Crystal structure of
2-ethyl-3-(4-fluoro-
phenylsulfinyl)-5,7-dimethyl-1-benzo-furan
Hong Dae Choiaand Uk Leeb*
aDepartment of Chemistry, Dongeui University, San 24 Kaya-dong, Busanjin-gu,
Busan 614-714, Republic of Korea, andbDepartment of Chemistry, Pukyong
National University, 599-1 Daeyeon 3-dong, Nam-gu, Busan 608-737, Republic of Korea. *Correspondence e-mail: uklee@pknu.ac.kr
Received 21 August 2014; accepted 22 August 2014
Edited by W. T. A. Harrison, University of Aberdeen, Scotland
In the title compound, C18H17FO2S, the dihedral angle between the planes of the benzofuran ring system (r.m.s. deviation = 0.004 A˚ ) and the 4-fluorophenyl ring is 86.38 (6). The terminal C atom of the ethyl substituent is displaced by 1.444 (3) A˚ from the benzofuran ring system to the same side of the molecule as the 4-fluorophenyl ring. In the crystal, molecules are linkedviapairs of C—H interactions into inversion-related dimers. These dimers are further linked by –interactions between the benzene rings of neighbouring molecules [centroid–centroid distance = 3.715 (3) A˚ ] and between the furan rings of neighbouring molecules [centroid–centroid distance = 3.598 (3) A˚ ]. The molecules are stacked along thea-axis direction. In the sulfinyl group, the S and O atoms are disordered over two sets of sites, with site-occupancy factors of 0.797 (3) and 0.213 (3).
Keywords:crystal structure; benzofuran; 4-fluorophenyl;–interactions; sulfinyl group; natural products.
CCDC reference:1020650
1. Related literature
For pharmaceutical properties of benzofuran compounds, see: Aslam et al.(2009); Galalet al.(2009); Howlettet al.(1999); Khanet al.(2005); Onoet al.(2002). For natural products with a benzofuran ring, see: Akgul & Anil (2003); Soekamtoet al.
(2003). For the synthesis of the starting material 2-ethyl-3-(4-fluorophenylsulfanyl)-5,7-dimethyl-1-benzofuran, see: Choiet al.(1999). For a related structure, see: Choiet al.(2010).
2. Experimental
2.1. Crystal data
C18H17FO2S
Mr= 316.38
Triclinic,P1
a= 9.1523 (2) A˚
b= 9.5503 (2) A˚
c= 10.3099 (2) A˚ = 65.666 (1)
= 81.636 (1)
= 70.782 (1)
V= 775.29 (3) A˚3
Z= 2
MoKradiation = 0.22 mm1
T= 173 K
0.450.410.27 mm
2.2. Data collection
Bruker SMART APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009)
Tmin= 0.907,Tmax= 0.942
14476 measured reflections 3874 independent reflections 3481 reflections withI> 2(I)
Rint= 0.028
2.3. Refinement R[F2> 2(F2)] = 0.063
wR(F2) = 0.170
S= 1.08 3874 reflections 209 parameters
17 restraints
H-atom parameters constrained
max= 0.94 e A˚ 3 min=1.64 e A˚ 3
Table 1
Hydrogen-bond geometry (A˚ ,).
Cg1 is the centroid of the C13–C18 phenyl ring.
D—H A D—H H A D A D—H A
C10—H10B Cg1i
0.98 2.89 3.822 (2) 159
Symmetry code: (i)xþ2;y;zþ1.
Data collection: APEX2 (Bruker, 2009); cell refinement: SAINT (Bruker, 2009); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 2008); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008); molecular graphics: ORTEP-3 for Windows (Farrugia, 2012) and DIAMOND (Bran-denburg, 1998); software used to prepare material for publication: SHELXL97.
Acknowledgements
The X-ray Centre of the Gyeongsang National University is acknowledged for providing access to the single-crystal diffractometer.
data reports
o1058
Choi and Lee doi:10.1107/S1600536814019023 Acta Cryst.(2014).E70, o1058–o1059Supporting information for this paper is available from the IUCr electronic archives (Reference: HB7276).
References
Akgul, Y. Y. & Anil, H. (2003).Phytochemistry,63, 939–943.
Aslam, S. N., Stevenson, P. C., Kokubun, T. & Hall, D. R. (2009).Microbiol. Res.164, 191–195.
