Abstract
Salm, Jeffrey Richard. Ligands from Combinatorial Peptide Libraries for Virus Removal.
(Under the direction of Dr. Ruben G. Carbonell and Dr. Dennis T. Brown)
Small peptides were investigated as affinity ligands for virus removal from human blood plasma (HBP). Sindbis virus (SV) was radiolabeled with 35S and screened against a solid phase peptide library. The screening identified 9 hexapeptides that were
synthesized on a TosoBioseparations Toyopearl 650M Amino Resin. The peptide sequences SGKPVA and IATDGG were found to remove approximately 3 logs and 2.4 logs of SV from buffer respectively and approximately 1.5 logs of SV from 50% (HBP). Toyopearl Amino bound only 0.6 logs of SV in both buffer and 50% HBP. Good agreement was seen between infectious quantification methods and quantification of the virus using radiation. Injections of several batches of SV showed variations between the batches.
A similar screening procedure was also applied to radiolabeled canine parvovirus (CPV). Screening against a solid phase library identified 25 leads through to be specific to CPV. Tests with a portion of these leads found less than 0.5 logs of CPV clearance in both buffer and 50% HBP. Electron microscopy of the CPV used in these experiments showed that large aggregates of virus were present in the radiolabeled virus. Infectious assays for CPV were also problematic and inconsistent.
Ligands from Combinatorial Peptide Libraries
for Virus Removal
by
Jeffrey R. Salm
A dissertation submitted to the Graduate Faculty of North Carolina State University
In partial fulfillment of the requirements for the Degree of Doctor of Philosophy
Department of Chemical Engineering May, 2004
Approved By:
__________________________ __________________________ Dr. Peter K. Kilpatrick Dr. Jan Genzer
__________________________ __________________________ Dr. Dennis T. Brown Dr. Ruben G. Carbonell
Dedication
Biography
Acknowledgements
I would like to thank the American Red Cross for the financial support for this work. Specifically David Hammond for always supporting the Bioseparations Group at NCSU.
I would like to thank the professors and researchers at North Carolina State University who helped me obtain my degree. Specifically, thank you to Dr. Raquel Hernandez for her expertise, patience, and belief in the abilities of a chemical engineer, Dr. Brown for his open door, his knowledge, and belief in this project, and Dr. Carbonell for his direction, his belief in me, and his ability to brighten the darkest periods of this research.
I would like to thank the members of the Bioseparations lab group, Honglue, Viterose and Guan, and the Biochemistry lab group Steevenson, Chris II, John, and Katie. Special thanks to Patrick and Patrick for their help and friendship, and to Chris for always having the time and the answers to help me.
Table of Contents
List of Tables... viii
List of Figures... ix
1. Introduction and Overview 1.1. Introduction...1
1.1.1. Motivation...1
1.1.2. Overview...2
1.1.3. Review of Viral Clearance...3
1.1.3.1. Virus Inactivation Techniques ...3
1.1.3.2. Virus Removal Techniques...4
1.1.3.3. Affinity Adsorption for Virus Removal...5
2. Identification and Characterization of Peptide Ligands from Combinatorial Peptide Libraries that Bind Sindbis Virus 2.1. Introduction...12
2.1.1. Background...12
2.1.2. SV Background...16
2.2. Experimental ...17
2.2.1. Materials ...17
2.2.2. Virus Growth ...18
2.2.2.1. Cell Passing Procedure ...18
2.2.2.2. Radio-labeling Procedure...18
2.2.2.3. Sindbis Virus Purification...19
2.2.3. Sindbis Virus Characterization ...20
2.2.3.1. Plaque Assay...20
2.2.3.2. Protein and Radiation Characterization ...21
2.2.4. Library and Peptide Synthesis ...22
2.2.5. Library Screening (Primary Screening) ...23
2.2.6. Binding Confirmation (Secondary Screening)...24
2.2.7. Column Screening (Tertiary Screening) ...25
2.3. Results and Discussion ...26
2.3.1. Preparation and Characterization of SV ...26
2.3.2. Lead Identification (Primary Screening) ...33
2.3.3. SV Binding to Resins (Secondary Screening) ...35
2.3.4. Clearance of SV from Plasma (Tertiary Screening) ...39
2.3.5. Resin Regeneration ...49
2.4. Conclusions...49
2.A.1 Secondary Screening...52
References...54
3.1.1. Background...60
3.1.2. CPV Background ...60
3.2. Experimental ...61
3.2.1. Materials ...61
3.2.2. Virus Growth ...63
3.2.2.1. Radio-Labeling Procedure ...63
3.2.2.2. Canine Parvovirus Purification...63
3.2.3. Canine Parvovirus Characterization ...65
3.2.3.1. Plaque assay...65
3.2.3.2. Protein and Radiation Characterization ...66
3.2.4. Library Screening...66
3.3. Results and Discussion ...67
3.3.1. Preparation and Characterization of CPV...67
3.3.2. CPV Primary Screening...73
3.3.3. CPV Binding to Resins (Secondary Screening)...77
3.3.4. Column Screening of CPV (Tertiary Screening) ...80
3.4. Conclusions...87
References...89
4. Characterization of Affinity Peptide Ligands that Bind Parvoviruses 4.1. Introduction...93
4.1.1. Background...93
4.1.2. ARC Primary Screening ...94
4.1.3. ARC Binding Verification ...96
4.1.4. Discussion of ARC Work ...96
4.2. Experimental Methods ...97
4.2.1. Materials ...97
4.2.2. Stock Parvoviruses...98
4.2.3. Parvovirus Quantification ...99
4.2.4. Column Preparation ...100
4.2.5. Binding Conformation ...101
4.2.6. Comparison of Column Format ...101
4.2.6.1. PolyPrep® and PIKSI Column Experiments...101
4.2.6.2. Omega Column Experiments...102
4.2.7. Large Volume 10 ml Experiments...102
4.3. Results and Discussion ...103
4.3.1. Preparation of Viruses...103
5. Conclusions and Recommendations
5.1. Summary of Conclusions...119
5.1.1. Sindbis Virus...119
5.1.2. Canine Parvovirus...119
5.1.3. Parvoviruses...120
5.2. Recommendations for the Future...121
5.2.1. Improve Primary Screening Technique ...121
5.2.2. Improve Library Design...122
5.2.3. Libraries with Longer Peptides...122
5.2.4. Improved Virus Quantification Methods ...123
5.2.5. Resin Regeneration Procedure...123
5.2.6. Equilibrium Isotherm...124
Appendix A Small Peptide Ligands for Affinity Separations of Biological Molecules Downstream Processing in Biopharmaceutical Production...126
Affinity Chromatography...127
Advantages of Small Peptide Ligands ...128
Combinatorial Peptide Libraries ...132
Phage Displayed Libraries ...132
Combinatorial Libraries: Libraries on Chromatographic Resins...134
Screening of One-bead-one-peptide Libraries ...136
Protein Purification by Small Peptide Ligands...143
Characterization of Peptide Ligands...143
Single and multi-point attachment and the effect of peptide density...143
Ligand-target interactions ...145
Role of peptide amino acid sequence...148
Role of peptide density ...150
Rates of adsorption ...151
Future challenges and opportunities ...152
References...156
Appendix B Rapid Preparative Purification of West Nile and Sindbis Virus PCR Products Utilizing a Microbore Anion-Exchange Column Abstract...163
Introduction...164
Materials and Method ...167
PCR amplifications, PCR reaction parameters and instrumentation ...167
DNA purification ...169
HPLC Instrumentation and chromatogram analysis ...170
Results...171
Template DNA and PCR product analysis ...171
Chromatography calibration and analysis of PCR products ...172
Sindbis virus mixed product PCR, HPLC purification and product analysis ...174
Discussion...176
List of Tables
Table 2.1 SV Plaque Counts and Titers for Figure 1...28
Table 2.2 Criteria for Batches of SV...29
Table 2.3 Sequences Identified from Screening 35S-SV Against a Solid Phase Peptide Library ...34
