organic papers
Acta Cryst.(2006). E62, o1281–o1283 doi:10.1107/S1600536806007628 Andreas Fischer C
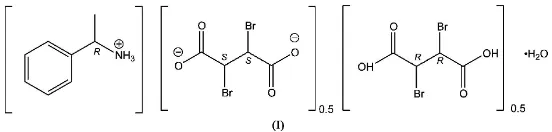
8H12N+0.5C4H2Br2O420.5C4H4Br2O4
o1281
Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
(
R
)-1-Phenylethanaminium–(
S
,
S
)-2,3-dibromo-succinate–(
R
,
R
)-2,3-dibromosuccinic acid–
water (2/1/1/2)
Andreas Fischer
Inorganic Chemistry, School of Chemical Science and Engineering, Royal Institute of Technology (KTH), 100 44 Stockholm, Sweden
Correspondence e-mail: andif@inorg.kth.se
Key indicators
Single-crystal X-ray study
T= 299 K
Mean(C–C) = 0.013 A˚
Rfactor = 0.059
wRfactor = 0.120
Data-to-parameter ratio = 20.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 15 February 2006 Accepted 2 March 2006
#2006 International Union of Crystallography
All rights reserved
From an aqueous solution of racemic 2,3-dibromosuccinic acid and (R)-1-phenylethanamine, crystals of the title compound, C8H12N
+
0.5C4H2Br2O4 2
0.5C4H4Br2O4H2O, were obtained
in almost quantitative yield. The structure contains both enantiomers of the starting material, dibromosuccinic acid. The S,S enantiomer is present as a dianion and the R,R enantiomer as the neutral acid; both of these components lie on twofold rotation axes. The structure features a complex two-dimensional network of hydrogen bonds.
Comment
Recently, we reported the crystal structure of (2R,3S )-2,3-dibromosuccinic acid (dbs) (Erikssonet al., 2006). The struc-ture of racemic 2,3-dibromosuccinic acid had been reported earlier (Bolte & Degen, 2000). To determine the structure of the pure enantiomer, (R,R)- or (S,S)-dibromosuccinic acid, we tried to crystallize the dibromosuccinate with the enanti-omerically pure base (R)-1-phenylethanamine. The result of this crystallization was, however, the title compound, (I), which is almost insoluble in water and whose structure is described here.
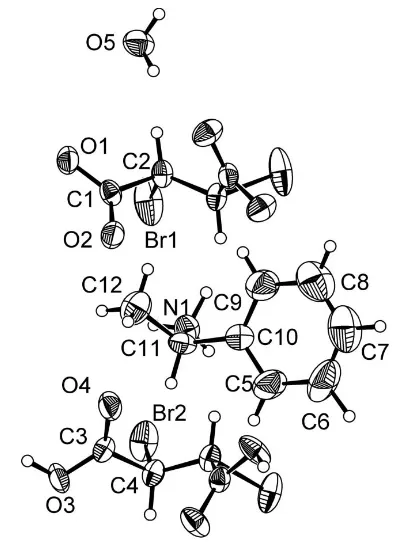
Compound (I) features phenylethanaminium cations, anions of the 2S,3Senantiomer of dbs, the 2R,3Renantiomer of the neutral H2dbs, and water of crystallization. In both the
anion and the neutral acid, a twofold rotation axis passes through the mid-point of the central C—C bond (Fig. 1 and Fig. 2). The bond lengths and angles in (I) are unexceptional. The molecule of water of crystallization participates in hydrogen bonding, both as donor and acceptor. The hydrogen-bonding pattern is shown in Fig. 3, with numeric details in Table 1.
A similar crystallization behaviour has been reported recently (Li et al., 2005) in the structure of the hydroxy-quinolinium salt of the meso form of dibromosuccinic acid, where both the protonated and deprotonated forms of dibromosuccinic acid are present.
Experimental
[image:1.610.204.480.434.500.2]ether (100 ml) and bromine (12.1 g, Aldrich, 99.5%) was added. After complete dissolution of the maleic acid, the solution was evaporated and the residue was recrystallized from water. The yield after recrystallization was 13.3 g (67%). The racemate (276 mg) was dissolved in water (5 ml). Upon addition of (R)-1-phenylethylamine (0.13 ml, Fluka, purum, enantiomeric purity verified by measuring the optical rotation), the solution was kept at 323 K for 12 h. After this time, the title compound had crystallized in almost quantitative yield as crystals of up to several mm in size.