Brandenburg, K. (1998).DIAMOND. Crystal Impact GbR, Bonn, Germany. Bruker (2009).APEX2,SADABSandSAINT. Bruker AXS Inc., Madison,
Wisconsin, USA.
Choi, H. D., Seo, P. J. & Son, B. W. (1999).J. Korean Chem. Soc.43, 606–608.
Farrugia, L. J. (2012).J. Appl. Cryst.45, 849–854.
Galal, S. A., Abd El-All, A. S., Abdallah, M. M. & El-Diwani, H. I. (2009).
Bioorg. Med. Chem. Lett.19, 2420–2428.
Howlett, D. R., Perry, A. E., Godfrey, F., Swatton, J. E., Jennings, K. H., Spitzfaden, C., Wadsworth, H., Wood, S. J. & Markwell, R. E. (1999).
Biochem. J.340, 283–289.
Khan, M. W., Alam, M. J., Rashid, M. A. & Chowdhury, R. (2005).Bioorg. Med. Chem.13, 4796–4805.
Ono, M., Kung, M. P., Hou, C. & Kung, H. F. (2002).Nucl. Med. Biol.29, 633– 642.
Sheldrick, G. M. (2008).Acta Cryst.A64, 112–122.
supporting information
sup-1
Acta Cryst. (2014). E70, o1058–o1059
supporting information
Acta Cryst. (2014). E70, o1058–o1059 [doi:10.1107/S1600536814019023]
Crystal structure of
2-ethyl-3-(4-fluorophenylsulfinyl)-5,7-dimethyl-1-benzo-furan
Hong Dae Choi and Uk Lee
S1. Comment
Benzofuran compounds show various pharmacological properties such as antibacterial and antifungal, antitumor and
antiviral, antimicrobial activities (Aslam et al.. 2009, Galal et al., 2009, Khan et al., 2005), and potential inhibitor of β
-amyloid aggregation (Howlett et al., 1999, Ono et al., 2002). These benzofuran compounds are widely occurring in
nature (Akgul & Anil, 2003, Soekamto et al., 2003). As a part of our ongoing project of
3-(4-fluorophenylsulfinyl)-5,7-dimethyl-1-benzofuran derivatives containing methyl substituent in 2-position (Choi et al., 2010), we report herein on the
crystal structure of the title compound.
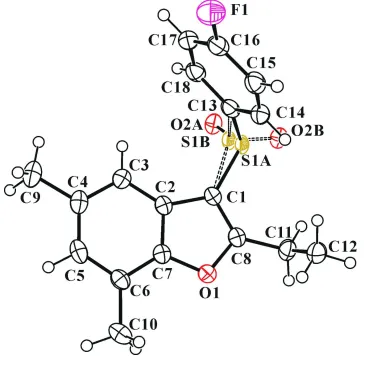
In the title molecule (Fig. 1), the benzofuran unit is essentially planar, with a mean deviation of 0.004 (2) Å from the
least-squares plane defined by the nine constituent atoms. The 4-fluorophenyl ring is essentially planar, with a mean
deviation of 0.005 (2) Å from the least-squares plane defined by the six constituent atoms. In the sulfinyl group, the S1
and O2 atoms are disordered over two positions with site-occupancy factors, from refinement of 0.797 (3) (part A) and
0.213 (3) (part B). The dihedral angle formed by the benzofuran ring and the 4-fluorophenyl ring is 86.38 (6)°. In the
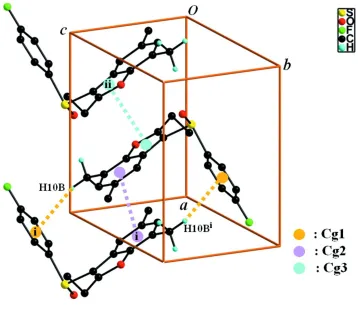
crystal structure (Fig. 2), molecules are linked via pairs of C–H···π interactions (Table 1, Cg1 is the C13-C18
4-fluoro-phenyl ring) into inversion-related dimers. These dimers are further linked by π–π interactions between the benzene rings
of neighbouring molecules with a Cg2···Cg2i distance of 3.715 (3) Å and an interplanar distance of 3.464 (3) Å resulting
in a slippage of 1.342 (3) Å (Cg2 is the C2-C7 benzene ring), and between the furan rings of neighbouring molecules
with a Cg3···Cg3ii distance of 3.598 (3) Å and an interplanar distance of 3.507 (3) Å resulting in a slippage of 0.804 (3) Å
(Cg3 is the C1/C2/C7/O1/C8 furan ring). The molecules are stacked along the a-axis direction.