Table 2.4 Sequences Identified from Screening 35S-SV Against a Solid Phase Peptide Library with a 1 M NaCl Wash...35
Table 2.5 Tertiary Screening of Resins Using 35S-labeled Sindbis Virus ...42
Table 2.6 Log Reduction from Four Batches of SV ...45
Table 2.A.1 Secondary Screening of Resins Using 35S-labeled Sindbis Virus ...53
Table 3.1 Criteria for Well-Growth Batches of CPV...72
Table 3.2 Sequences Identified from Screening 35S-CPV Against a Solid Phase Peptide Library ...73
Table 3.3 Sequences Identified from Screening 35S-CPV Against a Solid Phase Peptide Library with a 1 M NaCl Wash...74
Table 3.4 Secondary Screening of Resins Using 35S-labeled Canine Parvovirus...77
Table 3.5 Secondary Screening of Resins with Varied Peptide Densities and Ligand Length Using 35S-labeled Canine Parvovirus...79
Table 3.6 Secondary Screening of TosoBioSep Resins Using 35S-labeled Canine Parvovirus ...80
List of Figures
Figure 2.1 SV Plaques on BHK Cell Monolayers Observed, A. 10-8 Dilution, B. 10-7
Dilution, C. 10-6 Dilution...28
Figure 2.2 Effect of Radioactive Label on Stability of SV ...31
Figure 2.3 Effect of Radioactive Label on Stability of SV ...32
Figure 2.4 Effect of Specific Activity of Radio-labeled SV ...33
Figure 2.5 Loads of SV on SGKPVA at Varied Flow Rates ...37
Figure 2.6 Effect of Flow Rate on Log Removal ...38
Figure 2.7 Injections of Varied Quantities of SV on SGKPVA...39
Figure 2.8 Injections of SV in PBS on Several Resins ...40
Figure 2.9 Injections of SV in 50% HBP on Several Resins ...41
Figure 2.10 Comparison of Plaque Assay Quantification and Radiation Quantification for SV in PBS on SGKPVA ...43
Figure 2.11 Comparison of Plaque Assay Quantification and Radiation Quantification for SV in 50% HBP on SGKPVA ...44
Figure 2.12 Comparison of Chromatograms from Batches of SV on SGKPVA in PBS...46
Figure 2.13 Comparison of Chromatograms from Batches of SV on SGKPVA in 50% HBP.47 Figure 2.14 a, b, HBP Protein Adsorption to SGKPVA. A. Chromatograms of SV in PBS and HBP B. SDS-PAGE of Fractions from HBP Chromatogram...48
Figure 3.1 Negative Stain Electron Microscopy of CsCl Purified CPV ...68
Figure 3.2 Negative Stain Electron Microscopy of Purified CPV ...70
Figure 3.3 Scintillation Counts of CPV Dilutions...71
Figure 3.4 Comparison of CPV Monoclonal Antibody and Sequences from Primary Screening ...75
Figure 3.6 Injections of Two Concentrations of CPV on RYNDWA...87
Figure 3.7 Injections of CPV on NIIVQR at Varied Flow Rates...83
Figure 3.8 Re-injection of Unbound Fraction on RYNDWA ...84
Figure 3.9 Re-injection of Unbound Fraction on Toyopearl Amino...85
Figure 3.10 Effect of Prefiltering CPV on GFYGAH Adsorption...86
Figure 3.11 Effect of Prefiltering CPV on RYNDWA Adsorption ...87
Figure 4.1 Log Clearance from 10 ml Injection of PPV in Varied HBP Concentrations on Amino Resin ...109
Figure 4.2 Log Clearance from 10 ml Injection of PPV in Varied HBP Concentrations on ARC30 Resin ...110
Figure 4.3 Log Clearance from 10 ml Injection of PPV in Varied HBP Concentrations on ARC49 ...111
Figure 4.4 Log Clearance from 10 ml Injection of PPV in Varied HBP Concentrations on ARC40 Resin ...112
Figure 4.5 Log Clearance from 10 ml Injection of PPV in Varied HBP Concentrations on FLLHPI ...113
Appendix B Figure 1 Electrophoretic Analysis of Ethidium Bromide Stained Agarose Gels of Template DNA...186
Appendix B Figure 2 Chromatograms of the Test PCR Mixtures and the Complete PCR Reactions...187
Appendix B Figure 3 Peak Fraction Analysis of the [α-32] P Labeled PCR Reaction ...188
Chapter 1
Introduction and Overview
1.1. Introduction
1.1.1 Motivation
Biological products derived from either human or animal origins contain an
inherent risk of viral contamination. As a result, the United States Food and Drug
Administration has mandated that all biological products be treated with two virus
removal or inactivation steps. In response to federal regulations, manufacturers have
incorporating steps to determine the level of viral contamination and processes to remove
or inactivate viruses. These are referred to as viral validation and viral clearance
respectively.
Generally, viral clearance processes are one of the last steps in a production
process. This is a result of a lack of specificity demonstrated by most clearance
strategies. For example, membranes are commonly used for viral clearance as most
biologicals are substantially smaller than potential viral contaminants. However,
membranes are prone to fouling when challenged with complex feed streams such as cell
culture supernatant or human blood plasma. The cost of replacing or cleaning the
membranes generally relegates them to a late stage viral clearance process.
A viral clearance process that is specific to a virus would be of great value to the
biotechnology industry. For product streams such as human blood plasma that are
viral clearance strategy that is used once in the beginning of a purification process instead
of multiple times for each product at the end of the process. A clearance strategy that is
virus specific could also target viruses such as human B-19 parvovirus that are not
adequately addressed by currently available clearance processes.
Affinity adsorption is a separation method that could meet the required specificity
needed for virus removal. However, this method has been largely ignored in viral
clearance. Current affinity ligands such as monoclonal antibodies are not suitable for
large scale processes because of the cost of producing the antibodies, the inability of such
ligands to withstand harsh solvent treatments, and the potential for ligand leakage. For an
affinity chromatography method to be industrially applicable, it must demonstrate
specificity to the target virus and overall cost efficiency. Small peptide affinity ligands
are inexpensive and robust which could make them ideal for a viral clearance method.
1.1.2 Overview
Several viral clearance strategies are currently used by the biotechnology industry
that are non-specific to virus particles. This work describes the development of a viral
clearance method that uses affinity peptides for virus removal from human blood plasma.
Combinatorial peptide libraries were screened to identify peptides specific to different
model viruses. After screening, peptides were tested for their ability to bind target
and in the literature. Chapter 2 describes the identification and characterization of
affinity ligands specific sindbis virus. Chapter 3 details efforts to discover affinity
ligands for canine parvovirus. Chapter 4 focuses on affinity ligands specific to three
viruses from the parvovirus family. Overall conclusions and recommendations for future
work are summarized in Chapter 5. Appendix A has a review of the use of affinity
peptide ligands for protein purification. Appendix B is a manuscript detailing a
purification process for PCR products.
Introduction
1.1.3 Review of Viral Clearance
1.1.3.1 Virus Inactivation Techniques
Heat inactivation [4-7] and radiation inactivation [8, 9] are common inactivation
strategies that involve exposing the virus to harsh environmental conditions. These
methods balance the inactivation of the virus with maintaining product viability [3, 8, 10,
11]. For viruses like human immunodeficiency virus (HIV) and hepatitis B which are
denatured with treatment at 60ºC, heat inactivation strategies work extremely well
because the virus particles are significantly less stable than product proteins [4, 12].
Other viruses like human B-19 parvovirus, which is stable to 80ºC, require alternative
clearance strategies.
