Yet Another Marked Difference among Impurities as Modi
fi
er Elements
for Re
fi
nement of Eutectic Si in Al-Si Alloys
Yosuke Suzuki-Yamamoto
1,+, Ryota Ozaki
1, Masato Yoshiya
1,2,
Tomoya Nagira
1and Hideyuki Yasuda
1,31Department of Adaptive Machine Systems, Osaka University, Suita 565-0871, Japan 2Nanostructures Research Laboratory, Japan Fine Ceramics Center, Nagoya 456-8587, Japan 3Department of Materials Science and Engineering, Kyoto University, Kyoto 606-8501, Japan
First principles calculations have been carried out in conjunction with lattice dynamics to evaluate free energies of ternary intermetallic phases, Al2Si2X (X=Ca, Sr, Ba, Eu, Y, Yb). The most stable crystal structure is identified for each X as a function of temperature. It is found
that some of the intermetallic phases are stable over wide temperature range, thereby giving ability to modify and refine Al-Si eutectic phases with better mechanical properties. Substitution energy of the intermetallic phase is evaluated to aiming at examining the magnitude tolerance of the intermetallic phases with respect to deviation of chemical composition. Finally, the role of Al2Si2X as a modifier is examined from a solely
geometric viewpoint, which provides yet another possible factors among many behind the X species dependence on modification of eutectic Si without using existing theories. [doi:10.2320/matertrans.MA201576]
(Received May 1, 2015; Accepted June 2, 2015; Published July 17, 2015)
Keywords: aluminiumsilicon alloys, eutectic silicon, twin, modification, lattice matching, thermodynamics, intermetallics, crystal structure
1. Introduction
Al alloys are widely used in industry due to its excellent mechanical properties in spite of their small molar weight, low material cost, and abundance.1)It is known that its alloys
with Si are abrasion resistant, which provides wide spectrum of applications. In addition to the well-known importance of controlling microstructure in general, it is crucial to control microstructure not only of coarser dominant matrix phase but alsofiner secondary phases to further improve materials properties, especially mechanical properties, since, while coarser matrix grains allow dynamics of dislocations to respond to external load, finer grains would play roles to inhibit unwanted further dislocation motion.2)
In the case of binary system of Al-Si, plate-like eutectic Si grains are formed in between coarser primary grains, which has a detrimental impact on the mechanical properties since local stress is concentrated at the edge of the plate-like Si grains, eventually leading to crack opening under external load. Attempts have been made to avoid this weakening micro-process. Those include an easily accessible process, that is, addition of small amount of a third element to the Al-Si alloys, such as Na or Sr, to modify the morphology of eutectic Si from plate-like structure tofine equiaxed one.36)
Taken into account the experimental fact that total amount of modifier element needed for the modification is on the ppm order, it is plausible to assume that the modification takes place on the nanometer scale at largest even if resultant microstructure of eutectic Si is on the micrometer scale. This process has been applied for practical use.7) If the modification mechanism is revealed completely, production of Al-Si alloys could be optimized, or the mechanism could be applied even to other alloy systems, thereby improving materials properties for wider applications. In practice, however, the mechanism behind the modification of eutectic Si is still controversial.
Theories have been proposed so far to elucidate the modification mechanism. Among them, two major theories are widely used to explain the phenomena observed in experiments. One is restriction TPRE (twin plane re-entrant edge) growth mechanism.4)TPRE is a concaved site where
Si atom can be attached more easily without two-dimen-sional nucleation which requires greater driving force. As a result, Si growth along ©211ª direction is promoted. A doped impurity atom of the third element is supposedly attached at TPRE and then deteriorate the preferential Si growth advantage, leading to formation of an equiaxed structure instead of plate-like one. The other is called IIT (impurity induced twinning) mechanism,3) where atomic size is a key factor. An atom of which size is 1.65 times larger than Si atom attached to Si growth plane {111} and distort the plane. As a result, twinning takes place and twin-led growth is promoted and Si growth orientation become diverse to form fine equiaxed grains. These two theories were originally discussed considering the third elements as monoatomic state, instead of as an element of any phase.
From experiments, Nogitaet al.have studied hypoeutectic Al-Si alloy to classify impurity elements as modifiers, and revealed element species having appropriate atom size for the IIT mechanism for the modification of eutectic Si with their magnitudes. They reported that Na, Ca, Sr, Ba, or Eu triggered Si grain modification from plate-like to fibrous while Y, Yb, and that some rare earth elements caused refinement of plate-like structure.6,8,9)
Spatial distribution of the modifier elements is also studied along with the microstructures with an aid of spectroscopic methods. Nogita et al.also showed that presence of the two modifier elements, Sr and Eu, are confined to fine eutectic Si grains in hypoeutectic Al-Si alloy,10,11) while Yb is not
distributed in eutectic Si grains.11)Timpelet al.have reported
where exactly in the eutectic Si phase Sr atoms exist in hypoeutectic Al-Si; Sr co-segregation with Al and Si within the eutectic Si grains has been indicated.12)
Ozaki et al. has proposed a thermodynamics-based mechanism for the modifiers.13)Instead of monoatomic states
of Sr or Eu, a quasi-ternary system of Al-Si-Al2Si2Sr or
Al-Si-Al2Si2Eu is considered and the intermetallic phase of
Al2Si2Sr or Al2Si2Eu is assumed to be a key factor of the
modification. This thermodynamics-based theory success-fully explained experimental facts that could not be explained by monoatomic-based earlier theories. Those include a fact that eutectic Si grains are modified while primary Si grains are not modified in hyper-eutectic Al-Si and a fact that eutectic temperature is decreased by the third element, implying ternary eutectic solidification occur instead of binary eutectic solidification.
