organic papers
Acta Cryst.(2007). E63, o221–o223 doi:10.1107/S1600536806051488 Melanie Rademeyer C
8H12N+Br
o221
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
2-Phenylethylammonium bromide
Melanie Rademeyer
School of Chemistry, University of KwaZulu-Natal, Pietermaritzburg Campus, Private Bag X01, Scottsville 3209, South Africa
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 295 K
Mean(C–C) = 0.005 A˚ Rfactor = 0.032 wRfactor = 0.059
Data-to-parameter ratio = 32.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 7 November 2006 Accepted 28 November 2006
#2007 International Union of Crystallography All rights reserved
2-Phenylethylammonium bromide, C8H12N +
Br, adopts a layered structure consisting of alternating hydrophilic and hydrophobic regions. The ammonium groups and bromide anions interact through N+—H Br hydrogen bonds, forming transoid one-dimensional ladders, which are further linked by electrostatic N+ Br interactions into two-dimensional sheets.
Comment
The crystal structure of 2-phenylethylammonium bromide, (I) (Fig. 1), was determined as part of an ongoing study of the structural characteristics and hydrogen-bonding interactions of organic salts consisting of arylammonium cations and halide anions. The unit-cell parameters of (I) have been reported previously by Tsoucaris (1961), but fractional coordinates were not given in that report. The crystal structure of the analogous chloride salt has also been described previously (Tsoucaris, 1961; Hornet al., 1990), and (I) is isostructural to it. Compared to the unit-cell parameters of the chloride salt [a
= 4.603 (1) A˚ , b= 5.906 (1) A˚ , c= 32.360 (1) A˚ ; Horn et al., 1990], the bparameter in (I) is slightly elongated and the c
parameter is slightly shortened. As expected, the unit-cell volume of (I) is larger than that of the chloride salt [880 (1) A˚3].
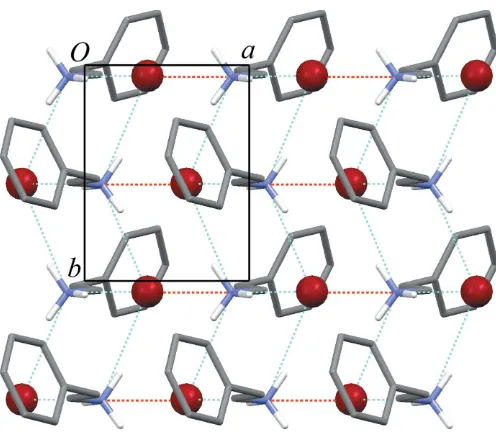
The crystal structure of (I) consists of layers lying parallel to theabplane, with alternating hydrophobic regions (containing the ammonium groups and bromide anions) and hydrophilic regions (containing the rest of the organic cations) (Fig. 2). In the organic cation, the ethylammonium chain is close to a fully extended all-transconformation, with the N1—C1—C2—C3 torsion angle equal to 173.6 (2). The benzene rings pack in a
non-interdigitated fashion, with the ring plane forming a dihedral angle of 55.68 (7)to theabplane. In this region, the shortest centroid-to-centroid distance between benzene rings is 5.590 (2) A˚ .
charge-assisted hydrogen bonds to three different bromide anions, with N+ Brdistances ranging from 3.3186 (19) A˚ to 3.370 (2) A˚ . These interactions define atransoid one-dimen-sional hydrogen-bonded ladder (Fig. 2). A fourth, longer N+ Brcontact [N1 Br1i= 3.459 (2) A˚ ; symmetry code: (i)
1 + x, y, z] links these hydrogen-bonded ladders into corrugated two-dimensional sheets (Fig. 3). A similar sheet structure has been reported for ethylammonium bromide (Jellinek, 1958; Bond, 2005). In both that structure and (I), adjacent sheets are stacked in a parallel offset fashion, with Br N+ Brangles close to 90.