Photoinactivation [11, 13, 14] and solvent detergent inactivation [3, 15-17] rely
on the addition of denaturing chemical agents. Chemicals such as tri(n-butyl) phosphate
mixed with Triton X-100 or Tween 80 can easily destroy the lipid membrane of a virus
making it an excellent chemical inactivation strategy for enveloped viruses [18]. For
concentration, virus specific chemicals such as Inactine™ have been shown to inactivate
viruses such as B-19 parvovirus and West Nile virus [19]. Naturally occurring molecules
such as the fatty acid caprylate have also been shown to result in significant viral
clearance [20]. Like heat inactivation, chemical inactivation methods can result in the
loss of the target molecule. Chemical inactivation methods have the added drawback of
requiring additional processing and validation steps to remove or neutralize potentially
toxic chemical additives [11, 15, 21, 22].
1.1.3.2 Virus Removal Techniques
Viral removal methods including filtration [22, 23], adsorption [24, 25],
precipitation [18], and chromatography [1, 4, 5, 22, 25-27] are also common industrial
viral clearance methods. Filtration methods use membranes to retain virus particles while
allowing protein products to pass through the membrane pores [23, 28]. Ultipor®
polyvinylidene fluoride membranes from Pall (East Hills, NY) have been shown to retain
a variety of virus particles including polio, Semliki Forest Virus, Herpes Simplex virus,
and HIV [28-30]. Viresolve NFP filters from Millipore (Billerica, MA) also have been
shown to clear viruses including porcine parvovirus, hepatitis A virus, murine
encephalomyocarditis virus, bovine viral diarrhoea virus, and bovine herpes virus [31].
Membrane removal systems are somewhat limited in the types of solutions they
Adsorption to filters is also used to remove viral contaminants [24, 25]. These
techniques rely on the physiological properties such as charge or hydrophobicity of the
product and the potential contaminants. As a result, shifts in the feed stream composition
can result in dramatically different adsorptive properties and ineffective viral clearance
[24]. Precipitation can be used for viral clearance, but separating a solution into a liquid
and solid fraction usually generates a fraction with less virus and a fraction with more
virus [18, 32]. For a solution such a human blood plasma that will be separated into
many products, this result is of limited value.
Chromatography is another common method used for viral clearance [1, 4, 5, 22,
25-27]. Darling et al. reported a range of 1-6 log[10] removal of polio virus and murine
leukemia virus using a Protein A Sepharose resin, Q-Sepharose resin, and S-Sepharose
resin [1]. Adcock et al. used a combination of ion exchange chromatography, gel
filtration chromatography, and heat inactivation to remove both hepatitis A and B viruses
from a modified plasma stream [4, 5]. Biescas et al. reported a 3.8 log[10] removal of
HIV-1 and a 2.9 log[10] removal of Bovine Diarrhea virus from Cohn’s plasma fraction
IV1 using a heparin-agarose gel chromatography column [33]. A significant problem
with all of the chromatographic methods used to date is that they work by selectively
binding the product and allowing the virus to flow through. This method can actually
result in the concentration of the contaminating virus. For a solution such as human
blood plasma, virtually all of the molecules in solution have value. By separating a
specific protein from a solution while retaining the viral contaminant, the viral clearance
problem has not been answered, but saved for another step.
The use of an affinity resin that targets the virus is a new viral clearance strategy.
A process that specifically targets the virus particle is advantageous for viral clearance as
it could treat a complex solution containing several products early in the production
process. Such a process would then serve as a viral clearance step for all of the products.
An addition advantage is that an affinity method should not result in product inactivation
or the need for the addition of chemicals that would require additional processing.
Affinity ligands such as monoclonal antibodies exist for many viruses, but are of
limited value from a viral clearance. Antibody columns are expensive to produce, and
are prone to ligand leakage and degradation. Other affinity ligands such as dyes or
metals generally lack the specificity that would be needed to bind a virus particle and
introduce potential toxicity issues if ligand leakage occurs.
Small peptide affinity ligands are reviewed in Appendix A of this thesis. Small
peptide affinity ligands offer many potential advantages for viral clearance. These
ligands are extremely robust allowing harsh solutions to be used for regeneration and
elution. Combinatorial screening techniques allow small peptide ligands to be identified
with specific viral interactions efficiently and with relatively low expense.
Several small peptides have been identified that are capable of inactivating
specific viruses in solution [34, 35]. Lutzke et al. reported that a hexameric peptide
References
1. Darling, A.J. and J.J. Spaltro, Process validation for virus removal
-Considerations for design of process studies and viral assays. Biopharm-the Technology & Business of Biopharmaceuticals, 1996. 9(9): p. 42-&.
2. (CPMP), C.f.P.M.P., Note for Guidance on Virus Validation Studies: The Design,
Contribution and Interpretation of Studies Validating the Inactivation and Removal of Viruses. 1996, European Agency for the Evaluation of Medical Products.
3. Roberts, P., Resistance of vaccinia virus to inactivation by solvent/detergent
treatment of blood products. Biologicals, 2000. 28(1): p. 29-32.
4. Adcock, W.L., et al., Chromatographic removal and heat inactivation of hepatitis
B virus during the manufacture of human albumin. Biotechnology and Applied Biochemistry, 1998. 28: p. 169-178.
5. Adcock, W.L., et al., Chromatographic removal and heat inactivation of hepatitis
A virus during manufacture of human albumin. Biotechnology and Applied Biochemistry, 1998. 28: p. 85-94.
6. Horwith, G. and D.R. Revie, Efficacy of viral clearance methods used in the
manufacture of activated prothrombin complex concentrates: focus on AUTOPLEX (R) T. Haemophilia, 1999. 5: p. 19-23.
7. Nissen, E., et al., Inactivation of hepatitis A and other enteroviruses during heat
8. Reid, B.D., The Sterways Process: a new approach to inactivating viruses using
gamma radiation. Biologicals, 1998. 26(2): p. 125-130.
9. Degiorgi, C.F., E.E. Smolko, and J.H. Lombardo, Cryo-gamma radiation
inactivation of bovine herpesvirus type-1. Radiation Physics and Chemistry, 1999. 55(4): p. 469-471.
10. Smales, C.M., D.S. Pepper, and D.C. James, Protein modification during antiviral
heat bioprocessing. Biotechnology and Bioengineering, 2000. 67(2): p. 177-188. 11. Rywkin, S., et al., New Phthalocyanines For Photodynamic Virus Inactivation in
Red- Blood-Cell Concentrates. Photochemistry and Photobiology, 1994. 60(2): p. 165-170.
12. Valdes, R., et al., Chromatographic removal combined with heat, acid and
chaotropic inactivation of four model viruses. Journal of Biotechnology, 2002. 96(3): p. 251-258.
13. Wagner, S.J., et al., Factors affecting virus photoinactivation by a series of
phenothiazine dyes. Photochemistry and Photobiology, 1998. 67(3): p. 343-349. 14. Skripchenko, A., D. Robinette, and S.J. Wagner, Comparison of methylene blue
451-16. Blesert, L. and H. Suhartono, Solvent/detergent treatment of human plasma - A
very robust method for virus inactivation. Validated virus safety of OCTAPLAS (R). Vox Sanguinis, 1998. 74: p. 207-212.
17. Lazo, A. Pathogen Inactivation of Red Cells with Inactine. in Biological Safety
and Production. 2000. The Ronald Reagan Building and International Trade Center, Washington, D.C.: Internation Business Communications, IBC.
18. Organization, W.H. Guidelines on Viral Inactivation and Removal Procedures
Intended to Assure the Safety of Human Blood Plasma Products. in Expert Committee on Biological Standardization. 2001.
19. Lazo, A., et al. NACTINE™ Technology Inactivates Clinical Isolates of
Parvovirus B19 in RBC Concentrates As Demonstrated by PCR and Infectivity Assays. in American Society of Hematology (ASH) Meeting. 2002.
