Acta Cryst.(2003). E59, o699±o700 DOI: 10.1107/S1600536803008274 R. F. Henryet al. C20H21ClO4
o699
organic papers
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Fenofibrate
R. F. Henry,a* G. Z. Zhang,b Y. Gaocand I. S. Bucknerd
aAbbott Laboratories, Global Pharmaceuticals
Division, Department 418, Building AP9, 100 Abbott Park Road, Abbott Park, IL 60064, USA,
bAbbott Laboratories, Global Pharmaceuticals
Division, Department R4P3, Building AP9, 100 Abbott Park Road, Abbott Park, IL 60064, USA,
cAbbott Laboratories, Global Pharmaceuticals
Division, Department D4P#, Building AP9, 100 Abbott Park Road, Abbott Park, IL 60064, USA, anddAbbott Laboratories, Global
Pharmaceuticals Division, Department D4P3, Building AP9, 100 Abbott Park Road, Abbott Park, IL 60064, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 193 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.052
wRfactor = 0.147
Data-to-parameter ratio = 18.4
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of 1-methylethyl 2-[4-(4-chlorobenzoyl)-phenoxy]-2-methylpropanoate, also known as feno®brate, C20H21ClO4, has been determined and is presented here.
The compound crystallizes in space group P1 and is notable for its lack of hydrogen-bond donors and thus a lack of hydrogen bonding.
Comment
Feno®brate belongs to a class of compounds, ®bric acid derivatives, which are used to treat hypercholesterolemia or mixed dyslipidemia (Kloer, 1987). The physicochemical properties of feno®brate, including solubility, hygroscopicity, distribution coef®cient, and solid-state characterization, have been studied in detail (Shoji et al., 1995). Recently, a meta-stable polymorph was reported (Di Martinoet al.2000); in that paper the original polymorph and the newly discovered polymorph were designated forms I and II, respectively. In this paper, we report the molecular structure of feno®brate form I.
Feno®brate form I (see Scheme) crystallizes in the
centro-symmetric triclinic space group P1. The molecule lacks
hydrogen-bond donating groups, making it impossible for the structure to contain any type of hydrogen bonding. In the absence of hydrogen-bonding interactions, the molecules are arranged head-to-head and tail-to-tail, producing aliphatic and aromatic layers. These layers are perpendicular to thec
axis. An interesting feature of the conformation of the mol-ecule is the symmetrical nature of the isopropyl ester. A survey of the CSD (Allen, 2002) found 115 structures containing isopropyl esters. These 115 structures contained a total of 171 isopropyl ester fragments. The symmetry of the isopropyl ester was measured as the torsion angle between the carbonyl carbon, estericsp3oxygen, isopropyl methine carbon
and the centroid of the two methyl groups. Values near zero or
180 would indicate a highly symmetric orientation of the
isopropyl group. In this orientation, the isopropyl group's bisecting mirror plane coincides with the plane of the two O atoms and one carbon of the carbonyl group. The mean value found for this torsion angle was 150.7. The value nearest 180
was 174.6(Newkomeet al., 1985). The corresponding torsion
angle in feno®brate is 178.0, making it the most symmetric
crystallographically characterized isopropyl ester.
Experimental
Crystals were grown by slow evaporation of an ethanol solution.
Crystal data
C20H21ClO4
Mr= 360.82
Triclinic,P1
a= 8.1605 (16) AÊ
b= 8.2664 (16) AÊ
c= 14.511 (3) AÊ = 93.951 (3)
= 105.664 (3)
= 96.002 (3)
V= 932.5 (3) AÊ3
Z= 2
Dx= 1.285 Mg mÿ3
MoKradiation Cell parameters from 6105
re¯ections = 2.5±28.3
= 0.23 mmÿ1
T= 193 K
Parallelepiped, colourless 0.40.40.4 mm
Data collection
Bruker SMART Apex CCD diffractometer
!scans
Absorption correction: none 6105 measured re¯ections 4225 independent re¯ections
3694 re¯ections withI> 2(I)
Rint= 0.065 max= 28.3
h=ÿ9!10
k=ÿ10!10
l=ÿ19!19
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.052
wR(F2) = 0.147
S= 1.06 4225 re¯ections 230 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.1706P)2
+ 0.7796P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.38 e AÊÿ3 min=ÿ0.34 e AÊÿ3
H atoms were treated as riding atoms (CÐH = 0.93 and 0.97 AÊ).