S2. Experimental
The starting material 2-ethyl-3-(4-fluorophenylsulfanyl)-5,7-dimethyl-1-benzofuran was prepared by literature method
(Choi et al. 1999). 3-Chloroperoxybenzoic acid (77%, 224 mg, 1.0 mmol) was added in small portions to a stirred
solution of 2-ethyl-3-(4-fluorophenylsulfanyl)-5,7-dimethyl-1-benzofuran (270 mg, 0.9 mmol) in dichloromethane (30
ml) at 273 K. After being stirred at room temperature for 8h, the mixture was washed with saturated sodium bicarbonate
solution (2 X × 10 ml) and the organic layer was separated, dried over magnesium sulfate, filtered and concentrated at
reduced pressure. The residue was purified by column chromatography (hexane-ethyl acetate, 2:1 v/v) to afford the title
compound as a colorless solid [yield 74% (234 mg); m.p. 378-379 K; Rf = 0.59 (hexane-ethyl acetate, 2:1 v/v)].
Colourless blocks were prepared by slow evaporation of a solution of the title compound (20 mg) in acetone (15 ml) at
All H atoms were positioned geometrically and refined using a riding model, with C–H = 0.95 Å for aryl and 0.98 Å for
methylene and 0.99 Å for methyl H atoms, respectively. Uiso (H) = 1.2Ueq (C) for aryl and methylene, and 1.5Ueq (C) for
methyl H atoms. The positions of methyl and methylene hydrogens were optimized using the SHELXL-97's command
AFIX 137 (Sheldrick, 2008).The S1 and O2 atoms of the sulfinyl group is disordered over two positions with
site-occupancy factors, from refinement of 0.797 (3) (part A) and 0.213 (3) (part B). The distance of equivalent S═O,
C(pheny)–S and C(furan)–S pairs were restrained to 1.488 (1), 1.762 (1) and 1.788 (1) Å using command DFIX and
[image:4.610.125.494.200.567.2]DELU, and displacement ellipsoids of S1 and O2 set were restrained using SHELXL command EADP, respectively.
Figure 1
The molecular structure of the title compound with displacement ellipsoids drawn at the 50% probability level. The S1
and O2 atoms of the sulfinyl group are disordered over two positions with site-occupancy factors, from refinement of
supporting information
sup-3
[image:5.610.126.484.70.382.2]Acta Cryst. (2014). E70, o1058–o1059
Figure 2
A view of the C–H···π and π–π interactions (dotted lines) in the crystal structure of the title compound. H atoms
non-participating in hydrogen-bonding and disordered part B atoms were omitted for clarity. [Symmetry codes: (i) -x+2, -y,
-z+1; (ii) -x+1, -y, -z+1.]
2-ethyl-3-(4-fluorophenylsulfinyl)-5,7-dimethyl- 1-benzofuran
Crystal data
C18H17FO2S
Mr = 316.38
Triclinic, P1
Hall symbol: -P 1
a = 9.1523 (2) Å
b = 9.5503 (2) Å
c = 10.3099 (2) Å
α = 65.666 (1)°
β = 81.636 (1)°
γ = 70.782 (1)°
V = 775.29 (3) Å3
Z = 2
F(000) = 332
Dx = 1.355 Mg m−3 Melting point = 379–378 K
Mo Kα radiation, λ = 0.71073 Å
µ = 0.22 mm−1
T = 173 K
Block, colourless 0.45 × 0.41 × 0.27 mm
Data collection
Bruker SMART APEXII CCD diffractometer
Radiation source: rotating anode Graphite multilayer monochromator
Detector resolution: 10.0 pixels mm-1
φ and ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2009)
Tmin = 0.907, Tmax = 0.942 14476 measured reflections 3874 independent reflections 3481 reflections with I > 2σ(I)
h = −12→12 l = −13→13
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.063
wR(F2) = 0.170
S = 1.08
3874 reflections 209 parameters 17 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: difference Fourier map H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0799P)2 + 0.8614P]
where P = (Fo2 + 2Fc2)/3 (Δ/σ)max < 0.001
Δρmax = 0.94 e Å−3 Δρmin = −1.64 e Å−3
Special details
Experimental. 1H NMR (δ p.p.m., CDCl3, 400 Hz): 7.62-7.67 (m, 2H), 7.15-7.21 (m, 2H), 6.85 (s, 2H), 3.13 (q, J =7.52 Hz, 2H), 2.43 (s, 3H), 2.24 (s, 3H), 1.44 (t, J = 7.52 Hz, 3H).
