organic papers
Acta Cryst.(2006). E62, o739–o741 doi:10.1107/S1600536806002388 Odabas¸ogˇlu and Bu¨yu¨kgu¨ngo¨r C
6H18N2O22+2C2Cl3O2
o739
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
3,6-Dioxaoctane-1,8-diammonium
bis(trichloroacetate)
Mustafa Odabas¸ogˇlua* and Orhan Bu¨yu¨kgu¨ngo¨rb
a
Department of Chemistry, Faculty of Arts and Sciences, Ondokuz Mayıs University, TR-55139 Kurupelit Samsun, Turkey, andbDepartment of
Physics, Faculty of Arts and Sciences, Ondokuz Mayıs University, TR-55139 Kurupelit Samsun, Turkey
Correspondence e-mail: muodabas@omu.edu.tr
Key indicators
Single-crystal X-ray study
T= 296 K
Mean(C–C) = 0.007 A˚
Rfactor = 0.069
wRfactor = 0.204
Data-to-parameter ratio = 18.1
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, C6H18N2O2 2+
2C2Cl3O2
, the cation possesses a twofold rotation axis passing through the mid-point of the central C—C bond. Hydrogen-bonded rings are formed by two cations and two anions. The ions further form a three-dimensional network via N—H O and N—H Cl hydrogen bonds.
Comment
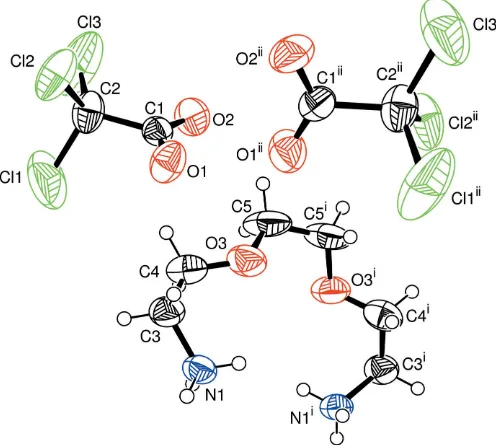
Intermolecular interactions, especially hydrogen bonds, are probably the most widely used interaction to generate supramolecularly organized organic systems with a variety of novel structural features (Prins et al., 2001; Desiraju, 1995). Hydrogen bonding has been used effectively to predict and design supramolecular assemblies in one and two dimensions (Karle et al., 1997). We have been interested in supramole-cularly hydrogen-bonded systems formed by organic amines and carboxylic acids (Odabas¸ogˇlu, Bu¨yu¨kgu¨ngo¨r & Lo¨nnecke, 2003; Odabas¸ogˇlu, Bu¨yu¨kgu¨ngo¨r, Turgutet al., 2003; Odaba-s¸ogˇlu & Bu¨yu¨kgu¨ngo¨r, 2006). The present work is part of a structural study of compounds of organic ammonium systems with hydrogen-bond donors and we report here the structure of the title compound, (I) (Fig. 1).
In compound (I), the cation possesses a twofold rotation axis passing through the mid-point of the central C—C bond. In (I), the 3,6-dioxaoctane-1,8-diammonium ions are linked to the trichloroacetate ions through N—H O and N—H Cl hydrogen bonds, resulting in the formation of hydrogen-bonded rings (Fig. 2 and Table 2). The hydrogen-hydrogen-bonded rings are formed within a three-dimensional network. It is generally recognized that N—H O bonds of the type N+ O with matching pKa values of hydrogen-bond donor and acceptor
groups are stronger than N—H O bonds involving neutral acceptors and donors. These bonds traditionally are consid-ered to be salt bridges and are termed low barrier hydrogen bonds or charge-assisted hydrogen bonds (Hess & Reinhardt, 1999). Accordingly, the strongest N—H O hydrogen bonds are found in amine–carboxylates which possess linear hydrogen bonds, with an average N O bond distance of 2.811 A˚ and an average N O bond angle of 158. The title
compound has a slightly longer N—H O bond distance and a slightly smaller N—H O bond angle (average N O bond distance = 2.835 A˚ and average bond angle N—H O = 151).
