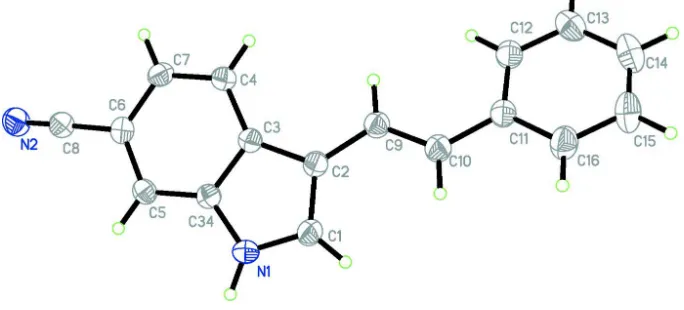
3 [(E) 2 Phenylethenyl] 1H indole 6 carbonitrile
7
0
0
Full text
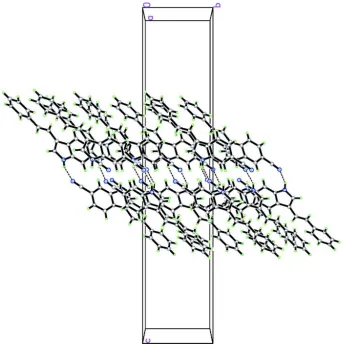
Figure
Related documents