It should be noted that the atom-based theories and the thermodynamic theories are not exclusive but shed light on the two aspects of the same physics although information directly obtained are different. Atomic radius is a conse-quence of the interatomic bonding of solid and thus is different from mere a spatial distribution of electronic cloud of an isolated atom. On the other hand, a thermodynamically stable intermetallic phase is another consequence of the interatomic bonding, with more realistic environment that the third element encounters upon solidification of Al-Si alloys. While geometric picture that causes the modification is clear in the atom-based theories, it is less clear in the thermody-namics-based theory since our knowledge about the stable crystal structures is limited, especially the ones at the elevated temperature for solidification of eutectic Si. Furthermore, in order to discuss attachment of the interme-tallic phase or atoms on growing eutectic Si and its modification, exact crystal structure needs to be clarified for both categories of theories. Although pressure dependence of the intermetallic phases of Al2Si2Sr are reported,14)stability
of the intermetallic phase and the most stable crystal structures remain unclear. Lack of it impedes to fully utilize the modifier-induced mechanism to refine eutectic Si in Al-Si alloys, thereby improving their mechanical properties.
In this study, focusing on several possible modifier elements that have already experimentally studied, thermo-dynamically stable crystal structures for each third element at elevated temperatures is identified in ternary system of Al-Si-X where Al-Si-X is the third element through ab initio lattice dynamics calculations to obtain free energy. Chemistry of the intermetallic phases, Al2Si2X, is also examined as the
chemical composition can deviate from its chemical formula at elevated temperature. Finally, based on the difference in the stable crystal structures for each element, the impact of the stable crystal structure on the growth of eutectic Si is examined solely from a geometric viewpoint.
2. Methodology
For calculating free energies of various phases examined in this study, first-principles electronic structure calculations were carried out at first to obtain an optimized unitcell or supercell and atomic coordinates in it and to obtain internal energy for each crystal structure. Plane waves are used as a basis set with enough high cut off energies for plane waves depending on elements contained so that total internal energy of a material is converged within a few meV per atom at
most. Generalized gradient correction approximation (GGA) parametrized by Perdew, Burke and Ernzerhof (PBE)15,16)for
exchange-correlation term is used with an aid of projector augmented wave (PAW) method.17) Convergence with
respect to Monkhorst-Pack k-point mesh18) and to fast Fourier transformation mesh is also achieved with the accuracy better than a few meV per atom. All the electronic structure calculations were performed using VASP code.19,20) Iteration for electronic structure for a given set of atomic coordinates and a unitcell and that for structure optimization are repeated until total internal energy and the largest among the sums of forces exerted on each atom are numerically converged within 1.0©10¹8eV and 1.0©10¹4eV/¡,
respectively. Since lattice dynamics requires forces exerted on each atom which is numerically less accurate than energy due to derivation, far better accuracy for ordinary electronic structure calculations is imposed.
After optimized structure of each phase is obtained, lattice dynamics calculations within the harmonic approximation were carried out to obtain Helmholtz free energy including contribution from vibrational entropy. Using phonopy code21) that is based on a finite displacement method proposed by Parlinski et al.,22)a small displacement from its equilibrium position is given to an atom by 0.01¡and then forces exerted on atoms were evaluated by the aforementioned electronic structure calculation without the structure optimization. From sets of forces for a given displacement, a dynamical matrix
D(k) is calculated and by solving the governing equation of phonons,23)
DðkÞ eðk; vÞ ¼½ðk; vÞ2eðk; vÞ;
phonon states were specified by its eigen vector eðk; vÞand frequency½ðk; vÞ, wherekis reciprocal vector andvis band index. From the phonon states, Helmoltz free energy was then calculated as a function of temperature23) for a given crystal structure. Sufficiently fine Monkhorst-Pack k-point mesh for the phonon calculations is chosen so that mesh size dependence is negligibly small. This method in combination with aforementioned electronic structure calculations relies on the size of a supercell, integer multiples of a unitcell, to avoid drawbacks of three dimensional periodic boundary conditions. Interaction between atoms decay with the distance between them, but its convergence is slower than we wish. Thus, to minimize the artificial effect by the choice of supercells, supercells are prepared so that shortest lattice constant of the supercell is longer than 10¡beyond which distance interaction between atoms can be neglected within the accuracy needed in this study.
Free energy of a structure at a given composition was then calculated as
FtotalðxAl; xSi; xX; TÞ ¼UtotalðxAl; xSi; xXÞ þFZPðxAl; xSi; xXÞ þFphononðxAl; xSi; xX; TÞ where Utotal is total internal energy of a structure obtained from electronic structure calculation, Fphonon is phonon part of free energy as a function of temperature,FZPis free energy
due to zero-point vibration, xAl, xSi, and xX, are molar
fractions of Al, Si, and the third element X, respectively, and
member phases of Al-Si-X ternary system was calculated as,
FtotalðxAl; xSi; xX; TÞ ¼FtotalðxAl; xSi; xX; TÞ fxAlFAltotalðTÞ þxSiFSitotalðTÞ þxXFXtotalðTÞg
whereFitotalðTÞis free energy of an end member phaseias a function of temperature.
Substitution energy or free energy change required for substitution of an atom M for an atom N in the matrix phase Z,FZsubðMNÞ, in the matrix phase is defined as follows.