20. BayerHealthcare, Bayer Announces Innovation for Viral Inactivation of
Plasma-based Product. 2003.
21. Burnouf, T. and M. Radosevich, Reducing the risk of infection from plasma
products: specific preventative strategies. Blood Reviews, 2000. 14(2): p. 94-110. 22. Stern, M.D., Viral safety of biotech products - Developments and challenges of
biopharmaceutical viral clearance. Genetic Engineering News, 1998. 18(10): p. 26-+.
23. Aranha-Creado, H., J. Peterson, and P.Y. Huang, Clearance of murine leukaemia
membrane filter. Biologicals, 1998. 26(2): p. 167-172.
24. Lukasik, J., et al., Influence of salts on virus adsorption to microporous filters.
Applied and Environmental Microbiology, 2000. 66(7): p. 2914-2920.
25. Powell, T., et al., Investigating the effect of carbon shape on virus adsorption.
Environmental Science & Technology, 2000. 34(13): p. 2779-2783.
26. Anderson, R., et al., A method for the preparation of highly purified
adeno-associated virus using affinity column chromatography, protease digestion and solvent extraction. Journal of Virological Methods, 2000. 85(1-2): p. 23-34. 27. Richieri, S.P., et al., Characterization of highly purified, inactivated HIV-1
particles isolated by anion exchange chromatography. Vaccine, 1998. 16(2-3): p. 119-129.
28. Aranha-Creado, H., Filtration virus-removal in process validation - Clearance of
viruses with ultipor VF filters. Genetic Engineering News, 2000. 20(6): p. 64-64. 29. AranhaCreado, H., et al., Virus retention by a hydrophilic triple-layer PVDF
microporous membrane filter. Pda Journal of Pharmaceutical Science and Technology, 1997. 51(3): p. 119-124.
14(1): p. 140-147.
32. Azari, M., et al., Evaluation and validation of virus removal by ultrafiltration
during the production of diaspirin crosslinked haemoglobin (DCLHb). Biologicals, 2000. 28(2): p. 81-94.
33. Biescas, H., et al., Characterization and viral safety validation study of a
pasteurized therapeutic concentrate of antithrombin III obtained through affinity chromatography. Haematologica, 1998. 83(4): p. 305-311.
34. Heinz, B.A., et al., Genetic and Molecular Analyses of Spontaneous Mutants of
Human Rhinovirus-14 That Are Resistant to an Antiviral Compound. Journal of Virology, 1989. 63(6): p. 2476-2485.
35. Mosser, A.G. and R.R. Rueckert, Win 51711-Dependent Mutants of Poliovirus
Type-3 - Evidence That Virions Decay after Release from Cells Unless Drug Is Present. Journal of Virology, 1993. 67(3): p. 1246-1254.
36. Lutzke, R.A.P., et al., Identification of a Hexapeptide Inhibitor of the
Chapter 2
Identification and Characterization of Peptide Ligands
from Combinatorial Peptide Libraries that Bind
Sindbis Virus
2.1. Introduction
2.1.1 Background
Protein therapeutics are an increasingly important sector of the health care and
biotechnology industry. Recombinant proteins have been developed from information
garnered from the human genome project and used to treat conditions ranging from
inflammation to cancer. Protein therapeutics derived directly from natural sources are
commonly used to treat a variety of conditions ranging from genetic disorders to blood
deficiencies.
With all protein therapeutics, there exists an inherent risk of viral contamination
[1-8]. Potential sources of contamination include the mammalian cells lines or animals
used to produce the proteins, materials derived from living sources such as blood plasma
or fetal calf serum, or contamination from workers involved in the production process.
strategies include the use of heat and radiation to denature the virus particles [1, 2, 10,
11]. This strategy works well for several viruses such as Hepatitis A and B Viruses that
are not heat stable [1, 2]. More stable viruses, such as B19 parvovirus, denature only in
conditions that also result in significant protein degradation. Another common
inactivation strategy involves the addition of chemicals to the production process that
denature the virus [12]. Enveloped viruses such as HIV are commonly treated with
detergents such as tri(n-butyl) phosphate that destroy the lipid bilayer of the virus [13,
14]. Other viruses are treated with chemical agents, such as Inactine (Vitex Corp.) to
denature the viral proteins. One drawback of adding chemicals to a process is that they
must be removed due to their potential toxicity to patents [15, 16]. This results in
additional processing steps that can substantially add to the overall processing cost.
These chemicals can also modify or denature desirable target proteins or other
biomolecules in the mixture.
The other primary method of viral clearance used in the biotechnology industry is
virus removal. Removal strategies have the advantage that they generally do not result in
product inactivation. Ultra-porous and nano-porous membranes often composed of
polyvinylidene fluoride are used in many viral clearance processes as they are extremely
effective at removing viruses such as HIV that are significantly larger than other
molecules in the production stream [17-19]. For smaller virus particles such as B-19
parvovirus that are not easily captured by a membrane removal, adsorption methods such
as ion-exchange chromatography can be effective [20]. These methods are of limited
value as they lack viral specificity in complex feed streams such as cell culture
Affinity adsorption has also been suggested as a method for viral clearance [21].
Antibodies have the ability to specifically target viruses in complex feed streams. In
principle, affinity adsorption offers a significant advantage over other removal strategies
because a single adsorption step could be used to remove viruses in the beginning of a
purification process that makes several products. However, purification and use of an
antibody specific to a virus particle is cost prohibitive. Antibody adsorption columns are
prone to ligand degradation and leakage resulting from washing conditions. Dye ligands
are more stable than antibodies allowing them to be treated with harsh elution conditions.
However, dye ligands lack the specificity needed for a viral clearance process and they
can be toxic if they leak from the column. For virus removal by adsorption, the ligand
must be relatively inexpensive and safe since affinity supports could be used for a very
limited number of batches.
An alternative approach to antibodies and dyes involves using small peptide (3-8
amino acids) affinity ligands. Small peptides can be synthetically produced making them
significantly cheaper than antibodies. Small peptides also are not likely to exhibit the
toxicity issues of antibodies or dyes should ligand leakage occur. Peptide identification is
the greatest challenge to using small molecule affinity ligands. Rational design
approaches cannot realistically be applied to viruses because of the complexities of the
proteinase inhibitor [22], von Willibrand Factor [23], fibrinogen [24], and α lactalbumin
[25]. Appendix A contains an extended review of the literature on the use of small
peptides as ligands for affinity chromatography. Random library screening should
identify peptides that bind various sites on the virus surface.
The two major types of peptide libraries used in screening are synthetic libraries
and phage display libraries [26-29]. Phage libraries use bacteriophages with peptides
expressed on their surface. Peptides are produced by genetic manipulation of the phages
genome. The major advantages to phage libraries are that the phages can be propagated
through several rounds of screening to identify the strongest binders and that they can be
used to make large peptides. The disadvantages to phage libraries are that only natural
amino acids can be used, and that ligands identified on phages may not have the same
affinity properties when immobilized to a matrix suitable for a viral clearance step.
In synthetic libraries, peptides can be chemically synthesized onto a support
matrix such as a membrane or resin. One advantage of synthetic libraries is that they can
be synthesized on the matrix that will later be used in an industrial application. Synthetic
libraries also have the advantage of not being limited to naturally occurring amino acids.
One drawback to synthetic libraries is that the length of the peptide is limited to
approximately 8 amino acids by the physical size of the library and the cost associated
with sequencing the peptides.
Peptides have been identified that are specific to viruses [30, 31]. Lutzke et al.
reported that HCKFWW identified by screening a soluble combinatorial library bound
specifically to human immunodeficiency virus [32]. However, these are generally
small peptides attached to a solid phase matrix for viral adsorption is a new idea. A viral
clearance process that uses hexameric ligands could have the needed specificity to
remove viruses in one step while balancing issues such as cost and toxicity.
