organic papers
o1276
Samaset al. C11H15O4PH2O doi:10.1107/S1600536807006496 Acta Cryst.(2007). E63, o1276–o1278
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
2-Hydroxy-5,5-dimethyl-4-phenyl-1,3,2-dioxaphosphorinan-2-one monohydrate
Brian Samas,* Todd Groendyke, Anthony C Blackburn and Delara B Godrej
Pfizer Global Research and Devlopment, Pharmaceutical Sciences, Ann Arbor, MI 48105, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 173 K
Mean(C–C) = 0.002 A˚ Rfactor = 0.034 wRfactor = 0.085
Data-to-parameter ratio = 18.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 2 February 2007 Accepted 7 February 2007
#2007 International Union of Crystallography All rights reserved
The title compound, C11H15O4PH2O, commonly called (S )-phencyphos monohydrate, belongs to a class of phosphori-nanes. Molecules form a hydrogen-bond network of corru-gated ribbons.
Comment
Phosphorinanes have been popular in classical chiral resolu-tion (CCR) for several decades (Ten Hoeve & Wynberg, 1985). Although the crystal structure has not been deter-mined, the title compound, (I), has been used in CCR and also in Dutch Resolution (Lohet al., 2006).
One end of the molecule of compound (I) is polar and the other is non-polar. The hydrophilic end interacts with the water molecule. These interactions form a layer parallel to both theaaxis and thebaxis. The hydrophobic phenyl ring interleaves in a layer, also parallel to both theaaxis and theb
axss. These hydrophobic and hydrophilic layers alternate along the c axis. The phenyl rings are stacked, but none of these interactions are–in nature.
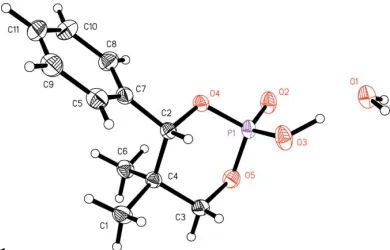
[image:1.610.251.413.314.421.2] [image:1.610.235.430.589.714.2]The structure has a contact present within the van der Waals radius, from a metahydrogen on the phenyl group to one of the O atoms of the phosphorinane. Although the phenyl ring has room to rotate, it is configured within the van der Waals
Figure 1
radius; hence this contact is considered to be a weak hydrogen bond (Table 1).
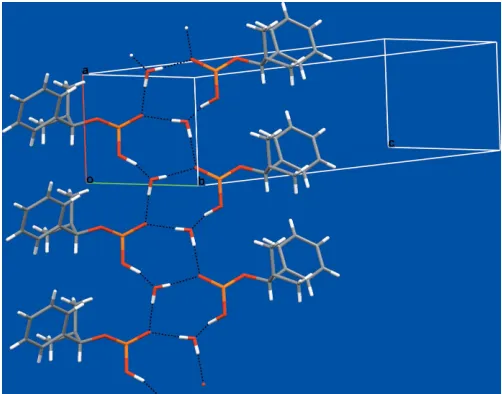
The most important close contacts are hydrogen bonds between the acid group and the water molecule. The hydrogen bonding between these groups forms an uncommon 10-membered ring (Table 1 and Fig. 2). The hydrogen-bonded rings stack back-to-back, forming a puckered ribbon propa-gating in the direction of both the b axis and the c axis. Molecules are hydrogen bonded to equivalent molecules generated by the 21 screw symmetry. All strong donors and acceptors are used in this crystal structure and form strong hydrogen bonds, which are almost linear.
The acidic proton forms a very strong hydrogen bond with the water molecule. This P O(—OH) group is chiral in the solid state (R); this chirality is not inherent to the compound as the proton is exchangable in the solution state.
The water molecule is in a very stable environment. The O atom accepts one strong hydrogen bond, along a lone pair, while the water also donates two hydrogen bonds. This is the most common hydrogen-bond conformation for a hydrate (Gillonet al., 2003). We measured the critical water activity of compound (I) at room temperature to be less than 0.2 (Golles, 1962). Both the hydrate and the anhydrous forms were observed in the course of our experiments.
The dioxaphosphorinane ring is in the expected chair conformation. This compound has the P O oxygen and the phenyl group in the expected equatorial positions.