In (I), the C3—N1 bonds are equal to a normal Csp3—Nsp3
single-bond length (Vaidhyanathanet al., 2002) but the C1— C2 bond is slightly longer than a normal C—C single bond. This elongation is attributed to the repulsive effect of elec-trons on the Cl and O atoms.
Experimental
The title compound was prepared by mixing 2-[2-(2-amino-ethoxy)ethoxy]ethanamine and trichloroacetic acid in a 1:2 molar ratio in water at 353 K. Crystals of (I) were obtained by slow evaporation of the solvent (m.p. 380–381 K).
Crystal data
C6H18N2O22+2C2Cl3O2
Mr= 474.96
Monoclinic,C2=c a= 22.1995 (19) A˚
b= 7.5606 (10) A˚
c= 11.9320 (12) A˚
= 91.188 (7)
V= 2002.3 (4) A˚3 Z= 4
Dx= 1.576 Mg m
3
MoKradiation Cell parameters from 9773
reflections
= 2.5–27.8 = 0.89 mm1 T= 296 (2) K Plate, colourless 0.670.450.11 mm
Data collection
Stoe IPDS-II diffractometer
!scans
Absorption correction: integration (X-RED32; Stoe & Cie, 2002)
Tmin= 0.611,Tmax= 0.898
9773 measured reflections 1976 independent reflections
1352 reflections withI> 2(I)
Rint= 0.052
max= 26.0
h=27!27
k=9!9
l=14!14
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.069 wR(F2) = 0.204
S= 1.07 1976 reflections 109 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.091P)2
+ 4.5621P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.71 e A˚
3
min=0.54 e A˚
[image:2.610.353.524.71.310.2]3
Table 1
Selected geometric parameters (A˚ ,).
C1—O2 1.216 (5) C1—O1 1.219 (5) C1—C2 1.565 (6) C2—Cl3 1.750 (5) C2—Cl1 1.757 (6)
C2—Cl2 1.765 (5) C3—N1 1.484 (5) C3—C4 1.486 (7) C5—C5i
1.461 (12) O2—C1—O1 128.7 (4)
O2—C1—C2 116.6 (4) C1—C2—Cl3 113.5 (3)
C1—C2—Cl1 107.2 (3) Cl3—C2—Cl1 109.6 (3)
Symmetry code: (i)xþ2;y;zþ3 2.
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H5C O2iii
0.89 1.95 2.788 (5) 157 N1—H5D O1iv 0.89 2.11 2.878 (5) 143 N1—H5E O1v
0.89 2.02 2.838 (5) 152 N1—H5E Cl2v
0.89 2.69 3.343 (4) 131
Symmetry codes: (iii)x;yþ1;z; (iv)xþ2;yþ1;zþ1; (v)x;yþ1;zþ1 2.
All H atoms were refined using a riding model, with C—H = 0.97 A˚ [Uiso(H) = 1.2Ueq(parent atom)] for methylene C atoms and N—H =
0.89 A˚ [Uiso(H) = 1.5Ueq(parent atom)] for ammonium N atoms. Data collection: X-AREA (Stoe & Cie, 2002); cell refinement:
X-AREA; data reduction:X-RED32(Stoe & Cie, 2002); program(s) used to solve structure: SHELXS97(Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:ORTEP-3 for Windows(Farrugia, 1997); software used to prepare material for publication:WinGX(Farrugia, 1999).
References
Desiraju, G. R. (1995).Angew. Chem. Int. Ed. Engl.34, 2311–2327. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
organic papers
o740
Odabas¸ogˇlu and Bu¨yu¨kgu¨ngo¨r C6H18N2O22+2C2Cl3O2 Acta Cryst.(2006). E62, o739–o741 Figure 2
[image:2.610.45.293.73.296.2]A partial packing diagram of the title compound, showing the hydrogen-bonding scheme (dashed lines).