In this work, a solid phase hexa-peptide library was synthesized on
TosohBiosciences Toyopearl AF Amino-650M resin. The library was screened with 35S
labeled Sindbis virus (SV) to identify affinity ligands capable of removing SV from
whole human blood plasma (HBP). While SV is not a known blood pathogen, it is a
good model for highly structured, enveloped RNA viruses like Hepatitis C since it is
relatively easy to work with and is fairly well characterized. Two ligands were identified
that had moderate affinity to SV in the presence of HBP.
2.1.2 SV Background
SV is a 60-70 nm diameter human pathogen from the Togaviridae family of
viruses in the alphavirus genera. A structure for SV has been determined to 11 Å
resolution [33] using known atomic level resolution structures of the virus proteins and
cryo-electron microscopy. The virus consists of a single RNA molecule, a protein
capsid, a lipid bilayer, and two surface glycoproteins designated E1 and E2.
The SV genome consists of a single positive sense RNA strand approximately 12
kb in length. The 5’ end of the genome codes for four non-structural proteins that are
virion. The spikes are formed by three copies of a dimer consisting of the 56 kDa E1 and
the 51 kDa E2 proteins.
2.2 Experimental
2.2.1 Materials
The American Red Cross and Vitex Corporation donated the HBP used in this
research. The HBP was received in frozen 15 to 50 ml aliquots. HBP aliquots from each
delivery were thawed in a 37°C water bath for approximately 5 min. The HBP was then
pooled and mixed with a bench top vortex mixer on a low setting. The plasma was then
aliquoted in 0.5 ml fractions and flash frozen in liquid nitrogen. The vials were then
stored at –20°C until needed. Before using, aliquoted HBP was thawed at 37°C,
vortexed, and then passed through a 0.22µm filter.
All cell culture work was done using proper flaming and sterilization techniques
in a laminar flow hood. Cells were grown in a NapCo E Series Model 5300 Incubator
saturated with water and kept at 5% CO2 and 37°C unless otherwise noted. Cells were
grown in Minimum Eagle Medium (MEM) with Earl’s salts purchased from Invitrogen
(Carlsbad, CA). ‘Supplemented MEM’ was made by adding 50 µg/ml gentamicin sulfate
and 2 mM glutamine form Invitrogen (Carlsbad, CA), 10% v/
v heat inactivated fetal calf
serum (FCS) from Hyclone (Logan, UT), and 29.5 g/L tryptose phosphate broth from
Becton Dickinson (Sparks, MD).
35S-cysteine and 35S-methionine were purchased from NEN Life Science Products
(Boston MA). Phosphate Buffer Saline (PBS) and Phosphate Buffer Saline with 0.1 v/v
Cells were observed using a Zeiss Phase Inversion Light Microscope from Carl
Zeiss Instruments (Dallas, TX). Cell confluence was estimated after observation of the
cell monolayer under the microscope. Cells were counted using a hemocytometer and the
light microscope.
Heat resistant strain SV was acquired from Elmer Pfefferkorn at Dartmouth
College (Hanover, NH). Stocks of this virus were produced at a titer of 109 plaque
forming units per ml (pfu/ml) and stored at -80ºC by Dr. Dennis Brown’s Lab. When SV
was needed, stock virus was thawed on ice and diluted as appropriate.
2.2.2 Virus Growth
2.2.2.1 Cell Passing Procedure
‘Cell passing’ involves releasing a confluent monolayer of cells from a culture
flask and reseeding the cells into new flasks. To ‘pass’ cells, a confluent monolayer was
initially washed with PBS to remove the culturing media. A solution containing 0.25%
trypsin was added so that the cells were covered and the monolayer was observed for
detachment of the cells from the flask. After detachment, an equal volume of
supplemented media was added to the flask to inactivate the trypsin. The media
containing the cells was transferred to a 15 ml centrifuge tube and spun at 1000 rpm in a
HN-S II International Equipment Benchtop Centrifuge (Vernon Hill, IL) to pellet the
SV was grown using a procedure adapted from Hernandez et al. [34]. Briefly,
baby hamster kidney cells strain 21 (BHK) were provided by Peter Faulkner (Queens
University, Kingston, Ontario). The cells were passed in supplemented MEM at least
twice prior to infection. The cells were seeded into 75 cm2 culturing flasks from Corning
(Corning, NY) at a density of approximately 7 x 106 BHK cell/flask. The cells were
grown overnight in the incubator to approximately 75% confluence. The medium was
decanted from the flasks, and 5 ml of fresh supplemented MEM containing 4 µg/ml
Actinomycin D (Calbiochem, San Diego CA) were added to the flasks of cells. The
flasks were covered and put in the incubator. After 1 hour, the medium was poured from
the flasks and the cells were infected by adding 1 ml of infection solution. The infection
solution contained 3% v/v FCS and approximately 1 x 108 infectious SV particles. The
flasks were placed on a Bellco Glass Inc. plate rocker at room temperature. After 1 hour
of gentle rocking, 4 ml of supplemented MEM were added to each flask. The flasks were
then placed in the incubator for 3 hours and 45 minutes. After incubation, the medium
was poured from the cells and a Pasteur pipette was used to insure complete removal of
the supplemented MEM. Five ml of supplemented MEM without Cysteine or
Methionine (starvation medium) were added to the cells. The cells were then placed in
the incubator for 1 hour. The starvation medium was poured from the flasks, and 5 ml of
fresh starvation medium with 35S-cysteine and methionine were added to each flask. The
flasks were incubated overnight at 37°C. After a cytopathic effect caused by the virus
was observed under the microscope, the cell supernatant was pooled from all the infected
flasks and stored at 4°C.
The SV cell supernatant was twice purified over a k-tartaric acid density gradient.
Before the gradients were run, the supernatant was spun at 3000-5000 rpm in a HN-S II
International Equipment Benchtop Centrifuge (Vernon Hill, IL) to pellet cell debris. The
supernatant was then poured into a fresh tube to isolate the virus from the cell debris. A
two step gradient (15% w/v and 35% w/v tartaric acid in PBS) was formed in Ultraclear
SW28 ultracentrifuge tubes from Beckmann with at least 5 ml of both the 15% and 35%
solutions. The cell supernatant was then layered on top of the gradient. The gradient was
spun for 16 hours at 4°C and 24,000 RPM in an Optima L-90K Beckmann Ultra
Centrifuge. After spinning, the virus band was visualized at the interface of the 15% and
35% layers. The bottom of the ultracentrifuge tube was pierced with a 20 gauge needle
and dripped. The band of virus was collected and stored at 4°C. A second linear gradient
(15% w/v to 35% w/v tartaric acid in PBS) was prepared in Ultraclear SW40
ultracentrifuge tubes from Beckmann with a total volume of at least 9 ml. The tartaric
acid in the collected band was diluted by adding an equal volume of PBS. The band was
then layered on top of the linear gradient. The linear gradient was spun for 3 hours at
4°C and 26,000 RPM in a Beckmann Ultra Centrifuge. After spinning, the virus band
was visualized and drip collected using the same technique applied to the step gradients.
The collected bands were combined and dialyzed to remove the tartaric acid against PBS
The infectivity of SV was assayed using an end point plaque assay adapted from
Knipfer et al. [35]. Briefly, ten fold serial dilutions of virus samples were prepared with
supplemented MEM containing 10 mM HEPES-HCl (Sigma) pH=7.4 in sterile glass test
tubes on ice. A 25 cm2 culturing flask from Corning (Corning, NY) was prepared for
each dilution by seeding with ~2 x 106 BHK cells. The flasks were grown overnight to
75% confluence in the incubator. The medium was poured from the flasks of cells, and
200 µl of a given virus dilution was pipetted onto a given flask. The virus was allowed to
adsorb to the monolayers for 1 hour at room temperature on a plate rocker. After viral
adsorption, the liquid in the flasks was removed using a Pasteur pipette. The cells were
overlaid with 1% w/v agarose from Sigma (St. Louis, MO) at 45°C in supplemented
MEM. The agarose was allowed to solidify before the flasks were returned to the
incubator. After 2 days, the flasks were overlaid with 1% w/v agarose in supplemented
MEM containing 60 µg/ml Neutral Red from Fisher (Pittsburgh, PA) at 45° C. The
flasks were kept in the dark and placed in the incubator. After 6 hours, the flasks were
viewed on a light box and the plaques on each monolayer were counted. Individual
circular plaques observed on the cell monolayer represent 1 pfu, which is equivalent to
one infectious virus particle that results in plaque formation.