FZsubðMNÞ
¼ ðFZsubstitutedþ®NÞ ðFZpristineþ®MÞ TSconf where®Nis chemical potential of an atom N,FZpristine is free energy of a pristine end member phase in the quasi-ternary system of Al-Si-Al2Si2X, and MNdenotes substitution of an
M atom for an N atom at an N atomic site using Kröger-Vink notation. Fsubstituted
Z is free energy of a supercell after the substitution and was approximated in this study as
FZsubstituted ¼FZpristineþ ðEsubstitutedZ EpristineZ Þ
where EZpristine and EZsubstituted are internal energies of the pristine supercell and the substituted supercell. This approx-imation is equivalent to the assumption that modification of atomic vibration, and, in turn, vibrational entropy, by the substitution is negligible. Instead, configurational entropy which is supposed to be dominant for the substitution was included in the free energy change. The chemical potential for three elemental species in a quasi-ternary equilibrium Al-Si-Al2Si2X are determined as follows:®Siand®Alare simply
free energies of respective monoatomic phases per atom and ®Xwas calculated as
®X¼ ðEpristineAl2Si2XnAl®AlnSi®SiÞ
where nAl and nSi are the numbers of atoms in an Al2Si2X
supercell. Needless to say, this is equivalent to assume Al or Si atom in Al2Si2X is in equilibrium with those in
monoatomic Si or Al phases, respectively.
Several crystal structures at the chemical composition of Al2Si2X, namely, tetragonal (I4/mmm), trigonal (P3m1),
orthorhombic (Cmcm), and orthorhombic (Pnma) crystal structures, are reported.2429) Those crystal structures were
examined in this study with possible modifier elements, Ca, Sr, Ba, Eu, Y, and Yb. Hereafter, these crystal structures are referred to as Ttr-str, Hxg-str, Oc-str, and Op-str, respectively.
3. Results and Discussion
3.1 Crystal structures
A monoatomic Si crystal forms diamond-structure in which{111}is the close-packed plane. Growth along©111ªis the slowest, and thus attachment of atoms on the{111}from liquid phase controls overall rate of crystal growth of the diamond-structured Si (d-Si). If lattice matching on the atomic level between Si planes of the intermetallic phase and the{111}plane in d-Si phase is good, then those two phases can be adjoined seamlessly sharing the Si plane, since the adjoining Si plane is a native Si plane in both phases. If this is the case, it lowers the energy barrier for the nucleation of the intermetallic phase on a growing front of d-Si. Thus, it is
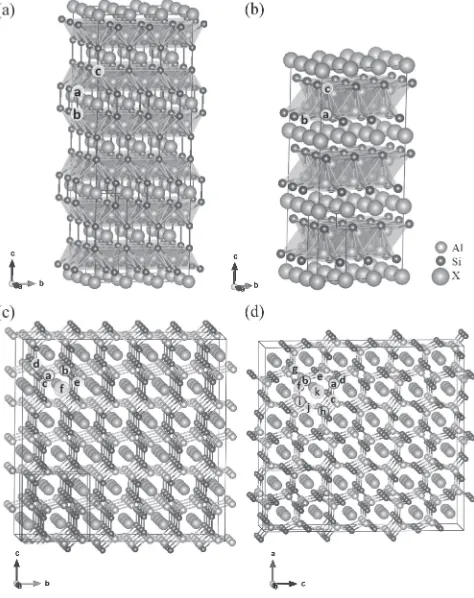
critically important to identify the crystal structure or precise atomic coordinates of nano-meter substances containing the third element. As mentioned earlier, four crystal structures are examined in this study and noticeable differences among the four crystal structures are clarified in this subsection before evaluating its stability at elevated temperature.
Figure 1 shows four crystal structures examined in this study. Optimized lattice constants are listed in Table 1 in comparison with experimental values if available. The errors of computed values with respect to experimental values are shown in parentheses on the experimental side of the table. Satisfactory agreements between computations and experi-ments were obtained and errors of computed values were below 1% with a few exceptions. Focusing on Si planes in these crystal structures,flat Si planes are found parallel toab -plane in Hxg-str and Ttr-str. Although its number density is low, Si planes are found also in Oc-str parallel toab-plane. In Op-str, however, no Si plane can be identified as clearly as in other structures.
Bond lengths for nearest-neighbor atomic pairs for each element in the four crystal structures of Al2Si2Sr are
summarized in Table 2. Although those bond lengths change with the third element, general difference among the four crystal structures can be found in the tableted values. No significant differences in the nearest-neighbor bond lengths themselves is found among the four structures. Carefully looking at three-dimensional crystal structures shown in Fig. 1 together with one-dimensional information of bond lengths in Table 2, one would notice that AlSi4 tetrahedra,
[image:3.595.309.546.72.371.2]form an AlSi layer which is then stacked alternately with an X layer along c-axis in Hxg-str and Ttr-str, resulting in layered structures. In contrast, while similar tetrahedra to the
[image:4.595.49.548.95.459.2]ones in Hxg-str or Ttr-str is found, the tetrahedra cannot be uniquely defined in Oc-str. In addition, configuration of Al and Si atoms cannot be described as tetrahedral network as in Table 1 Optimized lattice constants with experimental values if available. The errors of computed values with respect to experimental
values are shown in parentheses on the experimental.
Optimized lattice constants [¡] Experimental lattice constants [¡] with error of computed value
a b c a b c Ref.
Ca
Ttr-str 4.161 11.089
Hxg-str 4.155 7.101 4.141 +0.34% 7.133 ¹0.45% 29)
Oc-str 4.279 9.884 10.532
Op-str 9.929 4.061 10.614
Sr
Ttr-str 4.220 11.628
Hxg-str 4.206 7.446 4.183 +0.53% 7.410 +0.48% 26)
Oc-str 4.303 10.415 10.337
Op-str 10.006 4.156 10.793
Ba
Ttr-str 4.262 12.545 4.231 +0.73% 12.601 ¹0.45% 27)
Hxg-str 4.260 7.796
Oc-str 4.368 10.814 10.227 4.238 +3.06% 10.890 ¹0.70% 10.106 +1.20% 27)
Op-str 10.132 4.265 10.942 10.074 +0.58% 4.226 +0.93% 10.865 +0.71% 28)
Eu
Ttr-str 4.174 11.150
Hxg-str 4.179 7.220 4.180 ¹0.03% 7.252 ¹0.45% 24)
Oc-str 4.269 10.165 10.329
Op-str 9.865 4.115 10.665
Y
Ttr-str 3.991 11.424
Hxg-str 4.211 6.559 4.195 +0.38% 6.581 ¹0.33% 25)
Oc-str 4.349 9.021 10.931
Op-str 9.471 4.026 10.355
Yb
Ttr-str 4.132 11.026
Hxg-str 4.147 7.031 4.144 +0.07% 6.915 +1.68% 24)
Oc-str 4.272 9.817 10.489
Op-str 9.859 4.048 10.581
Table 2 Bonding length in Al2Si2X (X=Sr) specified by labeling in Fig. 1. For example,“a-b”stands for a bonding atom labeled“a”and
atom labeled“b”. Multiplication indicate the number of symmetric bonding.