Crystal data
C8H12N+0.5C4H2Br2O42
-0.5C4H4Br2O4H2O
Mr= 415.07
Orthorhombic,P21212
a= 12.609 (2) A˚
b= 13.235 (3) A˚
c= 9.689 (2) A˚
V= 1616.8 (6) A˚3
Z= 4
Dx= 1.705 Mg m3 MoKradiation Cell parameters from 39
reflections
= 8.0–20.2 = 5.03 mm1
T= 299 K Fragment, colourless 0.400.300.14 mm
Data collection
Bruker–Nonius KappaCCD diffractometer
’and!scans
Absorption correction: numerical (HABITUS; Herrendorf & Ba¨rnighausen, 1997)
Tmin= 0.265,Tmax= 0.759
12294 measured reflections
3652 independent reflections 2529 reflections withI> 2(I)
Rint= 0.075 max= 27.5
h=16!15
k=16!17
l=11!12
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.059
wR(F2) = 0.120
S= 1.21 3652 reflections 181 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0061P)2
+ 4.8215P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001 max= 0.65 e A˚3 min=0.60 e A˚3
Absolute structure: Flack (1983), 1550 Friedel pairs
Flack parameter: 0.00 (3)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O3—H3 O1i
0.95 1.48 2.431 (6) 177 N1—H1A O5ii
0.83 2.01 2.828 (6) 172 N1—H1B O2iii 0.99 2.02 2.947 (8) 155 N1—H1C O4iii
0.85 2.12 2.948 (8) 164 O5—H5A O4iv
0.88 1.97 2.833 (8) 167 O5—H5B O2iv 0.86 2.07 2.910 (8) 165
Symmetry codes: (i)xþ1
2;y12;z; (ii) xþ12;y12;zþ1; (iii)x;y;zþ1; (iv) x;yþ1;z.
The H atoms were located in a difference Fourier map and were refined in their as-found positions, using a riding model, withUiso(H)
= 1.2Ueqof the carrier atom, with C—H = 0.93–0.98 A˚ , N—H = 0.83–
0.99 A˚ , water O—H = 0.86–0.88 A˚ and carboxyl O—H = 0.95 A˚. Data collection: COLLECT (Nonius, 1999); cell refinement: DIRAX/LSQ(Duisenberget al., 2003); data reduction:EVALCCD (Duisenberg, 1992); program(s) used to solve structure:SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97 (Sheldrick, 1997); molecular graphics: DIAMOND (Brandenburg, 2005); software used to prepare material for publication: maXus (Mackayet al., 1999).
organic papers
o1282
Andreas Fischer C [image:2.610.68.265.68.339.2]8H12N+0.5C4H2Br2O420.5C4H4Br2O4 Acta Cryst.(2006). E62, o1281–o1283
Figure 1
The four components of the structure of (I). Displacement ellipsoids are drawn at the 50% probability level. Unlabelled atoms in the anion are related to labelled atoms by (x, 1y,z). Unlabelled atoms in the acid are related to labelled atoms by (x,y,z).
Figure 2
[image:2.610.44.294.583.713.2]The unit-cell contents of (I), viewed alongc.
Figure 3
The hydrogen-bonding (dashed lines) pattern in (I). Symmetry codes as in Table 1, plus (v)1
2x, 3
2y,z; (vi) 1x, 1y,1 +z; (vii)x,y, 1 +z; (viii)1
2x, 1
The Swedish Research Council (VR) is acknowledged for funding of the single-crystal diffractometer.
References
Bolte, M. & Degen, A. (2000).Acta Cryst.C56, e410.
Brandenburg, K. (2005).DIAMOND. Release 3.1. Crystal Impact GbR, Bonn, Germany.
Duisenberg, A. J. M. (1992).J. Appl. Cryst.25, 92–96.