2.2.3.2 Protein and Radiation Characterization
The amount of viral protein for pure virus samples was quantified using a
MicroBCA assay kit from Pierce. Virus samples were boiled for 5 minutes to denature
the virus before using the kit. The kit was capable of detecting protein concentrations
For virus samples containing plasma proteins, the virus was quantified using the
radiation incorporated into the virus. Samples smaller than one ml were put in 20 ml
glass scintillation vials with 15 ml of Ecolume® Scintillation Fluid from ICN (Costa
Mesa, CA). The vials were sealed and shaken to homogenize the virus sample and the
added fluid. After five minutes to insure the sample had dissolved into the scintillation
fluid, the vials were counted using a A2010 TriCarb Liquid Scintillation Counter from
Packard (Boston, MA). A standard curve relates the amount of radiation, the protein
from MicroBCA and the infectious plaque assay.
2.2.4 Library and Peptide Synthesis
The hexameric peptide library was synthesized on TosohBiosciences Toyopearl
AF Amino-650M resin (TosohBiosciences, Montgomerywille, PA) as has been done
previously in the literature [28]. The methacrylate-based resin has an average pore size
of 100 nm and a particle diameter of 65 µm. The surface area of the resin is 30 m2/g
while the external surface area is 0.094 m2/g. The resin was selected as the base support
because of its mechanical and chemical stability. The synthesis was performed by
Peptides International (Lexington, KT) using the ‘divide, couple, recombine’ technique.
Eighteen of the twenty naturally occurring amino acids (excluding cysteine and
methionine) were used to produce the one bead, one peptide library. The final library
Individual peptide leads were synthesized on Toyopearl AF Amino-650M resin
by Peptides International in a similar fashion to the peptide library. The leads were
synthesized to a final density of 100 µmols per gram of resin. Unreacted amino groups
on the surface of the resin were acetylated while the amino terminus of the peptide and
any functional groups on the peptide were maintained. 1 gram of resin expanded to
approximately 4 ml when swollen in methanol for 1 hour.
2.2.5 Library Screening (Primary Screening)
A detection scheme similar to the technique described by Mondorf et al. was used
to identify peptides specific to SV [36]. For each experiment, 20 mg of dry library
(approximately 20,000 beads) was weighed out and put in a 1.8 ml microcentrifuge tube.
The beads were swollen in methanol for 1 hour and then washed in PBS buffer. The
beads were allowed to settle and excess PBS was taken from the library. The beads were
mixed with 1 ml of whole human plasma overnight on an orbital rotator. After mixing,
100 µl of a solution containing approximately 108 purified SV pfu/ml in PBS was added
to the plasma/library mix. The library was then tumbled on an orbital rotator for 1 hour.
After tumbling, the library was removed from the rotator, and the resin was allowed to
settle to the bottle of the tube. Excess plasma and virus was pipetted from the resin and
the amount of unbound virus was quantified using the scintillation counter. The library
was then resuspended in one ml of PBS and transferred to a 10 ml BioRad wash column.
The library was washed with PBS containing Tween 20 until radiation was not detected
by scintillation in the flow through of the column. In later primary screening, a second
wash step of 1 M NaCl in PBS was used to remove non-specific or loosely bound SV
After all the washes, the library was resuspended in 2 ml of PBS. The beads were
transferred to a 50 ml conical tube and 38 ml of 1% w/v low melt agarose at
approximately 40° C were mixed with the library so that the beads were evenly
distributed throughout the agar. Approximately 20 ml of agarose was poured evenly onto
two 8” x 10” sheets of GelBond (FMC, Rockland, ME) forming a monolayer of the
library. The agarose gel was allowed to air dry overnight before being exposed to β-max
radiographic film (Kodak) for 1-4 days. After developing the film, positive beads were
identified by aligning the gel and film. Each gel was exposed to film at least twice to
confirm the positive signals. Selected beads were excised from the gel under a
microscope using a scalpel. Individual beads were placed in water and heated to 70°C to
remove agarose and bound SV. The beads were put in fresh water, and the heated wash
process was repeated. Library beads were sequenced at Texas A&M University or at
Iowa State University by Edman degradation.
2.2.6. Binding Confirmation (Secondary Screening)
Secondary screening was used to verify the ability of leads identified during
primary screening to bind specifically to SV. Leads were tested using the secondary
screening method previously described by Bastek et al. [22] and Gurgel et al. [37]. The
leads were synthesized directly onto Toyopearl AF Amino-650M resin at a substitution
was added to the resin. The resin and the virus were incubated for 1 hr at 25°C on an
orbital rotator. After one hour, the virus was collected by centrifugation. The resins were
then washed sequentially for 1 hour each with PBS, PBS with Tween 20, 1 M NaCl, 3 M
NaCl, 2% v/
v acetic acid, and 6 M guanidine hydrochloride. Each wash was collected and
quantified using the radiation and the scintillation counter.
2.2.7 Column Screening (Tertiary Screening)
Leads tested during secondary screening and untested leads identified during
primary screening were synthesized onto TosohBiosep Toyopearl 650M Amino resin by
Peptides International as detailed above. FractoGel ion exchange resins from E-Merck
Separations, Toyopearl amino resin, and acetylated Toyopearl amino resin were used as
controls. The resins were swollen in methanol for at least 1 hr and then washed with
water. The resins were wet packed into 5 x 0.4 cm Omega columns (Upchurch, Oak
Harbor, WA) and tested in a chromatographic format on a Waters 626 HPLC with a built
in UV detector. Samples were manually injected using a 100 µl or 1 ml sample loop.
The columns were maintained at 25° C by immersing the columns in a water bath.
For each injection, the column was equilibrated in PBS at pH 7.4. Samples were
prepared with 108 SV pfu/ml in PBS or 108 SV pfu/ml in 50% human blood plasma
(HBP) and PBS unless otherwise noted. PBS buffer was used to initially load the sample
at a flow rate of 0.05 ml/min unless otherwise noted. After 80 minutes, the buffer was
changed to 4 M Urea and 2.5 M KCl in PBS and the flow rate was changed to 0.2
ml/min. After 15 minutes, the buffer was switched back to PBS for 15 minutes and then
back to 4 M Urea and 2.5 M KCl in PBS for a second wash. After regenerated, the
Fractions for each sample were collected manually either in 1.8 ml
microcentrifuge tubes or in 20 ml glass scintillation vials. Samples collected in
microcentrifuge tubes were stored at 4 °C and tested for infectivity using the plaque assay
described above within 24 hours. The amount of radiation in the samples was then
quantified using the Scintillation Counter.
2.3 Results and Discussion
2.3.1 Preparation and Characterization of SV
Several factors can influence SV grown in BHK cells. The health of the cells,
fresh solutions, and a controlled cell growth environment all play a critical role in the
development of uniform virions [35, 38-40]. Early experiments found large variations in
the adsorption of a peptide lead to SV between different batches of virus. Several
experiments and observations were made that were found to influence the uniformity of
the SV grown.
