organic papers
o972
Graham Smithet al. C10H9N3O DOI: 10.1107/S1600536803012479 Acta Cryst.(2003). E59, o972±o974 Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
8-Quinolinylurea
Graham Smith,a* Urs D. Wermuthaand Jonathan M. Whiteb
aSchool of Physical and Chemical Sciences,
Queensland University of Technology, GPO Box 2434, Brisbane 4001, Australia, and
bSchool of Chemistry, University of Melbourne,
Parkville 3010, Australia
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.044
wRfactor = 0.098
Data-to-parameter ratio = 11.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of 8-quinolinylurea, C10H9N3O, shows
the urea moiety to be close to coplanar with the quinoline ring, due to intramolecular hydrogen bonding between the quino-line-N atom and the H atom on the nearest urea N, and between the urea O atom and a quinoline-ring H atom. The molecules associate, through hydrogen bonds involving all potential donor and acceptor sites, to form a two-dimensional sheet structure.
Comment
We have previously reported the synthesis and crystal struc-tures of unsymmetrically substituted ureas together with their adducts with a number of carboxylic acids. These ureas included phenylurea (Kashino & Haisa, 1977; Bottet al., 2000) and 1,1-diethylurea (Smith & Kennard, 2000; Smith et al., 2000). These unsymmetrically substituted ureas are of interest because of their potential herbicidal properties,e.g.monuron [3-(4-chlorophenyl)-1,1-dimethylurea; Baughman, Hembreet al., 1980] and diuron [3-(3,4-dichlorophenyl) 1,1-dimethyl-urea; Baughman, Samset al., 1980] are commercial herbicides. The structure of the 1:1 proton-transfer compound of the unsymmetrical Lewis base-substituted urea (8-quinolinyl)urea with 3,5-dinitrosalicylic acid has previously been reported (Smithet al., 2001) and we report here the crystal structure of the parent urea compound, (8-quinolinyl)urea, (I), which has also been investigated for its phytotoxic properties (Paganiet al., 1983; Smithet al., 1997).
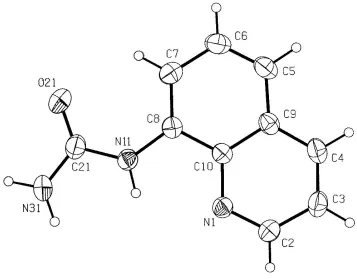
This determination shows only minor deviations from planarity in the overall molecule (Fig. 1), with the torsion angles C7ÐC8ÐN11ÐC21 and C8ÐN11ÐC21ÐN31 being 8.6 (3) and 171.0 (1) AÊ, respectively. This is largely due to the presence of intramolecular hydrogen bonds, on one side between the hetero-N atom of the quinoline residue and the H atom on the nearest urea-N atom [N11 N1 = 2.685 (2) AÊ], and on the other side between the urea O atom and a quinoline ring H atom [O21 C7 = 2.895 (2) AÊ]. The (8-quinolinium)urea cations in the 1:1 proton-transfer compound with 3,5-dinitrosalicylic acid are considerably different conformationally, with the urea side chain inverted and non-coplanar with the quinoline ring and with no intramolecular
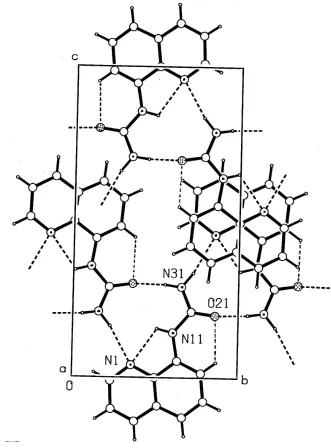
hydrogen bonds. The molecules of (I) also form a cyclic hydrogen-bonding association through the urea functional groups and the hetero-N atom of the quinoline ring (Fig. 2 and Table 1). These dimers are then extended into a convoluted two-dimensional sheet structurevia n-glide-related molecules, with a separation ofca3.8 AÊ [Cg1 Cg1 = 3.838 (3) AÊ (Cg1 is the centroid of the N11-containing ring) and = 0.02 (2)] between the sheets.