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is
used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based
on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
S1A 0.46524 (7) 0.39880 (7) 0.27750 (7) 0.0245 (2) 0.797 (3)
O2A 0.3828 (2) 0.4499 (2) 0.39425 (17) 0.0291 (4) 0.797 (3)
S1B 0.47017 (19) 0.40209 (16) 0.3147 (3) 0.0245 (2) 0.20
O2B 0.3618 (7) 0.4565 (9) 0.1973 (6) 0.0291 (4) 0.20
F1 0.9355 (2) 0.7389 (2) 0.05373 (19) 0.0497 (4)
O1 0.69201 (18) −0.05783 (19) 0.39017 (17) 0.0278 (4)
C1 0.5858 (2) 0.20046 (14) 0.3595 (2) 0.0255 (4)
C2 0.6924 (2) 0.1271 (3) 0.4764 (2) 0.0250 (4)
C3 0.7407 (3) 0.1777 (3) 0.5674 (2) 0.0278 (5)
H3 0.6994 0.2857 0.5605 0.033*
C4 0.8512 (3) 0.0653 (3) 0.6687 (2) 0.0312 (5)
C5 0.9101 (3) −0.0934 (3) 0.6775 (2) 0.0324 (5)
H5 0.9856 −0.1675 0.7476 0.039*
C6 0.8642 (3) −0.1485 (3) 0.5895 (2) 0.0286 (5)
C7 0.7543 (2) −0.0323 (3) 0.4904 (2) 0.0262 (4)
C8 0.5892 (2) 0.0857 (3) 0.3131 (2) 0.0264 (4)
C9 0.9111 (3) 0.1158 (4) 0.7667 (3) 0.0416 (6)
H9A 0.8504 0.2264 0.7538 0.062*
H9B 1.0201 0.1105 0.7439 0.062*
H9C 0.9016 0.0434 0.8657 0.062*
supporting information
sup-5
Acta Cryst. (2014). E70, o1058–o1059
H10A 0.8436 −0.3642 0.6110 0.057*
H10B 0.9986 −0.3831 0.6794 0.057*
H10C 0.9857 −0.3192 0.5104 0.057*
C11 0.5104 (3) 0.0881 (3) 0.1959 (3) 0.0334 (5)
H11A 0.4131 0.1784 0.1735 0.040*
H11B 0.4830 −0.0132 0.2281 0.040*
C12 0.6102 (3) 0.1066 (3) 0.0612 (3) 0.0388 (6)
H12A 0.6319 0.2102 0.0252 0.058*
H12B 0.5551 0.1027 −0.0113 0.058*
H12C 0.7078 0.0189 0.0832 0.058*
C13 0.61273 (19) 0.4976 (2) 0.2173 (2) 0.0284 (4)
C14 0.6924 (3) 0.4950 (3) 0.0925 (2) 0.0314 (5)
H14 0.6723 0.4362 0.0455 0.038*
C15 0.8004 (3) 0.5770 (3) 0.0365 (3) 0.0334 (5)
H15 0.8551 0.5759 −0.0489 0.040*
C16 0.8272 (3) 0.6608 (3) 0.1073 (3) 0.0333 (5)
C17 0.7502 (3) 0.6671 (3) 0.2305 (3) 0.0352 (5)
H17 0.7718 0.7258 0.2769 0.042*
C18 0.6402 (3) 0.5856 (3) 0.2857 (3) 0.0329 (5)
H18 0.5839 0.5898 0.3698 0.040*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
S1A 0.0258 (3) 0.0285 (3) 0.0181 (4) −0.0026 (2) −0.0041 (2) −0.0109 (2)
O2A 0.0216 (8) 0.0372 (10) 0.0300 (9) −0.0030 (7) −0.0004 (5) −0.0190 (8)