Figure 1
A view of the ions of (I), with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. [Symmetry codes: (i)xþ2;y;zþ3
[image:2.610.312.567.380.457.2]Hess, R. A. & Reinhardt, L. A. J. (1999).J. Am. Chem. Soc.121, 9867–9870. Karle, I. L., Ranganathan, D. & Haridas, V. (1997).J. Am. Chem. Soc.119,
2777–2783.
Odabas¸ogˇlu, M. & Bu¨yu¨kgu¨ngo¨r, O. (2006).Acta Cryst.E62, o236–o238. Odabas¸ogˇlu, M., Bu¨yu¨kgu¨ngo¨r, O. & Lo¨nnecke, P. (2003).Acta Cryst.C59,
o51–o52.
Odabas¸ogˇlu, M., Bu¨yu¨kgu¨ngo¨r, O., Turgut, G., Karadagˇ, A., Bulak, E. & Lo¨nnecke, P. (2003).J. Mol. Struct.648, 133–138.
Prins, L. J., Timmerman, P. & Reinhoudt, D. N. (2001).J. Am. Chem. Soc.123, 10153–10163.
Sheldrick, G. M. (1990).Acta Cryst.A46, 467–473.
Sheldrick, G. M. (1997).SHELXL97.University of Go¨ttingen, Germany. Stoe & Cie (2002).X-AREA(Version 1.18) andX-RED32(Version 1.04). Stoe
& Cie, Darmstadt, Germany.
Vaidhyanathan, R., Natarajan, S. & Rao, C. N. R. (2002).J. Mol. Struct.608, 123–133.
organic papers
Acta Cryst.(2006). E62, o739–o741 Odabas¸ogˇlu and Bu¨yu¨kgu¨ngo¨r C
supporting information
sup-1
Acta Cryst. (2006). E62, o739–o741
supporting information
Acta Cryst. (2006). E62, o739–o741 [https://doi.org/10.1107/S1600536806002388]
3,6-Dioxaoctane-1,8-diammonium bis(trichloroacetate)
Mustafa Odaba
ş
oǧlu and Orhan B
ü
y
ü
kg
ü
ng
ö
r
3,6-Dioxaoctane-1,8-diammonium bis(trichloroacetate)
Crystal data
C6H18N2O22+·2C2Cl3O2− Mr = 474.96
Monoclinic, C2/c
Hall symbol: -C 2yc
a = 22.1995 (19) Å
b = 7.5606 (10) Å
c = 11.9320 (12) Å
β = 91.188 (7)°
V = 2002.3 (4) Å3 Z = 4
F(000) = 968
Dx = 1.576 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 9773 reflections
θ = 2.5–27.8°
µ = 0.89 mm−1 T = 296 K Plate, colorless 0.67 × 0.45 × 0.11 mm
Data collection
Stoe IPDS-II diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 6.67 pixels mm-1 ω scans
Absorption correction: integration (X-RED32; Stoe & Cie, 2002)
Tmin = 0.611, Tmax = 0.898
9773 measured reflections 1976 independent reflections 1352 reflections with I > 2σ(I)
Rint = 0.052
θmax = 26.0°, θmin = 2.9° h = −27→27
k = −9→9
l = −14→14
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.069 wR(F2) = 0.204 S = 1.07 1976 reflections 109 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.091P)2 + 4.5621P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.71 e Å−3
Δρmin = −0.54 e Å−3
Special details
supporting information
sup-2