Uisovalues for H atoms were ®xed at 1.2 timesUeqof the parent atom.
Data collection:SMART(Bruker, 2001); cell re®nement: SAINT-Plus (Bruker, 1999); data reduction: SAINT-Plus (Bruker, 1999); program(s) used to solve structure: SHELXTL (Sheldrick, 2000); program(s) used to re®ne structure:SHELXTL; molecular graphics:
ORTEPII (Johnson, 1976).
References
Allen, F. H. (2002).Acta Cryst.B58, 380±388.
Bruker (1999). SAINT-Plus. Version 6.02. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (2001).SMART. Version 5.624. Bruker AXS Inc., Madison, Wisconsin, USA.
Di Martino, P., Palmieri, G. F. & Martelli S. (2000).Pharmazie,55, 625±626. Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National
Laboratory, Tennessee. USA.
Kloer, H. U. (1987).Am. J. Med.83(suppl 5B), 3±8.
Newkome, G. R., Puckett, W. E., Kiefer, G. E., Gupta, V. K., Fronczek, F. R., Pantaleo, D. C., McClure, G. L., Simpson, J. B. & Deutsch, W. A. (1985).
Inorg. Chem.24, 811.
Sheldrick, G. M. (2000).SHELXTL.Version 6.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Shoji, R., Watanabe, T., Tashiro, S. & Shi, S. (1995).Iyakuhin Kenkyu,26, 386± 397.
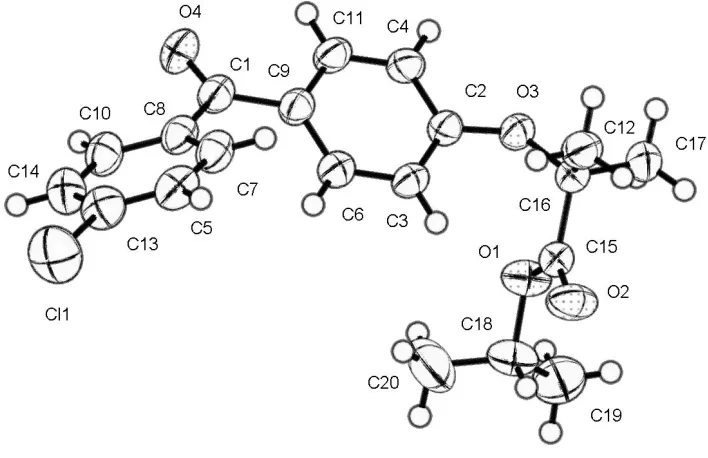
Figure 1
supporting information
sup-1
Acta Cryst. (2003). E59, o699–o700supporting information
Acta Cryst. (2003). E59, o699–o700 [doi:10.1107/S1600536803008274]
Fenofibrate
R. F. Henry, G. Z. Zhang, Y. Gao and I. S. Buckner
S1. Comment
Fenofibrate belongs to a class of compounds, fibric acid derivatives, which are used to treat hypercholesterolemia or
mixed dyslipidemia (Kloer 1987). The physicochemical properties of fenofibrate, that include solubility, hygroscopicity,
distribution coefficient, and solid-state characterization, have been studied in detail (Shoji et al., 1995). Recently, a
metastable polymorph was reported (DiMartino et al.. 2000), in which the original polymorph and the newly discovered
polymorph were designated Forms I and II, respectively. In this paper, we report the molecular structure of fenofibrate
Form I.
Fenofibrate Form I (see Scheme) crystallizes in the centrosymmetric triclinic space group P1. The molecule lacks
hydrogen bond donating groups making it impossible for the structure to contain any type of hydrogen bonding. In the
absence of hydrogen-bonding interactions, the molecules have arranged themselves head-to-head and tail-to-tail
producing aliphatic and aromatic layers. These layers are perpendicular to the c axis. An interesting feature of the
conformation of the molecule is the symmetrical nature of the isopropyl ester. A survey of the CCDC (Allen 2002) found
115 structures containing isopropyl esters. These 115 structures contained a total of 171 isopropyl ester fragments. The
symmetry of the isopropyl ester was measured as the torsion angle between the carbonyl carbon, esteric sp3 oxygen,
iso-propyl methine carbon and the centroid of the two methyl groups. Values near zero or 180° would indicate a highly
symmetric orientation of the isopropyl group. In this orientation, the isopropyl group's bisecting mirror plane coincides
with the plane of the two O atoms and one carbon of the carbonyl group. The mean value found for this torsion angle was
150.7°. The value nearest 180° was 174.6° (Newkome et al., 1985). The corresponding torsion angle in fenofibrate is
178.0°, making it the most symmetric crystallographically characterized isopropyl ester
S2. Experimental
Crystals were grown by slow evaporation from an ethanol solution.