S1B 0.0258 (3) 0.0285 (3) 0.0181 (4) −0.0026 (2) −0.0041 (2) −0.0109 (2)
O2B 0.0216 (8) 0.0372 (10) 0.0300 (9) −0.0030 (7) −0.0004 (5) −0.0190 (8)
F1 0.0540 (10) 0.0477 (9) 0.0512 (10) −0.0292 (8) 0.0032 (8) −0.0135 (8)
O1 0.0282 (8) 0.0260 (7) 0.0279 (8) −0.0071 (6) −0.0024 (6) −0.0094 (6)
C1 0.0242 (10) 0.0257 (7) 0.0229 (9) −0.0056 (6) −0.0007 (7) −0.0073 (8)
C2 0.0224 (9) 0.0283 (10) 0.0218 (9) −0.0083 (8) 0.0016 (7) −0.0073 (8)
C3 0.0278 (10) 0.0322 (11) 0.0253 (10) −0.0119 (9) 0.0034 (8) −0.0118 (9)
C4 0.0280 (11) 0.0438 (13) 0.0237 (10) −0.0168 (10) 0.0018 (8) −0.0110 (9)
C5 0.0266 (11) 0.0389 (12) 0.0236 (10) −0.0112 (9) −0.0033 (8) −0.0025 (9)
C6 0.0257 (10) 0.0282 (10) 0.0257 (10) −0.0087 (8) 0.0010 (8) −0.0045 (8)
C7 0.0245 (10) 0.0291 (10) 0.0238 (10) −0.0097 (8) 0.0006 (8) −0.0083 (8)
C8 0.0240 (10) 0.0291 (10) 0.0240 (10) −0.0081 (8) 0.0000 (8) −0.0083 (8)
C9 0.0429 (14) 0.0578 (17) 0.0311 (12) −0.0218 (13) −0.0025 (10) −0.0183 (12)
C10 0.0338 (12) 0.0280 (11) 0.0403 (13) −0.0046 (9) −0.0036 (10) −0.0043 (10)
C11 0.0333 (12) 0.0385 (12) 0.0318 (12) −0.0107 (10) −0.0057 (9) −0.0152 (10)
C12 0.0540 (16) 0.0366 (13) 0.0276 (11) −0.0157 (11) −0.0010 (11) −0.0126 (10)
C13 0.0288 (10) 0.0226 (10) 0.0265 (10) −0.0019 (7) −0.0031 (8) −0.0060 (8)
C14 0.0376 (12) 0.0295 (11) 0.0264 (10) −0.0074 (9) −0.0028 (9) −0.0113 (9)
C15 0.0375 (12) 0.0328 (11) 0.0259 (11) −0.0084 (10) 0.0011 (9) −0.0097 (9)
C16 0.0363 (12) 0.0262 (11) 0.0322 (12) −0.0087 (9) −0.0043 (9) −0.0057 (9)
C17 0.0461 (14) 0.0261 (11) 0.0330 (12) −0.0053 (10) −0.0081 (10) −0.0129 (9)
S1A—O2B 1.180 (3) C8—C11 1.484 (3)
S1A—O2A 1.4943 (9) C9—H9A 0.9800
S1A—C1 1.7597 (10) C9—H9B 0.9800
S1A—C13 1.7918 (10) C9—H9C 0.9800
O2A—S1B 1.182 (2) C10—H10A 0.9800
S1B—O2B 1.4869 (10) C10—H10B 0.9800
S1B—C1 1.7633 (10) C10—H10C 0.9800
S1B—C13 1.7865 (10) C11—C12 1.527 (4)
F1—C16 1.353 (3) C11—H11A 0.9900
O1—C8 1.366 (3) C11—H11B 0.9900
O1—C7 1.387 (3) C12—H12A 0.9800
C1—C8 1.355 (3) C12—H12B 0.9800
C1—C2 1.445 (3) C12—H12C 0.9800
C2—C7 1.391 (3) C13—C14 1.389 (3)
C2—C3 1.395 (3) C13—C18 1.395 (3)
C3—C4 1.391 (3) C14—C15 1.376 (4)
C3—H3 0.9500 C14—H14 0.9500