Experimental
2-Phenylethylammonium bromide was synthesized by dropwise addition of HBr (8.2 ml, 72.9 mmol, 48%, Fluka) to a solution of 2-phenylethylamine (5.3 ml, 42.2 mmol, Aldrich, 99%) in chloroform (30 ml). The resulting precipitate was filtered off. The crystal used for this structure determination was crystallized from an aqueous solu-tion of 2-phenylethylammonium bromide (0.500 g, 2.47 mmol) and
CoBr2(Aldrich, 99%, 0.271 g, 1.24 mmol) (2:1 molar ratio), open to the atmosphere, at room temperature over a period of eight weeks.
Crystal data
C8H12N+Br
Mr= 202.10
Orthorhombic,P212121
a= 4.6871 (4) A˚
b= 6.1419 (4) A˚
c= 32.047 (2) A˚
V= 922.56 (12) A˚3
Z= 4
Dx= 1.455 Mg m
3
MoKradiation
= 4.39 mm1
T= 295 (2) K Block, colourless 0.300.200.20 mm
Data collection
Oxford Diffraction Excalibur2 CCD diffractometer
!scans
Absorption correction: multi-scan (Blessing, 1995)
Tmin= 0.340,Tmax= 0.419
9448 measured reflections 2958 independent reflections 2063 reflections withI> 2(I)
Rint= 0.027 max= 32.0
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.032
wR(F2) = 0.059
S= 1.06 2958 reflections 91 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0257P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.003
max= 0.46 e A˚
3
min=0.86 e A˚
3
Absolute structure: Flack (1983), 1047 Friedel pairs
Flack parameter: 0.029 (15)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H1A Br1 0.89 2.49 3.360 (2) 166 N1—H1B Br1i
0.89 2.46 3.3186 (19) 162 N1—H1C Br1ii
0.89 2.51 3.370 (2) 163
Symmetry codes: (i)xþ1;y1 2;zþ
3
2; (ii)xþ1;yþ 1 2;zþ
3 2.
organic papers
o222
Melanie Rademeyer C [image:2.610.315.563.67.288.2]8H12N+Br Acta Cryst.(2007). E63, o221–o223
Figure 1
[image:2.610.99.240.74.302.2]The molecular structure of (I), showing displacement ellipsoids at the 50% probability level for non-H atoms.
Figure 2
View of (I) down theaaxis, showing layers lying parallel to theabplane and N+—H Brhydrogen bonds (blue dotted lines) forming extended one-dimensional ladders.
Figure 3
View of (I) down thecaxis, showing a single two-dimensional sheet in the
[image:2.610.59.279.349.458.2]H atoms were placed in calculated positions, with methylene C—H = 0.97 A˚ , aromatic C—H = 0.93 A˚, and N—H = 0.89 A˚, and were refined using a riding model, withUiso(H) = 1.2Ueq(C) or 1.5Ueq(N). Data collection: CrysAlis CCD (Oxford Diffraction, 2006); cell refinement: CrysAlis RED(Oxford Diffraction, 2006); data reduc-tion:CrysAlis RED; program(s) used to solve structure:SHELXS97
(Sheldrick, 1997); program(s) used to refine structure:SHELXL97
(Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows
(Farrugia, 1997) andMercury(Brunoet al., 2002); software used to prepare material for publication:PLATON(Spek, 2003) andWinGX
(Farrugia, 1999).
This work was funded by the University of KwaZulu-Natal Research Office, and the National Research Foundation (GUN:2054350).
References
Blessing, R. H. (1995).Acta Cryst.A51, 33–38. Bond, A. D. (2005).Cryst. Growth Des.5, 755–771.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838. Flack, H. D. (1983).Acta Cryst.A39, 876–881.
Horn, E., Tiekink, E. R. T., Jones, G. P., Naiola, B. P. & Paleg, L. G. (1990).Acta Cryst.C46, 1575–1576.