One of the most important aspects of growing consistent batches of virus was
found to involve having healthy cells in the log phase of growth. Before infecting, cells
were passed at least twice and examined carefully under a phase inversion light
microscope at 40x. Cells that did not have several mitotic cells in the viewing field were
not used to culture virus. Cells were only infected if the cell density was approximately
product. Dialysis in PBS buffer was used to remove the k-tartaric acid from the purified
SV. The radioactivity of samples before and after dialysis was quantified using the
scintillation counter. Approximately half of the virus was lost during dialysis.
Three methods were used to quantify the virus once it had been purified. An
assay was used to quantify the number of infectious particles. Plaques are white circles
observed on stained monolayers of cells. Figure 1 shows plaques caused by SV observed
on a light box for three serial dilutions of the SV plaque assay. A plaque is the result of
infected cells that cannot absorb the Neutral Red dye applied to the monolayer.
Uninfected cells turn red as they metabolize the dye. Only one virus particle is needed to
infect a cell. As the virus multiplies in the cell, neighboring cells become infected in a
radial manner resulting in a plaque. Therefore, each plaque represents one virus particle
that was infectious, or one plaque forming unit (pfu). If enough virus particles are added
to the monolayer simultaneously infecting all of the cells, dye will not be adsorbed into
any of the cells.
A general linear behavior is observed through the dilutions shown in Figure 2.1.
By counting the plaques on each dilution, the titer of the virus can be calculated using the
equation d
sv PC ml
pfu Titer
10
× =
= where PC is the plaque count, sv is the volume of sample
applied to the monolayer in ml, and d is the dilution of the sample being tested. For
Figure 1a, PC=4, d=-8, and sv=0.2 since 200 µl of sample was applied to the cells. The
A. B. C.
Figure 2.1 SV Plaques on BHK Cell Monolayers Observed, A. 10-8 Dilution, B. 10-7
Dilution, C. 10-6 Dilution. Serial dilutions of SV were prepared and applied to BHK cell
monolayers using the plaque assay procedure. Plaques were counted by visualization on a light box.
Table 2.1 shows plaque counts for each dilution shown in Figure 2.1 and the
resulting titers. The -7 and -8 dilutions result in titers that are almost identical, 2 x 109
and 1.6 x 109 respectively. Based on the -7 and -8 dilutions, the -6 dilution should have
approximately 400 plaques. However, the size of the flask used prevents this many
distinguishable plaques form forming on the monolayer. Virus particles from
neighboring plaques begin to merge resulting in smeared or non-circular plaques. As a
result of plaque crowding, only the -7 and -8 dilutions were used in determining the titer
of the virus in Figure 2.1 as approximately 2 x 109 SV pfu/ml. For a well-grown batch
of virus, the titer of the purified virus is expected to 108 to 109 pfu/ml.
A protein assay was used to detect the overall amount of viral protein for both
infectious and non-infectious purified virus. For well-grown virus, the concentration of
viral protein was approximately 5-30 µg/ml. Generally, batches of virus with less protein
also had lower titers while batches with more protein had higher titers. SV has a
molecular weight of 60 x 106, half of which is viral protein. Therefore, a well-grown
batch of virus has a particle to pfu ratio of approximately 10 to 1.
The radiation in the virus particle was also quantified using a scintillation counter
by the number of disintegrations per minute (dpm). Dilutions of radiolabeled canine
parvovirus demonstrated that radiation counts were linear within the range of 103 to 105
dpm (Chapter 3). Past experience indicates that linearity in the dpm extends from at least
102 to 107 dpm [42]. For well-grown virus, there were approximately 5 x 106 to 1 x 108
dpm/ml.
From the three characterization methods, a list of criteria was developed defining
SV that would be used in secondary and tertiary screening experiments. These criteria
are summarized in Table 2.2 and show the relationship between virus protein, infectivity,
and specific activity. This range of viral properties is consistent with SV described
elsewhere [41, 42].
Table 2.2 Criteria for Batches of SV. Several batches of SV were grown, and a list of
criteria was developed that established batches of virus that could be used in experiments.
Desired Characteristics of SV Batches
Infectivity Range 108 to 109 pfu/ml
Virus Protein Range 5-30 µg/ml
Radiation Range 5 x 106 to 1 x 108 dpm/ml
Particle to pfu Ratio 10:1
Viral incorporation of the 35S labeled cysteine (Cys) and methionine (Met) was
initially thought to play a role in the stability and uniformity of SV. Batches of SV were
grown with Cys, Met or both Cys and Met 35S label. Virus for each labeling condition
was purified and titered on a daily basis. Several experiments are shown in Figures 2-4.
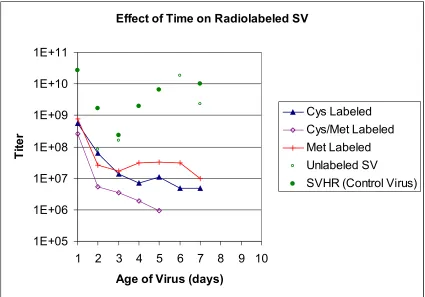
In Figure 2.2, all the SV batches that were radioactively labeled have a starting
titer of approximately 1 x 109 pfu. After three days of storing the virus at 4°C in PBS pH
7.4, the titers of the all of the viruses dropped to 1 x 107 pfu or less. Virus grown,
purified and stored in the same manner that was not radiolabled had a titer that varied
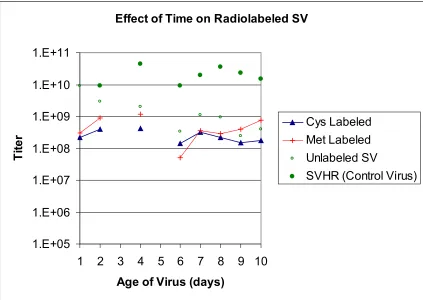
between 1 x 108 pfu/ml on day 3 to 1 x 1010 pfu/ml on day 6. Figure 2.3 shows a repeat
of the same experiment. In this case, the virus titer was 1 x 108 pfu for both labeled and
unlabeled virus. For all virus batches, the titer remained constant for 10 days. Figure 2.4
shows additional batches of SV that were grown with specific activities of 5 x 106
DPM/ml and 5 x 105 DPM/ml. The initial titer of the high specific activity virus and the
low activity virus were 1 x 1010 pfu and 1 x 109 pfu respectively. After ten days, both
batches had lost one log of titer when compared to the stock SVHR. Therefore, in this
run, the amount of 35S incorporation was not thought to have an effect on virus stability
and variability.
Several repetitions of this experiment failed to produce consistent titer results
Effect of Time on Radiolabeled SV
1E+05 1E+06 1E+07 1E+08 1E+09 1E+10 1E+11
1 2 3 4 5 6 7 8 9 10
Age of Virus (days)
Ti
te
r
Cys Labeled Cys/Met Labeled Met Labeled Unlabeled SV
SVHR (Control Virus)
Figure 2.2 Effect of Radioactive Label on Stability of SV. Several batches of SV
Effect of Time on Radiolabeled SV
1.E+05 1.E+06 1.E+07 1.E+08 1.E+09 1.E+10 1.E+11
1 2 3 4 5 6 7 8 9 10
Age of Virus (days)
Ti
te
r
Cys Labeled Met Labeled Unlabeled SV
SVHR (Control Virus)
Figure 2.3 Effect of Radioactive Label on Stability of SV. Several batches of SV
Effect of Time on Radiolabeled SV
1.E+05 1.E+06 1.E+07 1.E+08 1.E+09 1.E+10 1.E+11
1 2 3 4 5 6 7 8 9 10
Age of Virus (days)
Ti
te
r
High Low SV SVHR
Figure 2.4 Effect of Specific Activity of Radio-labeled SV. Batches of SV were
grown and labeled with Cys and Met. The batches were purified and stored in polypropylene tubes. The specific activity of the labeled batches differed by
approximately 1 order of magnitude. ‘High’ was SV with a specific activity of 4 x 106 CPM/ml. ‘Low’ was SV with a specific activity of approximately 5 x 106 CPM/ml. SV is virus that was grown and purified that was not radioactively labeled. SVHR is stock virus that normally titers at 1 x 109.
