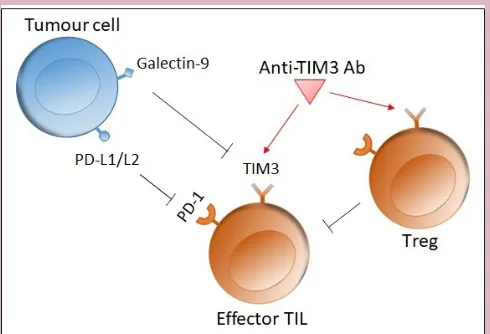
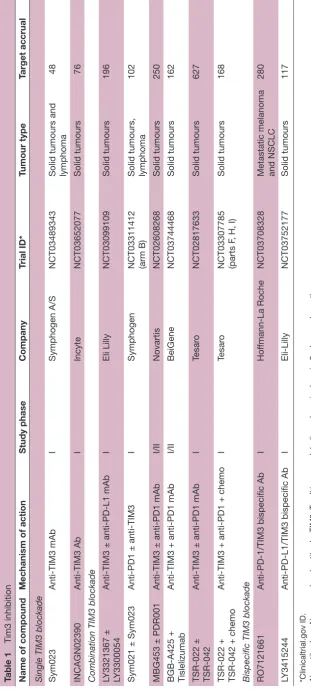
New emerging targets in cancer immunotherapy: the role of TIM3
Full text
Figure
Related documents
Once the data model has been defined, management screens can also be developed to allow operators to perform service provisioning and subscription management tasks.. 5
Scholz, Ann; Roseen, Robert M.; Ballestero, Thomas P.; Simpson, Michael; Lawson, Colin; Rubin, Fay; and Wake, Cameron P., "Climate and Land Use Consequences to
With the passage of the Presidential Libraries Act of 1955, the same system used to create and administer the Roosevelt Library became the official government procedure for the
A leaching column experiment was carried out to quantify the nitrate and heavy metal accumulation (Fe 2+ & Mn 2+ ) and leaching in sandy soil to assess the impact of
(III) surprisingly, renal function tests unlike liver parameters significantly were decreased in patients compared to healthy controls (IV) high levels of PAB
Table 1 shows the unstandardized means, standard deviations, and rank-order correlations among the six performance dimensions (amount of participation, impact, personal
experiences w/intersectionality influence their leadership practices? How do Black female school leaders describe their awareness of intersectionality as it relates to
In recent times, incessant upsurge in incidence of plagiarism among academic and students in Nigeria, among other factors, has immensely triggered partnership between the