Jellinek, F. (1958).Acta Cryst.11, 626–631.
Oxford Diffraction (2006). CrysAlis CCD and CrysAlis RED. Versions 1.171.29.9. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13. Tsoucaris, G. (1961).Acta Cryst.14, 909–914.
organic papers
Acta Cryst.(2007). E63, o221–o223 Melanie Rademeyer C
supporting information
sup-1
Acta Cryst. (2007). E63, o221–o223
supporting information
Acta Cryst. (2007). E63, o221–o223 [https://doi.org/10.1107/S1600536806051488]
2-Phenylethylammonium bromide
Melanie Rademeyer
2-Phenylethylammonium bromide
Crystal data
C8H12N+·Br− Mr = 202.10
Orthorhombic, P212121
Hall symbol: P 2ac 2ab
a = 4.6871 (4) Å
b = 6.1419 (4) Å
c = 32.047 (2) Å
V = 922.56 (12) Å3
Z = 4
F(000) = 408
Dx = 1.455 Mg m−3
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 4396 reflections
θ = 3.8–32.0°
µ = 4.39 mm−1
T = 295 K
Block, colourless 0.30 × 0.20 × 0.20 mm
Data collection
Oxford Diffraction Excalibur2 CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω–2θ scans
Absorption correction: multi-scan (Blessing, 1995)
Tmin = 0.340, Tmax = 0.419
9448 measured reflections 2958 independent reflections 2063 reflections with I > 2σ(I) Rint = 0.027
θmax = 32.0°, θmin = 3.8°
h = −6→6
k = −8→7
l = −46→45
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.032 wR(F2) = 0.059
S = 1.06
2958 reflections 91 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0257P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.003
Δρmax = 0.46 e Å−3 Δρmin = −0.86 e Å−3
Absolute structure: Flack (1983), 1047 Friedel pairs
Absolute structure parameter: 0.029 (15)
Special details
supporting information
sup-2
Acta Cryst. (2007). E63, o221–o223
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 0.88110 (6) 0.45078 (4) 0.792497 (8) 0.04480 (8)
N1 0.3964 (4) 0.4462 (3) 0.71525 (6) 0.0421 (4)
H1A 0.4992 0.4364 0.7385 0.063*
H1B 0.2841 0.3300 0.7130 0.063*
H1C 0.2897 0.5660 0.7162 0.063*
C1 0.5899 (5) 0.4568 (4) 0.67874 (7) 0.0419 (5)
H1D 0.7084 0.3274 0.6781 0.050*
H1E 0.7143 0.5822 0.6815 0.050*
C2 0.4263 (5) 0.4736 (5) 0.63826 (8) 0.0510 (7)
H2A 0.3185 0.3408 0.6339 0.061*
H2B 0.2920 0.5935 0.6400 0.061*
C3 0.6239 (6) 0.5098 (4) 0.60187 (8) 0.0475 (6)
C4 0.7563 (7) 0.7084 (6) 0.59673 (10) 0.0640 (9)
H4 0.7208 0.8207 0.6155 0.077*
C5 0.9434 (8) 0.7414 (6) 0.56346 (12) 0.0784 (11)
H5 1.0339 0.8751 0.5602 0.094*