Experimental
A stock solution of (I) was made in methanol (0.05M). A 1 ml aliquot of the stock solution was added to a 1.8 ml HPLC screw-capped vial. This was allowed to evaporate over 24 h. A crystal of (I) was removed from the vial and mounted on a mitogen loop with paratone-Noil. The water activity was measured by setting up slurries in mixtures of water and ethanol. Suspended solids were allowed to slurry for two weeks, filtered and then analysed. The powder X-ray patterns of
resulting solids from these slurries were measured and compared with the predicted powder X-ray pattern of compound (I).
Crystal data
C11H15O4PH2O
Mr= 260.22
Orthorhombic,P212121 a= 6.017 (2) A˚ b= 8.219 (3) A˚ c= 25.475 (10) A˚ V= 1259.7 (8) A˚3
Z= 4
Dx= 1.372 Mg m
3
MoKradiation
= 0.23 mm1 T= 173 (2) K Column, colourless 0.610.090.04 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2003) Tmin= 0.861,Tmax= 0.991
23045 measured reflections 3004 independent reflections 2899 reflections withI> 2(I) Rint= 0.030
max= 28.2
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.034 wR(F2) = 0.085
S= 1.11 3004 reflections 167 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0515P)2
+ 0.1668P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.45 e A˚
3
min=0.23 e A˚
3
Absolute structure: Flack (1983), 1213 friedel pairs
Flack parameter:0.03 (8)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H99B O2i
0.83 (3) 1.85 (3) 2.670 (2) 171 (2) O1—H99A O2ii 0.81 (3) 1.91 (3) 2.7179 (19) 176 (2) C9—H9 O4iii
0.95 2.68 3.625 (2) 174
O3—H99C O1iv
0.988 (10) 1.475 (11) 2.4395 (18) 164 (2)
Symmetry codes: (i) x;yþ1;z; (ii) xþ1 2;yþ
1
2;z; (iii) x;yþ 1 2;zþ
1 2; (iv)
x1;y1;z.
All H atoms were found in a Fourier difference map. H atoms bound to C atoms were placed in idealized positions and refined using a riding model, with C—H = 0.95 A˚ for Csp2, 0.98 A˚ for CH3, 0.99 A˚
for CH2and 1.00 A˚ for CH.Uiso(H) values were set at 1.5Ueq(C) for
the methyl groups and at 1.2Ueq(C) in all other cases. H atoms bound
to the water molecule and the acidic group were located in difference Fourier maps and their positions were refined freely with isotropic displacement parameters. The PO—H distance was restrained to 0.98 (1) A˚ .
Data collection:SMART(Bruker, 2003); cell refinement:SAINT (Bruker, 2003); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: Mercury(Brunoet al., 2002); software used to prepare material for publication:SHELXL97.
References
Bruker (2003).SMARTfor WNT/2000 (Version 5.630),SAINT-Plus(Version 6.45) andSHELXTL(Version 6.14). Bruker AXS Inc., Madison, Wisconsin, USA.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Flack, H. D. (1983).Acta Cryst.A39, 876–881.
organic papers
Acta Cryst.(2007). E63, o1276–o1278 Samaset al. C
[image:2.610.45.296.71.268.2]11H15O4PH2O
o1277
Figure 2
Gillon, A. L., Feeder, N., Davey, R. J. & Storey, R. (2003).Cryst. Growth Des.
3, 663–673.
Golles, F. (1962).Monatsh. Chem.93, 191–220.
Loh, J. S. C., Van Enckevort, W. J. P., Vlieg, E., Gervais, C., Grimbergen, R. F. P. & Kaptein, B. (2006).Cryst. Growth Des.6, 861–865.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Sheldrick, G. M. (2003).SADABS. Version 2.10. University of Go¨ttingen, Germany.