Duisenberg, A. J. M., Kroon-Batenburg, L. M. J. & Schreurs, A. M. M. (2003).
J. Appl. Cryst.36, 220–229.
Eriksson, M., Fischer, A., Lind, J. & Zazzi, A˚ . (2006).Acta Cryst.E62, o200– o201.
Flack, H. D. (1983).Acta Cryst.A39, 876–881.
Herrendorf, W. & Ba¨rnighausen, H. (1997). HABITUS. Universities of Giessen and Karlsruhe, Germany.
Li, D.-X., Xu, D.-J. & Xu, Y.-Z.(2005).Acta Cryst.E61, o402–o404. Mackay, S., Gilmore, C. J., Edwards, C., Stewart, N. & Shankland, K. (1999).
maXus. Bruker–Nonius, The Netherlands, MacScience, Japan, and The University of Glasgow, Scotland.
Nonius (1999).COLLECT. Nonius BV, Delft, The Netherlands.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
organic papers
Acta Cryst.(2006). E62, o1281–o1283 Andreas Fischer C
supporting information
sup-1
Acta Cryst. (2006). E62, o1281–o1283
supporting information
Acta Cryst. (2006). E62, o1281–o1283 [https://doi.org/10.1107/S1600536806007628]
(
R
)-1-Phenylethanaminium–(
S
,
S
)-2,3-dibromosuccinate–(
R
,
R
)-2,3-dibromo-succinic acid
–
water (2/1/1/2)
Andreas Fischer
(R)-1-Phenylethanaminium–(S,S)-2,3-dibromosuccinate– (R,R)-2,3-dibromosuccinic acid–water (2/1/1/2)
Crystal data
C8H12N+·0.5C4H2Br2O42−·0.5C4H4Br2O4·H2O
Mr = 415.07
Orthorhombic, P21212
Hall symbol: P 2 2ab
a = 12.609 (2) Å
b = 13.235 (3) Å
c = 9.689 (2) Å
V = 1616.8 (6) Å3
Z = 4
F(000) = 824
Dx = 1.705 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 39 reflections
θ = 8.0–20.2°
µ = 5.03 mm−1
T = 299 K
Fragment, colourless 0.40 × 0.30 × 0.14 mm
Data collection
Bruker–Nonius KappaCCD diffractometer
Radiation source: fine-focus sealed tube
φ and ω scans
Absorption correction: numerical
(HABITUS; Herrendorf & Bärnighausen, 1997)
Tmin = 0.265, Tmax = 0.759
12294 measured reflections
3652 independent reflections 2529 reflections with I > 2σ(I)
Rint = 0.075
θmax = 27.5°, θmin = 4.5°
h = −16→15
k = −16→17
l = −11→12
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.059
wR(F2) = 0.120
S = 1.21 3652 reflections 181 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0061P)2 + 4.8215P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.65 e Å−3
Δρmin = −0.60 e Å−3
Absolute structure: Flack (1983), 1550 Friedel pairs
Absolute structure parameter: 0.00 (3)
Special details
supporting information
sup-2
Acta Cryst. (2006). E62, o1281–o1283
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 0.13275 (8) 0.46912 (9) 0.39939 (7) 0.0718 (3)