C4—C5 1.399 (4) C15—C16 1.377 (4)
C4—C9 1.513 (3) C15—H15 0.9500
C5—C6 1.388 (3) C16—C17 1.374 (4)
C5—H5 0.9500 C17—C18 1.388 (4)
C6—C7 1.384 (3) C17—H17 0.9500
C6—C10 1.502 (3) C18—H18 0.9500
O2B—S1A—O2A 98.8 (4) H9A—C9—H9B 109.5
O2B—S1A—C1 132.7 (4) C4—C9—H9C 109.5
O2A—S1A—C1 106.79 (11) H9A—C9—H9C 109.5
O2B—S1A—C13 113.2 (4) H9B—C9—H9C 109.5
O2A—S1A—C13 104.60 (11) C6—C10—H10A 109.5
C1—S1A—C13 98.06 (10) C6—C10—H10B 109.5
S1B—O2A—S1A 11.31 (14) H10A—C10—H10B 109.5
O2A—S1B—O2B 99.1 (3) C6—C10—H10C 109.5
O2A—S1B—C1 124.2 (2) H10A—C10—H10C 109.5
O2B—S1B—C1 112.3 (3) H10B—C10—H10C 109.5
O2A—S1B—C13 121.5 (2) C8—C11—C12 112.6 (2)
O2B—S1B—C13 99.4 (3) C8—C11—H11A 109.1
C1—S1B—C13 98.13 (12) C12—C11—H11A 109.1
S1A—O2B—S1B 11.63 (11) C8—C11—H11B 109.1
C8—O1—C7 106.69 (17) C12—C11—H11B 109.1
C8—C1—C2 108.00 (14) H11A—C11—H11B 107.8
C8—C1—S1A 120.39 (16) C11—C12—H12A 109.5
C2—C1—S1A 131.60 (16) C11—C12—H12B 109.5
C8—C1—S1B 132.68 (19) H12A—C12—H12B 109.5
C2—C1—S1B 119.05 (19) C11—C12—H12C 109.5
S1A—C1—S1B 13.29 (10) H12A—C12—H12C 109.5
supporting information
sup-7
Acta Cryst. (2014). E70, o1058–o1059
C7—C2—C1 104.31 (18) C14—C13—C18 120.08 (16)
C3—C2—C1 136.2 (2) C14—C13—S1B 130.30 (19)
C4—C3—C2 118.0 (2) C18—C13—S1B 109.61 (19)
C4—C3—H3 121.0 C14—C13—S1A 117.84 (16)
C2—C3—H3 121.0 C18—C13—S1A 121.90 (17)
C3—C4—C5 120.1 (2) S1B—C13—S1A 13.08 (10)
C3—C4—C9 119.9 (2) C15—C14—C13 120.4 (2)
C5—C4—C9 120.0 (2) C15—C14—H14 119.8
C6—C5—C4 123.6 (2) C13—C14—H14 119.8
C6—C5—H5 118.2 C14—C15—C16 118.4 (2)
C4—C5—H5 118.2 C14—C15—H15 120.8
C7—C6—C5 114.2 (2) C16—C15—H15 120.8
C7—C6—C10 122.3 (2) F1—C16—C17 118.7 (2)
C5—C6—C10 123.5 (2) F1—C16—C15 118.4 (2)
C6—C7—O1 124.8 (2) C17—C16—C15 122.9 (2)
C6—C7—C2 124.7 (2) C16—C17—C18 118.5 (2)
O1—C7—C2 110.49 (18) C16—C17—H17 120.8
C1—C8—O1 110.49 (18) C18—C17—H17 120.8
C1—C8—C11 133.2 (2) C17—C18—C13 119.7 (2)
O1—C8—C11 116.23 (19) C17—C18—H18 120.2
C4—C9—H9A 109.5 C13—C18—H18 120.2
C4—C9—H9B 109.5
O2B—S1A—O2A—S1B −169.5 (5) C10—C6—C7—C2 −179.1 (2)
C1—S1A—O2A—S1B 50.7 (4) C8—O1—C7—C6 −179.7 (2)
C13—S1A—O2A—S1B −52.6 (4) C8—O1—C7—C2 −0.5 (2)
S1A—O2A—S1B—O2B 8.3 (4) C3—C2—C7—C6 −0.5 (3)