Experimental
The title compound, (I), was synthesized using the Vogel (1989) procedure. 8±Aminoquinoline (10 g, 0,069 mmol) was dissolved in 10 ml of hot glacial acetic acid and a solution of NaCNO (4.51 g, 0.069 mmol) in 50 ml of warm water was added with stirring, and the warmed solution stirred for a further 30 min. The mixture was cooled on an ice bath for 30 min and the precipitate of (I) removed by vacuum ®ltration and vacuum dried, giving 10 g of off-white product (77% yield). Recrystallization from absolute ethanol gave colourless crystals (m.p. 476±477 K) suitable for X-ray diffraction.
Crystal data
C10H9N3O
Mr= 187.20
Monoclinic,P21=n
a= 7.1436 (8) AÊ
b= 8.1394 (9) AÊ
c= 15.2836 (17) AÊ
= 93.333 (2) V= 887.16 (17) AÊ3
Z= 4
Dx= 1.402 Mg mÿ3
MoKradiation Cell parameters from 978
re¯ections
= 2.7±22.1
= 0.10 mmÿ1
T= 293 (2) K Block, colourless 0.100.160.32 mm
Data collection
Bruker CCD area-detector diffractometer
'and!scans
Absorption correction: none 4542 measured re¯ections 1566 independent re¯ections
1133 re¯ections withI> 2(I)
Rint= 0.050
max= 25.0
h=ÿ8!6
k=ÿ9!9
l=ÿ15!18
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.044
wR(F2) = 0.098
S= 0.97 1566 re¯ections 140 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.0391P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.13 e AÊÿ3
min=ÿ0.12 e AÊÿ3
Extinction correction:SHELXTL97 Extinction coef®cient: 0.0062 (18)
Table 1
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N11ÐH11 N1 0.859 (17) 2.227 (17) 2.685 (2) 113.3 (14) N31ÐH31A O21i 0.91 (2) 1.94 (2) 2.836 (3) 171 (2)
N31ÐH31B N1ii 0.90 (2) 2.19 (2) 3.072 (2) 166.1 (18)
C7ÐH7 O21 0.93 2.30 2.895 (2) 122
Symmetry codes: (i)1
2ÿx;ÿ12y;12ÿz; (ii)12ÿx;12y;12ÿz.
H atoms attached to N11 and N31 (H11, H31Aand H31B) were located in difference syntheses and their positional and isotropic displacement parameters were re®ned. Other H atoms were included in the re®nement in the riding-model approximation.
Data collection:SMART(Bruker, 2000); cell re®nement:SMART; data reduction: SAINT (Bruker, 1999); program(s) used to solve structure: SHELXTL97 (Bruker, 1997); program(s) used to re®ne structure:SHELXTL97; molecular graphics:PLATON for Windows (Spek, 1999); software used to prepare material for publication: SHELXTL97.
The authors acknowledge ®nancial support from The School of Physical and Chemical Sciences of the Queensland University of Technology and The University of Melbourne.
References
Baughman, R. G., Hembre, R. I., Helland, B. J. & Jacobson, R. A. (1980).
Cryst. Struct. Commun.9, 749±752.
Baughman, R. G., Sams, D. D., Helland, B. J. & Jacobson, R. A. (1980).Cryst. Struct. Commun.9, 885±889.
Bott, R. C., Smith, G., Wermuth, U. D. & Dwyer, N. C. (2000).Aust. J. Chem.
53, 767±777.
Bruker (1997).SHELXTL97. Bruker AXS Inc., Madison, Wisconsin, USA. Bruker (1999).SAINT. Version 6.02. Bruker AXS Inc., Madison, Wisconsin,
USA.
Bruker (2000).SMART. Version 5.55. Bruker AXS Inc., Madison, Wisconsin, USA.
Kashino, S. & Haisa, M. (1977).Acta Cryst.B33, 855±860.
Acta Cryst.(2003). E59, o972±o974 Graham Smithet al. C10H9N3O
o973
organic papers
Figure 1
The molecular con®guration and atom-labelling scheme for (I), with displacement ellipsoids drawn at the 30% probability level.
Figure 2
organic papers
o974
Graham Smithet al. C10H9N3O Acta Cryst.(2003). E59, o972±o974Pagani, G., Carmellino, M. L., Caccialanza, G. & Borgna, P. (1983).Dip. Chim. Farm.38, 128±134.
Smith, C. J., Hansch, C. & Morton, M. J. (1997).Mutat. Res.379, 167±175. Smith, G., Bott, R. C., & Wermuth, U. D. (2001).Acta Cryst.E57, o895±o897. Smith, G., Coyne, M. G. & White, J. M. (2000).Aust. J. Chem.53, 203±208.