O3 0.1973 (4) −0.0311 (4) 0.1058 (5) 0.0488 (15)
H3 0.2391 −0.0029 0.0338 0.059*
O4 0.0908 (4) 0.1007 (4) 0.0783 (6) 0.0485 (14)
C3 0.1185 (6) 0.0227 (5) 0.1366 (6) 0.0310 (15)
C4 0.0567 (5) −0.0154 (6) 0.2623 (6) 0.0338 (16)
H4 0.0612 −0.0893 0.2662 0.041*
O1 0.1979 (4) 0.5360 (4) 0.0837 (5) 0.0462 (13)
C1 0.1186 (6) 0.4810 (5) 0.1150 (6) 0.0316 (14)
O2 0.0919 (4) 0.4059 (4) 0.0548 (6) 0.0461 (14)
C2 0.0560 (5) 0.5188 (6) 0.2401 (6) 0.0319 (16)
H2 0.0688 0.5918 0.2358 0.038*
N1 0.2072 (4) 0.2464 (5) 0.9049 (5) 0.0393 (12)
H1A 0.2724 0.2510 0.8980 0.047*
H1B 0.1816 0.3129 0.9384 0.047*
H1C 0.1816 0.2088 0.9684 0.047*
C11 0.1695 (5) 0.2197 (5) 0.7626 (8) 0.0377 (17)
H11 0.1841 0.1479 0.7475 0.045*
C10 0.0511 (5) 0.2345 (6) 0.7540 (7) 0.0395 (17)
C9 0.0066 (9) 0.3289 (7) 0.7463 (11) 0.066 (3)
H9 0.0495 0.3861 0.7482 0.079*
C5 −0.0126 (8) 0.1520 (7) 0.7563 (11) 0.064 (3)
H5 0.0173 0.0878 0.7598 0.077*
C8 −0.1032 (10) 0.3392 (10) 0.7368 (12) 0.084 (4)
H8 −0.1333 0.4029 0.7272 0.100*
C6 −0.1231 (10) 0.1628 (10) 0.7534 (12) 0.088 (4)
H6 −0.1746 0.1013 0.7617 0.105*
C7 −0.1653 (7) 0.2561 (13) 0.7407 (11) 0.088 (3)
H7 −0.2385 0.2633 0.7342 0.106*
C12 0.2329 (6) 0.2793 (7) 0.6574 (9) 0.057 (2)
H12A 0.2181 0.2540 0.5666 0.069*
H12B 0.3072 0.2721 0.6767 0.069*
H12C 0.2136 0.3493 0.6624 0.069*
O5 0.0685 (3) 0.7490 (5) 0.0978 (5) 0.0528 (12)
H5A 0.0275 0.8026 0.0911 0.063*
H5B 0.0278 0.7009 0.0712 0.063*
supporting information
sup-3
Acta Cryst. (2006). E62, o1281–o1283 Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.0415 (5) 0.1352 (10) 0.0386 (4) 0.0213 (7) −0.0113 (4) 0.0044 (5)
O3 0.041 (3) 0.050 (3) 0.056 (3) 0.023 (3) 0.024 (2) 0.020 (3)
O4 0.038 (3) 0.049 (3) 0.059 (3) 0.014 (2) 0.014 (3) 0.020 (3)
C3 0.026 (4) 0.038 (4) 0.029 (3) −0.005 (3) −0.001 (3) 0.007 (3)
C4 0.027 (3) 0.043 (4) 0.031 (3) −0.006 (3) −0.001 (3) 0.002 (3)
O1 0.035 (3) 0.046 (3) 0.057 (3) −0.006 (2) 0.016 (2) −0.015 (3)
C1 0.024 (3) 0.038 (4) 0.033 (3) 0.005 (3) −0.004 (3) −0.007 (3)
O2 0.039 (3) 0.044 (3) 0.055 (3) −0.010 (2) 0.015 (3) −0.015 (3)
C2 0.025 (3) 0.044 (4) 0.026 (3) 0.000 (3) −0.002 (2) −0.003 (3)
N1 0.034 (2) 0.045 (3) 0.039 (3) 0.002 (3) −0.004 (2) −0.002 (3)
C11 0.043 (4) 0.026 (4) 0.045 (4) 0.003 (3) −0.005 (3) −0.003 (3)
C10 0.044 (4) 0.038 (5) 0.036 (4) 0.000 (4) −0.005 (3) 0.001 (4)
C9 0.057 (6) 0.055 (6) 0.086 (7) 0.000 (5) −0.006 (6) 0.015 (6)