S1A—O2A—S1B—C1 −116.6 (4) C1—C2—C7—C6 179.3 (2)
S1A—O2A—S1B—C13 115.3 (4) C3—C2—C7—O1 −179.77 (18)
O2A—S1A—O2B—S1B 8.1 (4) C1—C2—C7—O1 0.1 (2)
C1—S1A—O2B—S1B 130.9 (7) C2—C1—C8—O1 −0.7 (2)
C13—S1A—O2B—S1B −102.0 (5) S1A—C1—C8—O1 179.05 (14)
O2A—S1B—O2B—S1A −169.7 (5) S1B—C1—C8—O1 −174.62 (18)
C1—S1B—O2B—S1A −36.8 (6) C2—C1—C8—C11 −178.0 (2)
C13—S1B—O2B—S1A 66.1 (5) S1A—C1—C8—C11 1.8 (4)
O2B—S1A—C1—C8 13.3 (6) S1B—C1—C8—C11 8.1 (4)
O2A—S1A—C1—C8 133.10 (19) C7—O1—C8—C1 0.8 (2)
C13—S1A—C1—C8 −118.93 (19) C7—O1—C8—C11 178.54 (18)
O2B—S1A—C1—C2 −166.9 (5) C1—C8—C11—C12 96.1 (3)
O2A—S1A—C1—C2 −47.2 (2) O1—C8—C11—C12 −81.0 (3)
C13—S1A—C1—C2 60.8 (2) O2A—S1B—C13—C14 −158.4 (3)
O2B—S1A—C1—S1B −146.0 (6) O2B—S1B—C13—C14 −51.6 (4)
O2A—S1A—C1—S1B −26.3 (2) C1—S1B—C13—C14 62.8 (3)
C13—S1A—C1—S1B 81.7 (2) O2A—S1B—C13—C18 21.6 (3)
O2A—S1B—C1—C8 115.2 (3) O2B—S1B—C13—C18 128.4 (3)
O2B—S1B—C1—C8 −3.8 (4) C1—S1B—C13—C18 −117.2 (2)
C13—S1B—C1—C8 −107.5 (3) O2A—S1B—C13—S1A −139.2 (4)
C13—S1B—C1—C2 79.1 (2) O2B—S1A—C13—C14 −62.9 (4)
O2A—S1B—C1—S1A 139.6 (4) O2A—S1A—C13—C14 −169.35 (18)
O2B—S1B—C1—S1A 20.6 (4) C1—S1A—C13—C14 80.87 (19)
C13—S1B—C1—S1A −83.1 (2) O2B—S1A—C13—C18 112.2 (4)
C8—C1—C2—C7 0.4 (2) O2A—S1A—C13—C18 5.7 (2)
S1A—C1—C2—C7 −179.35 (18) C1—S1A—C13—C18 −104.0 (2)
S1B—C1—C2—C7 175.26 (16) O2B—S1A—C13—S1B 133.5 (4)
C8—C1—C2—C3 −179.8 (2) O2A—S1A—C13—S1B 27.1 (2)
S1A—C1—C2—C3 0.4 (4) C1—S1A—C13—S1B −82.7 (2)
S1B—C1—C2—C3 −4.9 (4) C18—C13—C14—C15 0.9 (3)
C7—C2—C3—C4 0.6 (3) S1B—C13—C14—C15 −179.1 (2)
C1—C2—C3—C4 −179.2 (2) S1A—C13—C14—C15 176.07 (18)
C2—C3—C4—C5 −0.2 (3) C13—C14—C15—C16 0.2 (4)
C2—C3—C4—C9 178.0 (2) C14—C15—C16—F1 178.7 (2)
C3—C4—C5—C6 −0.1 (4) C14—C15—C16—C17 −0.5 (4)
C9—C4—C5—C6 −178.4 (2) F1—C16—C17—C18 −179.4 (2)
C4—C5—C6—C7 0.2 (3) C15—C16—C17—C18 −0.2 (4)
C4—C5—C6—C10 179.5 (2) C16—C17—C18—C13 1.2 (3)
C5—C6—C7—O1 179.3 (2) C14—C13—C18—C17 −1.6 (3)
C10—C6—C7—O1 0.0 (3) S1B—C13—C18—C17 178.40 (19)
C5—C6—C7—C2 0.1 (3) S1A—C13—C18—C17 −176.58 (17)
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C13–C18 phenyl ring.
D—H···A D—H H···A D···A D—H···A
C10—H10B···Cg1i 0.98 2.89 3.822 (2) 159