Ten Hoeve, W. & Wynberg, H. (1985).J. Org. Chem.50, 4508–4514.
organic papers
o1278
Samaset al. Csupporting information
sup-1
Acta Cryst. (2007). E63, o1276–o1278
supporting information
Acta Cryst. (2007). E63, o1276–o1278 [https://doi.org/10.1107/S1600536807006496]
2-Hydroxy-5,5-dimethyl-4-phenyl-1,3,2-dioxaphosphorinan-2-one monohydrate
Brian Samas, Todd Groendyke, Anthony C Blackburn and Delara B Godrej
2-Hydroxy-5,5-dimethyl-4-phenyl-1,3,2-dioxaphosphorinan-2-one monohydrate
Crystal data
C11H15O4P·H2O Mr = 260.22
Orthorhombic, P212121
Hall symbol: P 2ac 2ab
a = 6.017 (2) Å
b = 8.219 (3) Å
c = 25.475 (10) Å
V = 1259.7 (8) Å3 Z = 4
F(000) = 552
Dx = 1.372 Mg m−3
Melting point: 86.5 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 983 reflections
θ = 2.6–27.3°
µ = 0.23 mm−1 T = 173 K
Columnar, colourless 0.61 × 0.09 × 0.04 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2003)
Tmin = 0.861, Tmax = 0.991
23045 measured reflections 3004 independent reflections 2899 reflections with I > 2σ(I)
Rint = 0.030
θmax = 28.2°, θmin = 1.6°
h = −7→7
k = −10→10
l = −32→33
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.034 wR(F2) = 0.085 S = 1.11 3004 reflections 167 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(Fo2) + (0.0515P)2 + 0.1668P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.45 e Å−3
Δρmin = −0.23 e Å−3
Absolute structure: Flack (1983), 1213 friedel pairs
supporting information
sup-2
Acta Cryst. (2007). E63, o1276–o1278
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-3
Acta Cryst. (2007). E63, o1276–o1278
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
P1 0.01937 (18) 0.02179 (18) 0.02406 (18) −0.00055 (14) −0.00106 (14) −0.00604 (14) O4 0.0261 (5) 0.0180 (5) 0.0208 (5) 0.0002 (4) −0.0004 (4) −0.0023 (4) O5 0.0345 (6) 0.0287 (6) 0.0185 (5) −0.0015 (5) −0.0003 (5) −0.0027 (4) O1 0.0240 (6) 0.0282 (6) 0.0333 (6) −0.0021 (5) 0.0020 (5) −0.0075 (5) O2 0.0264 (6) 0.0295 (6) 0.0350 (6) 0.0019 (5) −0.0021 (5) −0.0132 (5) O3 0.0201 (5) 0.0275 (6) 0.0527 (7) −0.0007 (5) −0.0012 (5) −0.0121 (6) C1 0.0363 (8) 0.0201 (7) 0.0297 (7) 0.0035 (7) −0.0010 (6) −0.0005 (6) C2 0.0186 (7) 0.0178 (6) 0.0210 (6) 0.0009 (5) 0.0016 (5) −0.0016 (5) C3 0.0331 (8) 0.0227 (7) 0.0218 (7) −0.0002 (7) −0.0029 (7) 0.0024 (5) C4 0.0234 (7) 0.0192 (7) 0.0194 (6) −0.0008 (6) 0.0007 (6) 0.0000 (5) C5 0.0330 (8) 0.0269 (7) 0.0250 (7) −0.0007 (7) 0.0066 (7) 0.0002 (6) C6 0.0251 (8) 0.0252 (7) 0.0293 (7) −0.0057 (6) 0.0021 (6) 0.0015 (6) C7 0.0270 (7) 0.0204 (7) 0.0187 (6) −0.0032 (6) 0.0014 (6) 0.0002 (5) C8 0.0337 (9) 0.0306 (8) 0.0249 (7) 0.0039 (7) −0.0017 (7) −0.0007 (6) C9 0.0538 (12) 0.0312 (9) 0.0205 (7) −0.0058 (8) 0.0083 (7) −0.0024 (6) C10 0.0401 (10) 0.0389 (9) 0.0311 (8) 0.0027 (9) −0.0108 (8) 0.0045 (7) C11 0.0565 (12) 0.0356 (9) 0.0212 (7) −0.0101 (9) −0.0062 (8) 0.0026 (7)