C5 0.053 (6) 0.052 (5) 0.086 (7) −0.015 (4) −0.013 (5) −0.003 (5)
C8 0.077 (9) 0.093 (9) 0.081 (7) 0.029 (7) −0.015 (7) 0.016 (7)
C6 0.056 (7) 0.107 (9) 0.101 (8) −0.041 (7) 0.001 (7) −0.014 (7)
C7 0.050 (5) 0.129 (10) 0.085 (7) 0.017 (9) −0.017 (5) 0.009 (10)
C12 0.058 (5) 0.068 (7) 0.046 (5) −0.008 (4) 0.001 (4) 0.012 (4)
O5 0.038 (2) 0.040 (2) 0.080 (4) 0.004 (3) −0.017 (2) −0.004 (4)
Br2 0.0392 (4) 0.0955 (7) 0.0389 (4) −0.0126 (5) −0.0101 (3) −0.0114 (4)
Geometric parameters (Å, º)
C1—C2 1.530 (9) C11—C12 1.516 (10)
C1—O1 1.273 (9) C11—H11 0.9800
C1—O2 1.201 (8) C10—C5 1.356 (11)
C2—C2i 1.497 (13) C10—C9 1.372 (12)
C2—H2 0.9817 C9—C8 1.393 (15)
C2—Br1 1.936 (6) C9—H9 0.9300
C3—O3 1.258 (8) C5—C6 1.401 (17)
C3—O4 1.229 (8) C5—H5 0.9300
C3—C4 1.531 (8) C8—C7 1.351 (16)
O3—H3 0.9517 C8—H8 0.9300
C4—C4ii 1.487 (13) C6—C7 1.350 (16)
C4—Br2 1.950 (7) C6—H6 1.0437
C4—H4 0.9800 C7—H7 0.9300
N1—C11 1.500 (8) C12—H12A 0.9601
N1—H1A 0.8265 C12—H12B 0.9600
N1—H1B 0.9934 C12—H12C 0.9600
N1—H1C 0.8541 O5—H5A 0.8801
C11—C10 1.508 (9) O5—H5B 0.8569
C3—O3—H3 112.9 N1—C11—H11 107.8
O4—C3—O3 126.3 (6) C10—C11—H11 107.7
supporting information
sup-4
Acta Cryst. (2006). E62, o1281–o1283
O3—C3—C4 113.8 (6) C5—C10—C9 119.5 (7)
C4ii—C4—C3 113.5 (5) C5—C10—C11 118.8 (8)
C4ii—C4—Br2 110.4 (4) C9—C10—C11 121.7 (8)
C3—C4—Br2 105.1 (4) C10—C9—C8 119.9 (10)
C4ii—C4—H4 109.3 C10—C9—H9 120.1
C3—C4—H4 109.3 C8—C9—H9 120.0
Br2—C4—H4 109.3 C10—C5—C6 120.5 (10)
O2—C1—O1 125.3 (7) C10—C5—H5 119.8
O2—C1—C2 120.7 (6) C6—C5—H5 119.8
O1—C1—C2 114.0 (6) C7—C8—C9 119.7 (10)
C2i—C2—C1 112.2 (5) C7—C8—H8 120.3
C2i—C2—Br1 111.0 (4) C9—C8—H8 120.0
C1—C2—Br1 105.2 (4) C7—C6—C5 119.1 (10)
C2i—C2—H2 118.8 C7—C6—H6 118.4
C1—C2—H2 101.7 C5—C6—H6 122.5
Br1—C2—H2 106.6 C6—C7—C8 121.2 (9)
C11—N1—H1A 105.0 C6—C7—H7 119.4
C11—N1—H1B 113.9 C8—C7—H7 119.3
H1A—N1—H1B 106.5 C11—C12—H12A 109.4
C11—N1—H1C 113.9 C11—C12—H12B 109.4
H1A—N1—H1C 118.6 H12A—C12—H12B 109.5
H1B—N1—H1C 99.1 C11—C12—H12C 109.6
N1—C11—C10 109.5 (6) H12A—C12—H12C 109.5
N1—C11—C12 109.1 (6) H12B—C12—H12C 109.5
C10—C11—C12 114.7 (6) H5A—O5—H5B 102.9
Symmetry codes: (i) −x, −y+1, z; (ii) −x, −y, z.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O3—H3···O1iii 0.95 1.48 2.431 (6) 177
N1—H1A···O5iv 0.83 2.01 2.828 (6) 172
N1—H1B···O2v 0.99 2.02 2.947 (8) 155
N1—H1C···O4v 0.85 2.12 2.948 (8) 164
O5—H5A···O4i 0.88 1.97 2.833 (8) 167
O5—H5B···O2i 0.86 2.07 2.910 (8) 165