44.
2. Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Oxidative stress in prostate cancer. Cancer Lett. 2009;282:125–36.
3. Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–54. 4. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation,
and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–16.
5. Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31:37–49.
6. Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–99.
7. Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125:376–93. 8. Morgan MJ, Kim YS, Liu ZG. TNFalpha and reactive oxygen species in
necrotic cell death. Cell Res. 2008;18:343–9.
9. Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–35.
10. Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. 2010;3:23–34. 11. Gorospe M, de Cabo R. AsSIRTing the DNA damage response. Trends Cell
Biol. 2008;18:77–83.
12. Yuan Z, Seto E. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle. 2007;6:2869–71.
13. Moore RL, Dai Y, Faller DV. Sirtuin 1 (SIRT1) and steroid hormone receptor activity in cancer. J Endocrinol. 2012;213:37–48.
14. He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997;43:69–77.
15. Korkmaz KS, Korkmaz CG, Ragnhildstveit E, Kizildag S, Pretlow TG, Saatcioglu F. Full-length cDNA sequence and genomic organization of human NKX3A - alternative forms and regulation by both androgens and estrogens. Gene. 2000;260:25–36.
16. Bowen C, Stuart A, Ju JH, Tuan J, Blonder J, Conrads TP, et al. NKX3.1 homeodomain protein binds to topoisomerase I and enhances its activity. Cancer Res. 2007;67:455–64.
17. Markowski MC, Bowen C, Gelmann EP. Inflammatory cytokines induce phosphorylation and ubiquitination of prostate suppressor protein NKX3.1. Cancer Res. 2008;68:6896–901.
18. Debelec-Butuner B, Alapinar C, Varisli L, Erbaykent-Tepedelen B, Hamid SM, Gonen-Korkmaz C, et al. Inflammation-mediated abrogation of androgen signaling: an in vitro model of prostate cell inflammation. Mol Carcinog. 2014;53(2):85–97.
19. Khalili M, Mutton LN, Gurel B, Hicks JL, De Marzo AM, Bieberich CJ. Loss of Nkx3.1 expression in bacterial prostatitis: a potential link between inflammation and neoplasia. Am J Pathol. 2010;176:2259–68.
20. Minelli A, Bellezza I, Conte C, Culig Z. Oxidative stress-related aging: A role for prostate cancer? Biochim Biophys Acta. 2009;1795:83–91.
21. Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76.
22. Mendrysa SM, O’Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20:16–21.
23. Reichardt JK. GEN: the genomic genetic analysis of androgen-metabolic genes and prostate cancer as a paradigm for the dissection of complex phenotypes. Front Biosci. 1999;4:D596–600.
24. Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–33.
25. Bookstein R, MacGrogan D, Hilsenbeck SG, Sharkey F, Allred DC. p53 is mutated in a subset of advanced-stage prostate cancers. Cancer Res. 1993;53:3369–73. 26. Brooks CL, Gu W. p53 regulation by ubiquitin. FEBS Lett. 2011;585:2803–9. 27. Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in
cancer: a clinical perspective. Onco Targets Ther. 2013;6:1399–416. 28. Erbaykent-Tepedelen B, Ozmen B, Varisli L, Gonen-Korkmaz C,
Debelec-Butuner B, Muhammed Syed H, et al. NKX3.1 contributes to S phase entry and regulates DNA damage response (DDR) in prostate cancer cell lines. Biochem Biophys Res Commun. 2011;414:123–8.