S3. Refinement
Molecule crystallized in the triclinic system; space group P1. H atoms were treated as riding atoms (C—H = 0.93 and
Figure 1
A view of fenofibrate with the atomic numbering scheme. Displacement ellipsoids are drawn at the 30% probability level.
1-methylethyl 2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoate
Crystal data C20H21ClO4 Mr = 360.82
Triclinic, P1 a = 8.1605 (16) Å b = 8.2664 (16) Å c = 14.511 (3) Å α = 93.951 (3)° β = 105.664 (3)° γ = 96.002 (3)° V = 932.5 (3) Å3 Z = 2
F(000) = 380 Dx = 1.285 Mg m−3 Melting point = 80–81 K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 6105 reflections θ = 2.5–28.3°
µ = 0.23 mm−1 T = 193 K
Parallelepiped, colourless 0.4 × 0.4 × 0.4 mm
Data collection
Bruker SMART Apex CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
6105 measured reflections 4225 independent reflections
3694 reflections with I > 2σ(I) Rint = 0.065
θmax = 28.3°, θmin = 2.5° h = −9→10
k = −10→10 l = −19→19
Refinement Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.052 wR(F2) = 0.147 S = 1.06 4225 reflections
0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
supporting information
sup-3
Acta Cryst. (2003). E59, o699–o700H-atom parameters constrained w = 1/[σ2(Fo2) + (0.1706P)2 + 0.7796P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001 Δρmax = 0.38 e Å−3 Δρmin = −0.34 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C17 2.0889 (2) 0.6045 (2) 0.87061 (15) 0.0440 (4) H17A 2.0985 0.6038 0.9380 0.066* H17B 2.1381 0.7090 0.8589 0.066* H17C 2.1489 0.5206 0.8509 0.066* C18 1.7095 (2) 0.8146 (2) 0.97655 (12) 0.0446 (4) H18 1.6866 0.9026 0.9347 0.054* C19 1.8357 (3) 0.8783 (3) 1.06991 (17) 0.0686 (6) H19A 1.8565 0.7910 1.1102 0.103* H19B 1.7909 0.9624 1.1012 0.103* H19C 1.9413 0.9228 1.0587 0.103* C20 1.5459 (3) 0.7347 (5) 0.9905 (3) 0.1158 (15) H20A 1.4666 0.6971 0.9289 0.174* H20B 1.4972 0.8123 1.0240 0.174* H20C 1.5691 0.6436 1.0274 0.174*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Cl1 0.0578 (3) 0.0836 (4) 0.0622 (3) 0.0194 (3) 0.0023 (2) 0.0244 (3) O3 0.0290 (5) 0.0236 (5) 0.0446 (6) 0.0027 (4) 0.0087 (4) 0.0058 (4) O1 0.0557 (7) 0.0389 (6) 0.0340 (5) 0.0203 (5) 0.0167 (5) 0.0070 (4) O4 0.0435 (6) 0.0311 (6) 0.0460 (6) −0.0064 (4) 0.0113 (5) 0.0067 (5) O2 0.0734 (9) 0.0330 (6) 0.0600 (8) 0.0217 (6) 0.0383 (7) 0.0177 (5) C1 0.0381 (7) 0.0281 (7) 0.0302 (7) −0.0029 (5) 0.0140 (6) −0.0007 (5) C2 0.0298 (6) 0.0243 (6) 0.0325 (7) 0.0016 (5) 0.0120 (5) 0.0024 (5) C3 0.0338 (7) 0.0211 (6) 0.0387 (7) 0.0026 (5) 0.0095 (6) 0.0034 (5) C4 0.0352 (7) 0.0245 (6) 0.0389 (7) 