Smith, G. & Kennard, C. H. L. (2000).Aust. J. Chem.53, 999±1001. Spek, A. L. (1999). PLATON for Windows. September 1999 Version.
University of Utrecht, The Netherlands.
supporting information
sup-1
Acta Cryst. (2003). E59, o972–o974supporting information
Acta Cryst. (2003). E59, o972–o974 [doi:10.1107/S1600536803012479]
8-Quinolinylurea
Graham Smith, Urs D. Wermuth and Jonathan M. White
S1. Comment
We have previously reported the synthesis and crystal structures of unsymmetrically substituted ureas together with their
adducts with a number of carboxylic acids. These ureas included phenylurea (Kashino & Haisa, 1977; Bott et al., 2000)
and 1,1-diethylurea (Smith & Kennard, 2000; Smith et al., 2000). These unsymmetrically substituted ureas are of interest
because of their potential herbicidal properties, e.g. monuron [3-(4-chlorophenyl)-1,1-dimethylurea; Baughman, Hembre
et al., 1980] and diuron [3-(3,4-dichlorophenyl) 1,1-dimethylurea; Baughman, Sams et al., 1980] are commercial
herbicides. The structure of the 1:1 proton-transfer compound of the unsymmetrical Lewis base-substituted urea
(8-quinolinyl)urea with 3,5-dinitrosalicylic acid has previously been reported (Smith et al., 2001) and we report here the
crystal structure of the parent urea compound, (8-quinolinyl)urea, (I), which has also been investigated for its phytotoxic
properties (Pagani et al., 1983; Smith et al., 1997). This determination shows only minor deviations from planarity in the
overall molecule (Fig. 1) with the torsion angles C7–C8–N11–C21 and C8–N11–C21–N31 being 8.6 (3) and 171.0 (1) Å,
respectively. This is largely due to the presence of intramolecular hydrogen bonds, on one side between the proton on the
first urea-N atom and the hetero-N atom of the quinoline residue [N11···N1 = 2.685 (2) Å], and on the other side between
the urea O atom and a quinoline ring H atom [O21···C7 = 2.895 (2) Å]. The (8-quinolinium)urea cations in the 1:1
proton-transfer compound with 3,5-dinitrosalicylic acid are considerably different conformationally, with the urea side
chain inverted and non-coplanar with the quinoline ring and with no intramolecular hydrogen bonds. The molecules of (I)
also give a cyclic hydrogen-bonding association through the urea functional groups and the hetero-N atom of the
quinoline ring (Fig. 2 and Table 1). These dimers then are then extended into a convoluted two-dimensional sheet
structure via similar peripheral n-glide-related molecules with a separation of ca 3.8 Å [Cg1···Cg1 = 3.838 (3) Å and α =
2.89 (2)°] between the sheets.
S2. Experimental
The title compound, (I), was synthesized using the Vogel (1989) procedure. 8-Aminoquinoline (10 g, 0,069 mmol) was
dissolved in 10 ml of hot glacial acetic acid and a solution of NaCNO (4.51 g, 0.069 mmol) in 50 ml of warm water was
added with stirring, and the warmed solution stirred for a further 30 min. The mixture was cooled on an ice bath for 30
min and the precipitate of (I) removed by vacuum filtration and vacuum dried, giving 10 g of off-white product (77%
yield). Recrystallization from absolute ethanol gave colourless data crystals (m.p. 476–477 K) suitable for X-ray
diffraction.