Bonding length [¡]
Al-Si Si-Si Al-Al X-Si X-Al
Ttr-str
2.558 (©4) 2.921 (©1) a-b 2.984 (©4) 3.322 (©8) 3.592 (©8)
4.156 (©4) a-c 4.220 (©4)
Hxg-str 2.536 (©3) a-b 4.123 (©3) b-c 3.065 (©3) 3.182 (©6) 3.698 (©6)
2.601 (©1) a-c 4.206 (©6) 4.206 (©6)
Oc-str
2.515 (©2) a-c 2.371 (©1) e-f 2.646 (©1) 3.238 (©2) f-c 3.663 (©4) f-b 2.570 (©1) a-d 3.691 (©2) c-d 4.303 (©2) 3.294 (©4) f-e 3.808 (©4) f-a
5.634 (©4) 4.303 (©2) c-e
Op-str
2.508 (©1) a-c 4.105 (©2) d-e 3.004 (©2) 3.210 (©1) k-f 3.324 (©1) k-i
2.531 (©2) a-d 4.273 (©1) c-e 4.156 (©2) 3.271 (©2) k-c 3.564 (©2) k-a
2.556 (©1) a-e 4.078 (©2) c-d 3.438 (©2) k-e 3.669 (©2) k-b
4.156 (©2) 3.514 (©2) k-j 3.683 (©2) k-h
2.491 (©2) b-f 3.793 (©2) f-g 3.336 (©2) 2.512 (©1) b-e 4.235 (©1) e-g 4.156 (©2) 2.560 (©1) b-g 4.249 (©2) e-f
[image:4.595.48.548.492.735.2]Hxg-str or Ttr-str. In Op-str, configuration of Al and Si atoms are even more complicated as variation of bond lengths shown in Table 2 suggest, reflecting two crystallographically different sites for both Al and Si.
Examining Hxg-str and Ttr-str more carefully, the AlSi4
tetrahedra are connected to each other by sharing their edges with their neighbors. However, orientation of the tetrahedra are different between the two crystal structures: In Hxg-str, one of bases of the tetrahedra is parallel toab-plane or the X layer while two edges of the tetrahedra are parallel to ab -plane or the X layer in Ttr-str. Consequently, the AlSi layer is terminated by triangle or rectangular Si sublattice in Hxg-str or Ttr-str, respectively. On the other hand, Al and Si atoms are distributed around X atoms in Oc-str and Op-str so that X atoms form one-dimensional X columns alonga-axis andb -axis, respectively, exhibiting significantly different atomic configuration of Al and Si from that in d-Si, in contrast to Hxg-str and Ttr-str which has tetrahedral network similar to that in d-Si.
3.2 Crystal structure stability
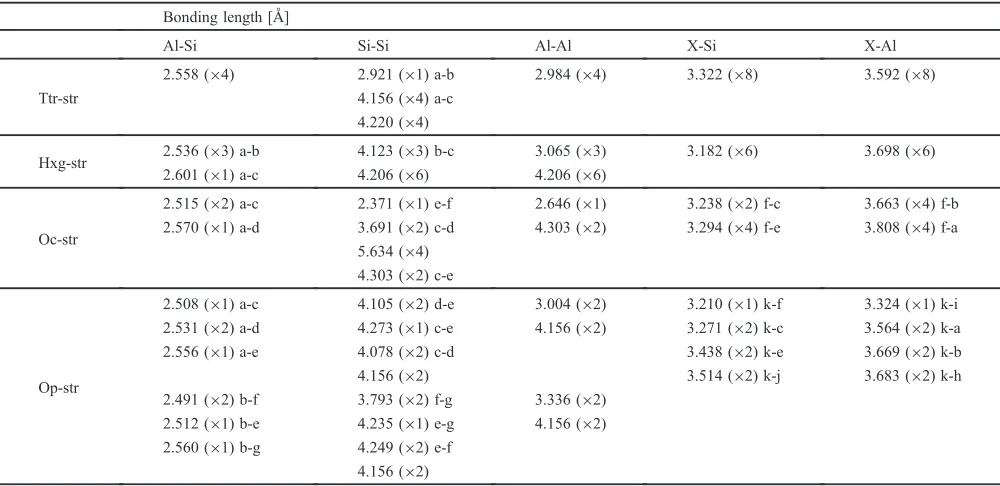
Thermodynamic stability of all of the four crystal structures mentioned above were examined for each third element, without accounting for any experimental findings other than initial crystal structures in prior to the calculations. Figure 2 shows formation free energy of intermetallic phases relative to unary phases of constituent elements, three end
member phases in ternary equilibrium, as a function of temperature.
Thermodynamic stability of intermetallic phase relative to monoatomic phase is shown in those plots in addition to relative stability of a crystal structure to others for a given X element species. Irrespective of X species, all the formation free energies are negative, indicating that all intermetallic phases and crystal structures are stable, more stable than macroscopic mixture of the unary phases. In other words, these intermetallic could exist as an equilibrium phase in the Al-Si-X ternary systems at the temperatures examined in this study.