0.0065 (5) 0.0140 (6) 0.0059 (5) C5 0.0545 (9) 0.0375 (8) 0.0293 (7) −0.0017 (7) 0.0112 (7) 0.0034 (6) C6 0.0319 (7) 0.0274 (7) 0.0361 (7) 0.0034 (5) 0.0089 (6) 0.0026 (5) C15 0.0283 (6) 0.0244 (6) 0.0356 (7) 0.0017 (5) 0.0100 (5) 0.0014 (5) C7 0.0430 (8) 0.0356 (7) 0.0308 (7) −0.0059 (6) 0.0149 (6) −0.0002 (6) C8 0.0377 (7) 0.0289 (7) 0.0285 (7) −0.0038 (5) 0.0111 (6) −0.0023 (5) C9 0.0336 (7) 0.0269 (6) 0.0301 (6) −0.0011 (5) 0.0129 (5) 0.0001 (5) C10 0.0402 (8) 0.0372 (8) 0.0348 (7) −0.0040 (6) 0.0136 (6) 0.0043 (6) C16 0.0299 (7) 0.0230 (6) 0.0419 (8) 0.0019 (5) 0.0138 (6) 0.0029 (5) C11 0.0403 (8) 0.0215 (6) 0.0384 (7) 0.0012 (5) 0.0154 (6) 0.0036 (5) C12 0.0492 (9) 0.0320 (7) 0.0462 (9) 0.0025 (6) 0.0258 (7) 0.0020 (6) C13 0.0451 (9) 0.0427 (8) 0.0347 (8) 0.0047 (7) 0.0055 (6) 0.0017 (6) C14 0.0374 (8) 0.0480 (9) 0.0415 (8) −0.0007 (7) 0.0122 (7) 0.0045 (7) C17 0.0288 (7) 0.0337 (8) 0.0681 (11) 0.0024 (6) 0.0120 (7) 0.0037 (7) C18 0.0576 (10) 0.0443 (9) 0.0379 (8) 0.0223 (8) 0.0181 (7) 0.0020 (7) C19 0.0758 (15) 0.0636 (13) 0.0610 (13) 0.0036 (11) 0.0181 (11) −0.0182 (10) C20 0.0498 (13) 0.131 (3) 0.157 (3) −0.0084 (15) 0.0428 (17) −0.080 (3)
Geometric parameters (Å, º)
supporting information
sup-5
Acta Cryst. (2003). E59, o699–o700O3—C16 1.4395 (16) C10—H10 0.9300 O1—C15 1.3181 (18) C16—C12 1.519 (2) O1—C18 1.4651 (18) C16—C17 1.526 (2) O4—C1 1.2187 (18) C11—H11 0.9300 O2—C15 1.1960 (18) C12—H12A 0.9600 C1—C9 1.490 (2) C12—H12B 0.9600 C1—C8 1.493 (2) C12—H12C 0.9600 C2—C3 1.3894 (19) C13—C14 1.379 (2) C2—C4 1.3922 (19) C14—H14 0.9300 C3—C6 1.390 (2) C17—H17A 0.9600 C3—H3 0.9300 C17—H17B 0.9600 C4—C11 1.375 (2) C17—H17C 0.9600 C4—H4 0.9300 C18—C19 1.486 (3) C5—C7 1.380 (2) C18—C20 1.496 (4) C5—C13 1.381 (3) C18—H18 0.9800 C5—H5 0.9300 C19—H19A 0.9600 C6—C9 1.3917 (19) C19—H19B 0.9600 C6—H6 0.9300 C19—H19C 0.9600 C15—C16 1.5322 (18) C20—H20A 0.9600 C7—C8 1.397 (2) C20—H20B 0.9600 C7—H7 0.9300 C20—H20C 0.9600 C8—C10 1.397 (2)
C8—C7—H7 119.8 O1—C18—H18 110.4 C10—C8—C7 118.85 (14) C19—C18—H18 110.4 C10—C8—C1 117.49 (13) C20—C18—H18 110.4 C7—C8—C1 123.60 (14) C18—C19—H19A 109.5 C6—C9—C11 118.25 (13) C18—C19—H19B 109.5 C6—C9—C1 123.73 (13) H19A—C19—H19B 109.5 C11—C9—C1 117.88 (13) C18—C19—H19C 109.5 C14—C10—C8 120.77 (14) H19A—C19—H19C 109.5 C14—C10—H10 119.6 H19B—C19—H19C 109.5 C8—C10—H10 119.6 C18—C20—H20A 109.5 O3—C16—C12 111.55 (12) C18—C20—H20B 109.5 O3—C16—C17 103.88 (11) H20A—C20—H20B 109.5 C12—C16—C17 111.06 (13) C18—C20—H20C 109.5 O3—C16—C15 111.82 (11) H20A—C20—H20C 109.5 C12—C16—C15 111.38 (12) H20B—C20—H20C 109.5