S3. Refinement
H atoms involved in hydrogen-bonding inetractions (H11, H31A and H31B) were located by difference syntheses and
their positional and isotropic displacement parameters were refined. Other H atoms were included in the refinement in the
supporting information
[image:5.610.126.483.71.350.2]sup-2
Acta Cryst. (2003). E59, o972–o974Figure 1
supporting information
[image:6.610.136.467.73.521.2]sup-3
Acta Cryst. (2003). E59, o972–o974Figure 2
Packing in the unit cell, viewed down a, showing hydrogen-bonding interactions as broken lines,
8-quinolinylurea
Crystal data
C10H9N3O
Mr = 187.20 Monoclinic, P21/n
Hall symbol: -P 2y n
a = 7.1436 (8) Å
b = 8.1394 (9) Å
c = 15.2836 (17) Å
β = 93.333 (2)°
V = 887.16 (17) Å3
Z = 4
F(000) = 392
Dx = 1.402 Mg m−3
Melting point = 476–477 K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 978 reflections
θ = 2.7–22.1°
µ = 0.10 mm−1
supporting information
sup-4
Acta Cryst. (2003). E59, o972–o974Data collection
Bruker CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
4542 measured reflections 1566 independent reflections
1133 reflections with I > 2σ(I)
Rint = 0.050
θmax = 25.0°, θmin = 2.7°
h = −8→6
k = −9→9
l = −15→18
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.044
wR(F2) = 0.098
S = 0.97 1566 reflections 140 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0391P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.13 e Å−3
Δρmin = −0.12 e Å−3
Extinction correction: SHELXTL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0062 (18)
Special details
Geometry. Bond distances, angles etc. have been calculated using the rounded fractional coordinates. All e.s.d.'s are estimated from the variances of the (full) variance-covariance matrix. The cell e.s.d.'s are taken into account in the estimation of distances, angles and torsion angles
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O21 0.3942 (2) 0.85917 (16) 0.19967 (8) 0.0791 (6)
N1 0.2027 (2) 0.35554 (16) 0.03980 (9) 0.0479 (5)
N11 0.3135 (2) 0.6068 (2) 0.14424 (10) 0.0530 (6)
N31 0.3128 (3) 0.6553 (3) 0.29036 (12) 0.0873 (9)
C2 0.1491 (3) 0.2315 (2) −0.01131 (12) 0.0554 (7)
C3 0.1322 (3) 0.2386 (2) −0.10303 (12) 0.0620 (8)
C4 0.1725 (3) 0.3823 (2) −0.14244 (12) 0.0603 (8)
C5 0.2810 (3) 0.6708 (2) −0.12906 (12) 0.0633 (8)
C6 0.3394 (3) 0.7968 (2) −0.07680 (13) 0.0657 (8)
C7 0.3541 (3) 0.7802 (2) 0.01482 (13) 0.0582 (7)
C8 0.3079 (2) 0.6349 (2) 0.05384 (11) 0.0465 (6)
C9 0.2336 (2) 0.5182 (2) −0.09216 (11) 0.0497 (7)
C10 0.2471 (2) 0.4995 (2) −0.00024 (11) 0.0430 (6)
C21 0.3420 (3) 0.7169 (2) 0.21112 (13) 0.0601 (8)
H2 0.120500 0.132600 0.015400 0.0660*
supporting information
sup-5
Acta Cryst. (2003). E59, o972–o974H4 0.159600 0.390900 −0.203200 0.0720*
H5 0.272000 0.684600 −0.189600 0.0760*
H6 0.370400 0.896400 −0.102000 0.0790*
H7 0.395600 0.868400 0.049400 0.0700*
H11 0.276 (3) 0.510 (2) 0.1571 (11) 0.065 (6)*
H31A 0.256 (3) 0.557 (3) 0.2977 (14) 0.108 (9)*
H31B 0.323 (3) 0.725 (2) 0.3359 (14) 0.092 (7)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O21 0.1126 (13) 0.0523 (9) 0.0718 (10) −0.0103 (8) 0.0004 (8) −0.0133 (7)