Acta Cryst. (2006). E62, o739–o741
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.90107 (19) 0.1138 (5) 0.3804 (3) 0.0484 (9) C2 0.8337 (2) 0.0914 (7) 0.3421 (4) 0.0632 (12) C3 0.9125 (2) 0.5894 (6) 0.5435 (4) 0.0639 (12)
H3A 0.9302 0.5530 0.4736 0.077*
H3B 0.8718 0.6302 0.5271 0.077*
C4 0.9105 (3) 0.4361 (6) 0.6212 (4) 0.0711 (13)
H4A 0.8912 0.4700 0.6902 0.085*
H4B 0.8874 0.3404 0.5871 0.085*
C5 0.9709 (3) 0.2364 (6) 0.7204 (5) 0.092 (2)
H5A 0.9654 0.1261 0.6800 0.111*
H5B 0.9387 0.2474 0.7737 0.111*
N1 0.94841 (16) 0.7360 (4) 0.5935 (3) 0.0546 (9)
H5C 0.9490 0.8266 0.5459 0.082*
H5D 0.9859 0.6991 0.6074 0.082*
H5E 0.9318 0.7704 0.6572 0.082*
O1 0.92925 (14) 0.2215 (5) 0.3264 (3) 0.0688 (9) O2 0.91773 (17) 0.0281 (5) 0.4615 (3) 0.0752 (10) O3 0.96880 (17) 0.3815 (4) 0.6437 (3) 0.0736 (10) Cl1 0.79681 (9) 0.2916 (4) 0.3707 (2) 0.1459 (10) Cl2 0.82866 (6) 0.0558 (2) 0.19599 (10) 0.0875 (6) Cl3 0.79694 (9) −0.0825 (3) 0.40921 (17) 0.1380 (10)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3
Acta Cryst. (2006). E62, o739–o741 Geometric parameters (Å, º)
C1—O2 1.216 (5) C4—O3 1.380 (7)
C1—O1 1.219 (5) C4—H4A 0.9700
C1—C2 1.565 (6) C4—H4B 0.9700
C2—Cl3 1.750 (5) C5—O3 1.429 (6)
C2—Cl1 1.757 (6) C5—C5i 1.461 (12)
C2—Cl2 1.765 (5) C5—H5A 0.9700
C3—N1 1.484 (5) C5—H5B 0.9700
C3—C4 1.486 (7) N1—H5C 0.8900
C3—H3A 0.9700 N1—H5D 0.8900
C3—H3B 0.9700 N1—H5E 0.8900
O2—C1—O1 128.7 (4) C3—C4—H4A 110.0
O2—C1—C2 116.6 (4) O3—C4—H4B 110.0
O1—C1—C2 114.6 (4) C3—C4—H4B 110.0
C1—C2—Cl3 113.5 (3) H4A—C4—H4B 108.4
C1—C2—Cl1 107.2 (3) O3—C5—C5i 109.0 (5)
Cl3—C2—Cl1 109.6 (3) O3—C5—H5A 109.9
C1—C2—Cl2 110.2 (3) C5i—C5—H5A 109.9
Cl3—C2—Cl2 108.5 (3) O3—C5—H5B 109.9
Cl1—C2—Cl2 107.7 (3) C5i—C5—H5B 109.9
N1—C3—C4 110.8 (4) H5A—C5—H5B 108.3
N1—C3—H3A 109.5 C3—N1—H5C 109.5
C4—C3—H3A 109.5 C3—N1—H5D 109.5
N1—C3—H3B 109.5 H5C—N1—H5D 109.5
C4—C3—H3B 109.5 C3—N1—H5E 109.5
H3A—C3—H3B 108.1 H5C—N1—H5E 109.5
O3—C4—C3 108.3 (4) H5D—N1—H5E 109.5
O3—C4—H4A 110.0 C4—O3—C5 111.8 (4)
O2—C1—C2—Cl3 −10.2 (5) O1—C1—C2—Cl2 50.7 (5)
O1—C1—C2—Cl3 172.6 (3) N1—C3—C4—O3 −58.7 (5)
O2—C1—C2—Cl1 111.0 (4) C3—C4—O3—C5 178.6 (4)
O1—C1—C2—Cl1 −66.2 (4) C5i—C5—O3—C4 −154.3 (6)
O2—C1—C2—Cl2 −132.1 (4)
Symmetry code: (i) −x+2, y, −z+3/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H5C···O2ii 0.89 1.95 2.788 (5) 157
N1—H5D···O1iii 0.89 2.11 2.878 (5) 143
N1—H5E···O1iv 0.89 2.02 2.838 (5) 152
N1—H5E···Cl2iv 0.89 2.69 3.343 (4) 131