Comparing dependence of stable crystal structure on X species, all the Al2Si2X phases were most stable in Hxg-str at
room temperature or lower except for Al2Si2Ba which is
stabilized in Op-str. The most stable structures were changed with increasing elevated temperature in some cases, indicat-ing phase transformation upon increasindicat-ing temperature. Ttr-str was stabilized at higher temperatures for Sr, Eu, and Ba, with transition temperatures of 1380, 720, and 530 K, respectively. On the other hand, while difference between free energies of Ttr-str and Hxg-str of Ca, Y, and Yb is decreased with increasing temperature, those curves will presumably cross at higher temperature, triggering the phase transformation even for those elements. However, the transformation is beyond the temperature range examined in this study, which suggests that those low-temperature phases undergo melting before the solid-state phase transformation takes place.
For all the X element species that are already known as modifier elements for refinement of eutectic Si, Ttr-str was stabilized at the temperature near binary Al-Si eutectic temperature, 850 K. In other words, a portion of residual melt after solidification of primary phase would be solidified to Ttr-str at ternary eutectic temperature which should not be significantly different from binary eutectic temperature of Al-Si, considering the experimental fact that only small amount of the modifier element is needed to modify the morphology of eutectic Si. It is generally known that symmetry of the stable crystal structure increases, though not linearly, as temperature is raised, which supports the phase trans-formation from Hxg-str or Op-str to Ttr-str at elevated temperature. However, thermal expansion was not taken into account upon calculating free energy in this study. Thus, phase transition temperature predicted in this study may be not very accurate. Nevertheless, these findings obtained in this study shed light on the modifier elements from different aspects from previous studies: The Ttr-str can be one of crucial factors that enables modification of eutectic Si morphology upon eutectic solidification.
It should be noted that, although lattice dynamics calculation in conjunctions withab initioelectronic structure calculations provides free energy which is an important quantity of macroscopic thermodynamics, it assumes the crystal state spans over all the macroscopic space based on three-dimensional periodic boundary conditions. However, one macroscopic state consists of many microscopic states according to statistical mechanics and what were computed in this study are just microscopic states. Thus, in reality, solid-state phase transformation may not be as simple as the ones predicted in this study, being affected by kinetics and Fig. 2 Free energy of formation for the four crystal structures of Al2Si2X,
[image:5.595.49.291.68.398.2]geometric constraints among many grains, and it is likely that many solid phases with different crystal structures coexist with its probability determined by statistical mechan-ics: When free energy of Ttr-str is close to that of Hxg-str, both crystallites are present with nearly equal probability while Ttr-str is dominant when its free energy is much lower than that of Hxg-str. Therefore, not only the most stable crystal structure at elevated temperature, but also the difference in free energies at elevated temperature is important since the latter can be a measure that quantify the magnitude or frequency of the modification of eutectic Si. Even though the most stable crystal structure at the temperature range examined was Hxg-str when X is Ca, Y or Yb, free energy of Ttr-str is close to Hxg-str at elevated temperature. This suggests that, even for Ca, Y, and Yb, a portion of the Ttr-structured intermetallic phase is present together with Hxg-structured phase at elevated temperature. Despite the fact that no experimentalfindings is prescribed in the calculation, it is found that the relative stability of Ttr-str to Hxg-str for these elements is in a good agreement with the experimentally-determined magnitude of the modification of eutectic Si by those elements.5,6,811)This result indicates that the crystal structure of Ttr-str itself, or atomic configuration therein, may have a capability of modification of eutectic Si.
3.3 Chemistry of intermetallic phase
Intermetallic Al2Si2X phases are found to be stable at
elevated temperature, and the most stable crystal structures for each X species at elevated temperatures are identified in the previous section. The next question one may have is whether the intermetallic phase is at that specific composition even at elevated temperature at which modification of eutectic Si takes place. To clarify the possible variation of the chemical composition of the stable intermetallic phases, further calculations were performed, by substituting one of constituent elements for another and calculating free energy change upon the substitution.
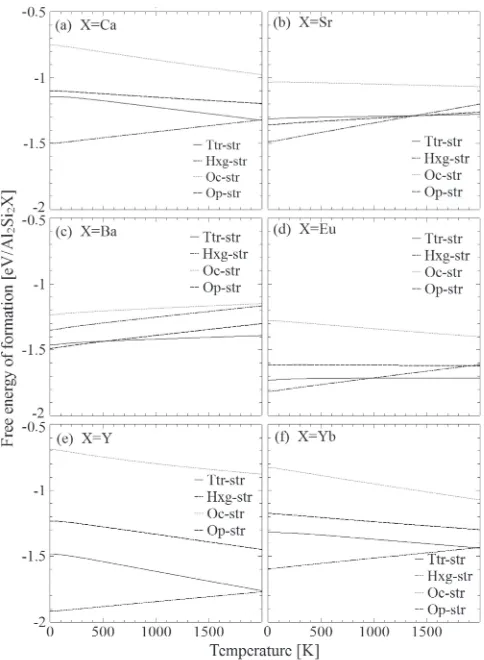
For that, a 3©3©1 Ttr-str supercell containing 24 Al and Si atoms and 12 X atoms was prepared for Sr, Ba, and Eu which are stabilized in Ttr-str at elevated temperatures. For Ca, Y, and Yb, a 3©3©2 Hxg-str supercell were prepared. Just for comparison, substitution in low-temperature struc-tures was also examined using 3©3©2 Hxg-str supercells for Sr and Eu and a 1©3©1 Op-str supercell for Ba, following to the results obtained in Sec. 3.2. Furthermore, supercells of the monoatomic Al and Si phases, containing 108 Al atoms and 64 Si atoms, respectively, were prepared so that similar variance in composition would be given upon substitution. In those supercells, only one atom of constituent elements was substituted for another atom of different species, once species at a time, followed by calculations of free energy change upon substitution.