Geometric parameters (Å, º)
P1—O2 1.4841 (13) C3—H3A 0.9900 P1—O3 1.5284 (13) C3—H3B 0.9900 P1—O5 1.5678 (12) C4—C6 1.539 (2) P1—O4 1.5778 (12) C5—C7 1.394 (2) O4—C2 1.4731 (17) C5—C9 1.395 (2) O5—C3 1.4564 (18) C5—H5 0.9500 O1—H99A 0.81 (3) C6—H6A 0.9800 O1—H99B 0.83 (3) C6—H6B 0.9800 O3—H99C 0.988 (10) C6—H6C 0.9800 C1—C4 1.531 (2) C7—C8 1.393 (2) C1—H1A 0.9800 C8—C10 1.392 (2) C1—H1B 0.9800 C8—H8 0.9500 C1—H1C 0.9800 C9—C11 1.383 (3) C2—C7 1.5080 (19) C9—H9 0.9500 C2—C4 1.548 (2) C10—C11 1.387 (3) C2—H2 1.0000 C10—H10 0.9500 C3—C4 1.535 (2) C11—H11 0.9500
supporting information
sup-4
Acta Cryst. (2007). E63, o1276–o1278
C3—O5—P1 116.29 (9) C9—C5—H5 119.9 H99A—O1—H99B 106 (2) C4—C6—H6A 109.5 P1—O3—H99C 115.1 (14) C4—C6—H6B 109.5 C4—C1—H1A 109.5 H6A—C6—H6B 109.5 C4—C1—H1B 109.5 C4—C6—H6C 109.5 H1A—C1—H1B 109.5 H6A—C6—H6C 109.5 C4—C1—H1C 109.5 H6B—C6—H6C 109.5 H1A—C1—H1C 109.5 C8—C7—C5 119.06 (15) H1B—C1—H1C 109.5 C8—C7—C2 121.32 (13) O4—C2—C7 107.06 (11) C5—C7—C2 119.61 (14) O4—C2—C4 109.18 (11) C10—C8—C7 120.29 (16) C7—C2—C4 115.63 (12) C10—C8—H8 119.9 O4—C2—H2 108.3 C7—C8—H8 119.9 C7—C2—H2 108.3 C11—C9—C5 120.37 (16) C4—C2—H2 108.3 C11—C9—H9 119.8 O5—C3—C4 112.25 (12) C5—C9—H9 119.8 O5—C3—H3A 109.2 C11—C10—C8 120.46 (18) C4—C3—H3A 109.2 C11—C10—H10 119.8 O5—C3—H3B 109.2 C8—C10—H10 119.8 C4—C3—H3B 109.2 C9—C11—C10 119.55 (16) H3A—C3—H3B 107.9 C9—C11—H11 120.2 C1—C4—C3 106.29 (12) C10—C11—H11 120.2
O2—P1—O4—C2 169.94 (10) C7—C2—C4—C3 178.55 (12) O3—P1—O4—C2 −62.95 (11) O4—C2—C4—C6 −63.82 (14) O5—P1—O4—C2 50.95 (11) C7—C2—C4—C6 56.93 (16) O2—P1—O5—C3 −167.49 (10) C9—C5—C7—C8 1.3 (2) O3—P1—O5—C3 65.40 (12) C9—C5—C7—C2 −177.73 (15) O4—P1—O5—C3 −48.74 (12) O4—C2—C7—C8 32.12 (19) P1—O4—C2—C7 175.61 (10) C4—C2—C7—C8 −89.78 (18) P1—O4—C2—C4 −58.53 (14) O4—C2—C7—C5 −148.88 (13) P1—O5—C3—C4 56.37 (16) C4—C2—C7—C5 89.22 (17) O5—C3—C4—C1 −175.98 (13) C5—C7—C8—C10 −0.9 (3) O5—C3—C4—C6 64.48 (16) C2—C7—C8—C10 178.12 (15) O5—C3—C4—C2 −57.92 (16) C7—C5—C9—C11 −0.5 (3) O4—C2—C4—C1 173.59 (12) C7—C8—C10—C11 −0.4 (3) C7—C2—C4—C1 −65.65 (17) C5—C9—C11—C10 −0.8 (3) O4—C2—C4—C3 57.79 (15) C8—C10—C11—C9 1.2 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H99B···O2i 0.83 (3) 1.85 (3) 2.670 (2) 171 (2)
O1—H99A···O2ii 0.81 (3) 1.91 (3) 2.7179 (19) 176 (2)
C9—H9···O4iii 0.95 2.68 3.625 (2) 174
O3—H99C···O1iv 0.99 (1) 1.48 (1) 2.4395 (18) 164 (2)