N1 0.0531 (10) 0.0437 (8) 0.0469 (9) 0.0044 (7) 0.0030 (7) −0.0024 (7)
N11 0.0683 (12) 0.0444 (10) 0.0461 (10) −0.0008 (8) 0.0005 (7) −0.0059 (8)
N31 0.144 (2) 0.0723 (14) 0.0451 (12) −0.0189 (14) 0.0019 (11) −0.0138 (11)
C2 0.0618 (13) 0.0473 (11) 0.0572 (12) 0.0006 (9) 0.0039 (9) −0.0041 (9)
C3 0.0723 (15) 0.0595 (13) 0.0537 (12) 0.0031 (10) 0.0003 (10) −0.0154 (10)
C4 0.0709 (15) 0.0692 (14) 0.0409 (11) 0.0097 (11) 0.0029 (9) −0.0059 (10)
C5 0.0754 (15) 0.0669 (13) 0.0484 (11) 0.0059 (11) 0.0115 (10) 0.0110 (10)
C6 0.0758 (15) 0.0568 (12) 0.0656 (14) −0.0006 (11) 0.0148 (11) 0.0131 (11)
C7 0.0595 (13) 0.0489 (11) 0.0666 (13) −0.0031 (9) 0.0063 (10) −0.0028 (9)
C8 0.0431 (11) 0.0491 (11) 0.0473 (11) 0.0059 (8) 0.0031 (8) −0.0026 (8)
C9 0.0512 (12) 0.0549 (11) 0.0434 (11) 0.0106 (9) 0.0074 (8) 0.0002 (9)
C10 0.0409 (11) 0.0448 (10) 0.0438 (10) 0.0091 (8) 0.0061 (7) 0.0005 (8)
C21 0.0699 (15) 0.0548 (12) 0.0546 (13) 0.0027 (10) −0.0052 (10) −0.0106 (10)
Geometric parameters (Å, º)
O21—C21 1.232 (2) C5—C9 1.413 (2)
N1—C2 1.320 (2) C5—C6 1.351 (3)
N1—C10 1.368 (2) C6—C7 1.405 (3)
N11—C8 1.399 (2) C7—C8 1.373 (2)
N11—C21 1.366 (2) C8—C10 1.430 (2)
N31—C21 1.338 (3) C9—C10 1.411 (2)
N11—H11 0.859 (17) C2—H2 0.9306
N31—H31A 0.91 (2) C3—H3 0.9298
N31—H31B 0.90 (2) C4—H4 0.9305
C2—C3 1.401 (3) C5—H5 0.9306
C3—C4 1.354 (2) C6—H6 0.9298
C4—C9 1.402 (2) C7—H7 0.9297
C2—N1—C10 117.22 (14) N1—C10—C8 118.18 (15)
C8—N11—C21 128.79 (16) N1—C10—C9 122.26 (15)
C8—N11—H11 112.6 (11) C8—C10—C9 119.56 (15)
C21—N11—H11 117.8 (11) O21—C21—N31 123.03 (19)
C21—N31—H31A 122.5 (14) N11—C21—N31 114.03 (17)
H31A—N31—H31B 118.6 (19) O21—C21—N11 122.93 (18)
supporting information
sup-6
Acta Cryst. (2003). E59, o972–o974N1—C2—C3 124.46 (16) C3—C2—H2 117.79
C2—C3—C4 118.19 (16) C2—C3—H3 120.86
C3—C4—C9 120.34 (17) C4—C3—H3 120.95
C6—C5—C9 120.27 (17) C3—C4—H4 119.84
C5—C6—C7 121.27 (16) C9—C4—H4 119.82
C6—C7—C8 120.65 (16) C6—C5—H5 119.82
C7—C8—C10 119.03 (16) C9—C5—H5 119.90
N11—C8—C10 115.80 (15) C5—C6—H6 119.33
N11—C8—C7 125.17 (16) C7—C6—H6 119.41
C4—C9—C10 117.50 (15) C6—C7—H7 119.69
C4—C9—C5 123.29 (16) C8—C7—H7 119.66
C5—C9—C10 119.21 (15)
C10—N1—C2—C3 −1.0 (3) C9—C5—C6—C7 −0.2 (3)
C2—N1—C10—C8 −179.20 (16) C5—C6—C7—C8 −0.5 (3)
C2—N1—C10—C9 1.2 (2) C6—C7—C8—N11 −178.49 (17)
C21—N11—C8—C7 8.6 (3) C6—C7—C8—C10 0.9 (3)
C21—N11—C8—C10 −170.82 (17) C7—C8—C10—N1 179.64 (15)
C8—N11—C21—O21 −10.6 (3) C7—C8—C10—C9 −0.8 (2)
C8—N11—C21—N31 170.96 (17) N11—C8—C10—N1 −0.9 (2)
N1—C2—C3—C4 −0.3 (3) N11—C8—C10—C9 178.71 (13)
C2—C3—C4—C9 1.5 (3) C4—C9—C10—N1 −0.1 (2)
C3—C4—C9—C5 178.91 (19) C5—C9—C10—C8 0.1 (3)
C3—C4—C9—C10 −1.3 (3) C4—C9—C10—C8 −179.70 (15)
C6—C5—C9—C4 −179.84 (19) C5—C9—C10—N1 179.72 (15)
C6—C5—C9—C10 0.3 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N11—H11···N1 0.859 (17) 2.227 (17) 2.685 (2) 113.3 (14)
N31—H31A···O21i 0.91 (2) 1.94 (2) 2.836 (3) 171 (2)
N31—H31B···N1ii 0.90 (2) 2.19 (2) 3.072 (2) 166.1 (18)
C7—H7···O21 0.93 2.30 2.895 (2) 122