First of all, for the substitutions in the monoatomic Si phase, substitution energy was positive irrespective of temperature examined as shown in Fig. 3, suggesting that compositional variance with those elemental species are energetically prohibitively unfavorable, in a good agreement with binary phase diagrams.3034)For the substitution in the
monoatomic Al phase, while some of substitution energies
turned to negative at elevated temperature, it is beyond the melting point of Al metal, indicating solubility limit of these elements in metal Al is quite low, in accordance with the binary phase diagram.3540)
For substitutions in Al2Si2X phases, large variation in
substitution energies and its temperature dependence was observed as shown in Fig. 4. In general, substitution energies were positive, independent of temperature examined, sug-gesting that compositional variance of the intermetallic phases are unlikely and the intermetallic phase is stable also in terms of chemistry. Looking carefully at details of the substitution energies, in the case of Sr, Eu, and Ba in Ttr-str, both substitutions of Al for Si and Si for Al, both of which form AlSi4 tetrahedra in AlSi layer, were not energetically
prohibitive, implying those substitution may be realized by thermalfluctuation. In the case of Ca, Y, and Yb, substitution of Al for Si was relatively possible while the opposite was more energetically expensive, unlike in Ttr-str. This trend was also observed in the same substitution in Hxg-str for Sr and Eu, implying that this mutual substitution is realized by the atomic configuration specific in Ttr-str, of which implications will be discussed in the next subsection. Substitution energy of Al for X was lower than that of Si for Al in Al2Si2Ca in whole temperature range and in
Al2Si2Yb at low temperature and it turned negative at high
temperature in Al2Si2Y. Energy penalty of those substitutions
are also determined by the atomic configuration in Hxg-str, though its implication remains unclear.
3.4 How the modifying elements affect eutectic Si phase morphology
[image:6.595.306.549.68.198.2]phases requires additional calculations using far larger supercells, but it is beyond the scope of this study. Thus, only geometric proximity between d-Si and the intermetallic phases is examined based on the optimized structures of the intermetallic phases for Sr as a representative of various X elements.
The intermetallic phases and d-Si phase have tetrahedral network in common, although they are different in that edges or vertices are shared by neighboring tetrahedra in the intermetallic phase or d-Si phase, respectively. While Al atoms reside at the center of the tetrahedron in the intermetallic phases, Si atoms occupy the center in d-Si phase. As discussed in the previous subsection, the Al could be replaced by Si in the intermetallic phases. If Al is replaced by Si, AlSi4 tetrahedra in the intermetallic phases become
SiSi4 tetrahedra as in d-Si. Taking these into account, we
discuss how the intermetallic phase can be adjoined at the growing front of d-Si{111}and its consequence upon further growth of eutectic Si. It is rational to assume that a Si plane is shared by both the intermetallic phase and d-Si phase at the junction since it is present in both phases and thus
it presumably lowers the energy penalty at the hetero-interface.
As a first step, it is a natural attempt to examine the lattice matching between Si planes discussed in Sec. 3.1, i.e., the base plane of the intermetallic phases and d-Si {111} plane. It was done by the quantity ¤ð111ÞSi
ðhklÞIM proposed by Bramfitt:41)
¤ð111ÞSi ðhklÞIM ¼
100 3
X3
i¼1
jðd½uvwiSicosªÞ d½uvwiIMj d½uvwiIM
where (hkl)y and ½uvwiy are a plane specified by Miller indices hkl in a phase y and d½uvwiy is the interatomic spacing along ½uvwiy, with a subscript y being either Si or IM which denotes d-Si phase or the intermetallic phases, respectively, and a superscript i indicating three different directions. This equation can be applied to different crystal structures such as triangle grid and square grid. Hxg-str has relatively better lattice matching with Si phase as shown in Table 3, below 10% for all the X species. Other crystal structures do not have better matching for all elements: The mismatch was around 12% or 19% for Ttr-str or Oc-str, Fig. 4 Substitution energy for stable crystal structures for (a) Ttr-str Al2Si2Sr, (b) Ttr-str Al2Si2Eu, (c) Ttr-str Al2Si2Ba, (d) Hxg-str
Al2Si2Sr, (e) Hxg-str Al2Si2Eu, (f ) Op-str Al2Si2Ba, (g) Hxg-str Al2Si2Ca, (h) Hxg-str Al2Si2Y, and (i) Hxg-str Al2Si2Yb.“A (right
[image:7.595.108.492.68.471.2]respectively. In Hxg-str, AlSi4tetrahedra are oriented so that
their bases parallel to{111}of d-Si, the slowest plane, with little difference along lateral directions on{111}of d-Si, and thus it is unlikely that growth of d-Si is promoted by the attachment of the intermetallic phase on its growing front, even if straining of the tetrahedra in the AlSi layer is neglected. On the other hand, poorer lattice matching between Ttr-str or Oc-str and d-Si {111} likely impede the advancement of the slowest{111}of d-Si even if it succeeds in nucleating on the growing front of d-Si, simply resulting in other parts of d-Si growing ahead being uninterrupted. Thus, what is expected for the d-Si growth on {111} upon the attachment of the intermetallic phases is just further suppression along this direction irrespective of the stable crystal structures of the intermetallic phases. Although thermal expansion is not taken into account in this study, it is expected that it would not significantly change these trends since thermal expansion would affect all the intermetallic crystal structures by a similar magnitude.