C6 0.9932 (8) 0.5778 (8) 0.53580 (12) 0.0832 (12)
H6 1.1163 0.6008 0.5135 0.100*
C7 0.8653 (10) 0.3822 (7) 0.54052 (11) 0.0827 (10)
H7 0.9010 0.2710 0.5215 0.099*
C8 0.6804 (7) 0.3472 (6) 0.57376 (11) 0.0666 (9)
H8 0.5939 0.2120 0.5770 0.080*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.04897 (14) 0.03200 (11) 0.05344 (14) 0.00065 (13) −0.00575 (15) 0.00154 (12)
N1 0.0466 (11) 0.0324 (9) 0.0473 (11) 0.0006 (14) −0.0066 (11) 0.0005 (9)
C1 0.0348 (13) 0.0423 (11) 0.0488 (13) 0.0021 (15) −0.0048 (12) 0.0031 (12)
C2 0.0418 (16) 0.0590 (16) 0.0520 (15) 0.0045 (14) −0.0028 (12) −0.0030 (13)
C3 0.0442 (13) 0.0514 (18) 0.0468 (14) 0.0051 (15) −0.0129 (14) 0.0027 (10)
C4 0.073 (2) 0.0584 (19) 0.061 (2) 0.0075 (16) −0.0054 (17) 0.0090 (16)
C5 0.074 (3) 0.075 (2) 0.086 (3) −0.006 (2) −0.010 (2) 0.030 (2)
C6 0.074 (2) 0.115 (3) 0.061 (2) 0.013 (2) 0.0072 (17) 0.022 (3)
C7 0.086 (3) 0.102 (3) 0.060 (2) 0.015 (3) 0.013 (2) −0.0136 (18)
supporting information
sup-3
Acta Cryst. (2007). E63, o221–o223 Geometric parameters (Å, º)
N1—C1 1.482 (3) C2—H2B 0.970
N1—H1A 0.890 C8—C7 1.390 (5)
N1—H1B 0.890 C8—H8 0.930
N1—H1C 0.890 C5—C6 1.360 (5)
C1—C2 1.511 (3) C5—C4 1.396 (5)
C1—H1D 0.970 C5—H5 0.930
C1—H1E 0.970 C6—C7 1.351 (6)
C3—C8 1.370 (4) C6—H6 0.930
C3—C4 1.379 (4) C4—H4 0.930
C3—C2 1.506 (4) C7—H7 0.930
C2—H2A 0.970
C1—N1—H1A 109.5 C3—C2—H2B 109.4
C1—N1—H1B 109.5 C1—C2—H2B 109.4
H1A—N1—H1B 109.5 H2A—C2—H2B 108.0
C1—N1—H1C 109.5 C3—C8—C7 120.8 (4)
H1A—N1—H1C 109.5 C3—C8—H8 119.6
H1B—N1—H1C 109.5 C7—C8—H8 119.6
N1—C1—C2 111.73 (19) C6—C5—C4 119.9 (4)
N1—C1—H1D 109.3 C6—C5—H5 120.0
C2—C1—H1D 109.3 C4—C5—H5 120.0
N1—C1—H1E 109.3 C7—C6—C5 120.5 (4)
C2—C1—H1E 109.3 C7—C6—H6 119.7
H1D—C1—H1E 107.9 C5—C6—H6 119.7
C8—C3—C4 118.6 (3) C3—C4—C5 120.2 (3)
C8—C3—C2 121.4 (3) C3—C4—H4 119.9
C4—C3—C2 120.0 (3) C5—C4—H4 119.9
C3—C2—C1 111.3 (2) C6—C7—C8 120.0 (3)
C3—C2—H2A 109.4 C6—C7—H7 120.0
C1—C2—H2A 109.4 C8—C7—H7 120.0
C8—C3—C2—C1 106.6 (3) C8—C3—C4—C5 0.0 (4)
C4—C3—C2—C1 −72.2 (3) C2—C3—C4—C5 178.9 (3)
N1—C1—C2—C3 173.6 (2) C6—C5—C4—C3 0.6 (5)
C4—C3—C8—C7 −0.6 (5) C5—C6—C7—C8 0.2 (6)
C2—C3—C8—C7 −179.4 (3) C3—C8—C7—C6 0.4 (6)
C4—C5—C6—C7 −0.8 (6)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1A···Br1 0.89 2.49 3.360 (2) 166
N1—H1B···Br1i 0.89 2.46 3.3186 (19) 162
N1—H1C···Br1ii 0.89 2.51 3.370 (2) 163