On the other hand, since an AlSi4 tetrahedron in Hxg-str
and Ttr-str has three other faces, attachment of those lateral faces of AlSi4tetrahedra to d-Si{111}is also possible. If this
is the case, there is a noticeable difference between Hxg-str and Ttr-str, which is originated from orientations of AlSi4
tetrahedra explained in Sec. 3.1. In this case, growth direction is presumably diverted so that AlSi4tetrahedra are
aligned as an extension of tetrahedral network of d-Si as depicted in Fig. 5. The tetrahedra form AlSi layer in both structures, with the diversion angles between ©111ª of d-Si and the AlSi layer being 36.1 deg. and 20.0 deg. in Ttr-str and Hxg-str, respectively. In both cases, tetrahedra are connected by sharing their edges. However, the AlSi layer in those structures are laterally terminated by edges and faces of the tetrahedra in Ttr-str and Hxg-str, respectively, as highlighted with lighter gray ovals in Fig. 5.
If we assume that the attachment of X atoms on an AlSi layer to form an X layer in the intermetallic structures is not a rate-controlling process,42) then formation tetrahedral
net-work of AlSi4, ahead of growing front of d-Si, along the AlSi
layer would not be significantly different from that of d-Si since local atomic coordination is similar to that in d-Si. Although it requires participation of Al atoms, it should be readily available at around eutectic composition and temper-ature. Besides, results in the previous subsection imply mutual substitution of Al and Si in Ttr-str. In the case of Hxg-str, termination of the AlSi layer by tetrahedral faces leads to its growth as slow as that of{111}of d-Si. On the other hand, termination of the AlSi layer only by tetrahedral edges, which correspond to {100} in d-Si, may allow the growth of the intermetallic phase across the termination of the AlSi layer, or
along its c-axis, and its speed would be higher than the one limited by the dense{111} of d-Si.
For this attachment to happen, ledges need to be introduced since faces of tetrahedra in different AlSi layer are not on a plane. Height and distance of the faces on ledges are 1.7 and 2.4¡in Ttr-str and 0.9 and 4.6¡or 2.5 and 3.6¡ in Hxg-str and 0.8 and 1.1¡ in d-Si, respectively. Here, possible distortion of the crystal structure near the hetero-interface is intentionally neglected for clarity. These might be the reason why twinning of takes place in eutectic Si. In addition, difference in tetrahedral network in Ttr-str and Hxg-str from that in d-Si, i.e., whether edges or vertices are shared by neighboring tetrahedra, might be another reason of the twinning since this difference could introduce distortion into tetrahedral network as well. Furthermore, the two crystal structures are different in symmetry with respect to growth axis of d-Si. As one of the examples, difference in tetrahedral stacking results in different diversion angles mentioned above as depicted in Fig. 5. In addition, bases of tetrahedra have three-fold rotation symmetry, if we assume it is regular triangle, thus the AlSi layer in Ttr-str and Hxg-str have three different direction of tetrahedral stacking. If d-Si can be attached again on a growing front of AlSi layer in Ttr-str or Hxg-str, these differences between Ttr-str and Hxg-str may eventually affect the growth of eutectic Si.
[image:8.595.307.546.71.402.2]Fig. 5 Relation between intermetallics, Ttr-str and Hxg-str, and d-Si based on geometric viewpoint. Connections of intermetallics and d-Si by shearing tetrahedra vertices are depicted in (a) and (b) for Ttr-str and Hxg-str, respectively. Lighter gray ovals highlight AlSi layer edge face. Ledges of tetrahedra are shown in (c), (d), and (e) for Ttr-str, Hxg-str, and d-Si, respectively.
Table 3 Mismatch between d-Si{111}plane and Si plane in Al2Si2X.
Mismatch (%)
Ca Sr Ba Eu Y Yb
Ttr-str 12.17 12.47 12.68 12.24 11.27 12.03
Hxg-str 6.94 8.05 9.23 7.47 8.18 6.75
[image:8.595.46.292.84.154.2]Although performing ab initio calculation of this kind of hetero-interface is beyond the scope of this study, consid-eration solely based on geometry using computed crystal structures and free energies shed light on detailed crystallo-graphic information and provides yet another possible factors among many behind the X species dependence on mod-ification of eutectic Si without using existing theories.
4. Conclusion
First principles calculations have been carried out in conjunction with lattice dynamics to evaluate free energies of ternary intermetallic phases, Al2Si2X (X=Ca, Sr, Ba, Eu, Y,
Yb) with different crystal structures, Tetragonal (I4/mmm), Trigonal (p3m1), Orthorhombic (Cmcm), and Orthorhombic (Pnma) those are referred to as Ttr-str, Hxg-str, Oc-str, and Op-str, respectively here. Substitution energies of the intermetallic phase were also calculated.
Hxg-str was most stable for Al2Si2X (X=Ca, Sr, Eu, Y,
Yb) at 0 K. Op-str was most stable for Al2Si2Ba at 0 K. Ttr-str
was most stable for Al2Si2X (X=Sr, Ba, Eu) at elevated
temperatures, indicating the crystal structure of Ttr-str itself, or atomic configuration therein, may have a capability of modification of eutectic Si.
For the substitutions in the monoatomic Si phase, substitution energy was positive irrespective of temper-ature examined, suggesting that compositional variance with those elemental species are energetically prohibitively unfavorable.
For the substitution in the monoatomic Al phase, while some of substitution energies turned to negative at elevated temperature, it is beyond the melting point of Al metal.
For substitutions in Al2Si2X phases, in general,
substitu-tion energies were positive independent of temperature examined, suggesting that compositional variance of the intermetallic phases are unlikely and the intermetallic phase is stable also in terms of chemistry. Mutual substitution between Si and Al is energetically possible in Ttr-str. Substitution of Al for Si was relatively possible while the opposite was more energetically expensive for Hxg-str.
Al centered AlSi4tetrahedra are aligned in Ttr-str and
Hxg-str similar to Si centered SiSi4 tetrahedra in
diamond-structured Si (d-Si). We discussed how Ttr-str and Hxg-str could affect eutectic Si phase morphology, focusing on tetrahedra connection among them.
Consideration solely based on geometry using computed crystal structures and free energies shed light on detailed crystallographic information and provides yet another possible factors among many behind the X species de-pendence on modification of eutectic Si without using existing theories.
Acknowledgments
This study was supported by Grant-in-Aid for Scientific Research (S) (Grant No. 24226018) and by a Grant-in-Aid for Scientific Research on Innovative Areas “Nano Infor-matics”(Grant No. 25106005) both from Japan Society for the Promotion of Science (JSPS).
REFERENCES
1) F. Yilmaz and R. Elliott:J. Mater. Sci.24(1989) 20652070.
2) Y. Wang, M. Chen, F. Zhou and E. Ma:Nature419(2002) 912915.
3) S.-Z. Lu and A. Hellawell:Metall. Trans. A18(1987) 17211733.
4) S.-Z. Lu and A. Hellawell:J. Crys. Growth73(1985) 316328.
5) K. Nogita and A. K. Dahle:Mater. Trans.42(2001) 393396.
6) K. Nogita, S. D. McDonald, J. W. Zindel and A. K. Dahle:Mater. Trans.42(2001) 19811986.
7) A. Pacz: US Patent No. 1387900, (1921).
8) K. Nogita, J. Drennan and A. K. Dahle:Mater. Trans.44(2003) 625 628.
9) K. Nogita, S. D. McDonald and A. K. Dahle:Mater. Trans.45(2004) 323326.
10) K. Nogita, H. Yasuda, K. Yoshida, K. Uesugi, A. Takeuchi, Y. Suzuki and A. K. Dahle:Scr. Mater.55(2006) 787790.
11) K. Nogita, H. Yasuda, M. Yoshiya, S. D. McDonald, K. Uesugi, A. Takeuchi and Y. Suzuki:J. Alloy. Compd.489(2010) 415420.
12) M. Timpel, N. Wanderka, R. Schlesiger, T. Yamamoto, N. Lazarev, D. Isheim, G. Schmitz, S. Matsumura and J. Banhart: Acta Mater. 60 (2012) 39203928.
13) R. Ozaki: Bachelor Thesis, Osaka University (2013).
14) A. M. Garay-Tapia, G. Trapaga and A. H. Romero:Phys. Rev. B83 (2011) 214111.
15) J. P. Perdew, K. Burke and M. Ernzerhof:Phys. Rev. Lett.77(1996) 38653868.
16) J. P. Perdew, K. Burke and M. Ernzerhof:Phys. Rev. Lett.78(1997) 13961396.
17) P. E. Blöchl:Phys. Rev. B50(1994) 1795317979.
18) H. J. Monkhorst and J. D. Pack:Phys. Rev. B13(1976) 51885192.
19) G. Kresse and J. Hafner:Phys. Rev. B49(1994) 1425114269.
20) G. Kresse and J. Furthmüller:Phys. Rev. B54(1996) 1116911186.
21) A. Togo, F. Oba and I. Tanaka:Phys. Rev. B78(2008) 134106.
22) K. Parlinski, Z. Q. Li and Y. Kawazoe:Phys. Rev. Lett.78(1997) 4063.
23) M. T. Dove:Introduction to Lattice Dynamics, (Cambridge University Press, Cambridge, UK, 1993) Ch. 6.
24) S. Bobev, P. H. Tobash, V. Fritsch, J. D. Thompson, M. F. Hundley, J. L. Sarrao and Z. Fisk:J. Solid State Chem.178(2005) 20912103.
25) C. Kranenberg, D. Johrendt and A. Mewis:Z. Anorg. Allg. Chem.625 (1999) 17871793.
26) S. M. Kauzlarich, C. L. Condron, J. K. Wassei, T. Ikeda and G. J. Snyder:J. Solid State Chem.182(2009) 240245.
27) S. Yamanaka, M. Kajiyama, S. N. Sivakumar and H. Fukuoka:High Pressure Res.24(2004) 481490.
28) C. L. Condron, H. Hope, P. M. B. Piccoli, A. J. Schultz and S. M. Kauzlarich:Inorg. Chem.46(2007) 45234529.
29) M. Imai, H. Abe and K. Yamada:Inorg. Chem.43(2004) 51865188.
30) J. Kim and I.-H. Jung:J. Chem. Thermodynamics81(2015) 273297.
31) H. Okamoto:J. Phase Equilibria Diffus.34(2013) 251263.
32) M. Pani and A. Palenzona:J. Alloy. Compd.454(2008) L1L2.
33) A. Palenzona, P. Manfrinetti, S. Brutti and G. Balducci: J. Alloy. Compd.348(2003) 100104.
34) M. Heyrman and P. Chartrand:J. Phase Equilibria Diffus.27(2006) 220230.
35) H. Okamoto: J. Phase Equilib.12(1991) 499500.
36) F. G. Meng, L. G. Zhang, H. S. Liu, L. B. Liu and Z. P. Jin:J. Alloy. Compd.452(2008) 279282.
37) Y.-B. Kang, A. D. Pelton, P. Chartrand and C. D. Fuerst:Comp. Coupl. Phase Diagrams Thermochem.32(2008) 413422.
38) B. Closset, H. Dugas, M. Pekguleryuz and J. E. Gruzleski: Metall. Trans. A17(1986) 12501253.
39) K. Ozturk, L.-Q. Chen and Z.-K. Liu:J. Alloy. Compd.340(2002) 199206.
40) V. P. Itkin and C. B. Alcock:J. Phase Equilib.14(1993) 518524.
41) B. L. Bramfitt:Metall. Mater. Trans. B1(1970) 19871995.