2.3.2. Lead Identification (Primary Screening)
Two different rounds of primary screening were performed to identify potential
binders of SV. In the first round of screening, the library was contacted with the virus in
HBP. The library was washed with saline buffer and plated out. Approximately 0.18
grams of the 18 g library was screened, representing roughly 340,000 sequences out of
the possible 34 million sequences. Five leads were identified and are summarized in
Table 2.3. Two of the five sequences ended in the same four amino acids (VIFLVR and
of the sequences contained a positively charged amino acid (Arg or Lys) in addition to
the terminal amine at the end of the sequence. The remaining amino acids appeared to be
randomly distributed.
Table 2.3 Sequences Identified from Screening 35S-SV Against a Solid Phase
Peptide Library. A solid phase peptide library was screened in whole human blood
plasma and 7 log10 pfu SV at 25°C for 1 hour. After washing, the library was exposed to
radiographic film and leads that bound SV were identified. Five sequences were isolated and sequenced using Edman chemistry. The sequences are listed from the amino to carboxy terminus.
Sequence Short Hand
Ala Tyr Phe Leu Val Arg AYFLVR
Phe Arg Ser Pro Asn His FRSPNH
Val Ile Phe Leu Val Arg VIFLVR
Asn Ile Ile Val Gln Arg NIIVQR
Lys Leu Tyr His Lys Ala KLYHKA
Characterization of these leads was done using secondary and tertiary screening.
None of the identified leads were found to consistently bind at least 2 log10 SV pfu/ml in
saline. A second round of screening was performed to try and identify additional leads
that might have greater adsorption to SV. An additional wash step of 1 M NaCl in PBS
buffer was added after contacting the library with the virus to try and avoid identification
of weak binders. Approximately 0.09 g of the 18 gram library was screened, representing
roughly 170,000 sequences out of the possible 34 million sequences. Four leads were
identified and are summarized in Table 2.4. No true consensus sequence was observed,
glutamic acid or aspartic acid. While the majority of the sequences from the first round
of screening had a net positive charge, the sequences of the second round of primary
screening were generally more neutral.
Table 2.4 Sequences Identified from Screening 35S-SV Against a Solid Phase
Peptide Library with a 1 M NaCl Wash. A solid phase peptide library was screened in
whole human blood plasma and 7 log10 pfu SV at 25°C for 1 hour. The library was
washed extensively with 1 M NaCl in PBS before exposing to radiographic film. Four sequences were isolated and sequenced using Edman chemistry. The sequences are listed from the amino to carboxy terminus.
Sequence Short Hand
Ile Ala Thr Asp Gly Gly IATDGG
Tyr Glu Trp Lys Trp Gly YEWKWG
Glu Trp Val Pro Thr Ile EWVPTI
Ser Gly Lys Pro Val Ala SGKPVA
One drawback to the primary screening method used in this work was the choice
of base resin. Toyopearl Amino resin has an average pore diameter of 100 nm. The
particle diameter of SV is 70 nm, so little if any pore penetration was expected in the
screening. This greatly reduces the surface area that is available to the virus to bind
during the screening process which could potentially result in missed leads. The impact
of the reduced surface area was somewhat nullified by the choice of 35S labeling. 35S has
a specific activity of 42,707 Ci/g compared to 4.46 Ci/g for 14C which was used in
previous work with proteins. A higher specific activity should allow smaller quantities of
SV to be detected, though the exact detection limits were not determined.
2.3.3 SV Binding to Resins (Secondary Screening)
Secondary screening was performed on several resins and the results are found in
Appendix 2.A.1. Secondary screening was unsuccessful because approximately 85% of
the virus added to the resin adsorbed to the reaction vessel holding the resin. Several
tertiary screening methods since the column system was found to adsorb less than 10% of
the virus.
Since a secondary screening system was not identified, equilibrium isotherm
experiments could not easily be performed. As a result, the resin capacity and
equilibrium-binding coefficient were not determined.
Experiments were performed that varied the flow rate through the column and the
concentration loaded onto the column. Figure 2.5 shows several chromatograms
generated by injections of SV in 50% HBP onto a 0.6 ml column of SGKPVA with flow
rates ranging from 0.05 ml/min to 0.8 ml/min. As the flow rate through the column was
reduced, the amount of SV found in the unbound fraction decreased. A flow rate of 0.05
ml was used for remaining injections to ensure adequate binding time in the column. A
second injection performed at 0.05 ml/ml demonstrated that the column was being
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35
0 1 2 3 4 5 6 7 8 9 10 11 12 13
Column Volumes
Fr
ac
ti
on of Tot
al
S
V
0.05 ml/min 0.1 ml/min 0.2 ml/min 0.8 ml/min 0.05 ml/min (2)
Figure 2.5 Loads of SV on SGKPVA at Varied Flow Rates. A column of SGKPVA
was challenged with SV at flow rates ranging from 0.05 ml/min to 0.8 ml/min. The flow through and eluted fractions were collected and quantified using radioactivity and the scintillation counter.
Figure 2.6 shows a plot of the log removal versus the flow rate through the
column. The log removal was calculated using the amount of radiation added to the
system and the amount of radiation recovered in the unbound fraction. For this batch of
virus, a maximum log reduction of 1.2 pfu was obtained at a flow rate of 0.05 ml/min.
As the flow rate was increased to 0.8 ml/min, the log reduction decreased to 0.36 pfu or
approximately the same log reduction found for the control resin. Since the flow rate
directly effects the residence time in the column, Figure 6 suggests that increasing the
0.00 0.20 0.40 0.60 0.80 1.00 1.20 1.40
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 Flow Rate (ml/min)
Log Removal
Figure 2.6 Effect of Flow Rate on Log Removal. The log removal for injections of
SV on SGKPVA is plotted versus the flow rate.
Figure 2.7 shows three different injections of SV with the total amount of viral
load ranging from 2.03 µg (~5 x 108 pfu) to 0.46 µg (~1 x 107 pfu). Figure 2.7 shows
that increasing the viral load results in increasing corresponding eluted fraction peaks.
The areas of these peaks are roughly proportional to the amount of SV initially loaded.
For the largest injection load tested, SV bound in a monolayer to the resin would cover a
surface area of 3.4 x 10-4 m2. In the 0.6 ml Omega columns used in these experiments,
the external surface area of the resin is 1.2 x 10-2 m2. Therefore, for the amount of SV
0 200000 400000 600000 800000 1000000 1200000 1400000 1600000
0 1 2 3 4 5 6 7 8 9 10 11 12 13
Column Volumes
DPM/ml
2.03 µg 1.06 µg
0.46 µg
Figure 2.7 Injections of Varied Quantities of SV on SGKPVA. A column of
SGKPVA was challenged with varied concentrations of SV. The flow through and eluted fractions were collected and quantified using radioactivity.
2.3.4 Clearance of SV from Plasma (Tertiary Screening)
Tertiary screening was performed to determine the selectivity of leads identified
for SV in the presence of 50% HBP. Toyopearl Amino resin, acetylated Toyopearl
Amino resin, two ion exchange resins from E-Merck separations and a peptide ligand
identified for Canine parvovirus were used as controls.
Representative chromatograms for several resins are shown in Figure 2.8 and 2.9.
The log clearances for these chromatograms along with clearances for other resins that
were tested are summarized in Table 2.5. The clearance was calculated using the amount
of radiation loaded onto each column and the amount of radiation found in the unbound
fraction. Figures 2.8 and 2.9 show the virus loaded onto the column was not completely
0.00 0.05 0.10 0.15 0.20 0.25 0.30
0 1 2 3 4 5
Column Volumes
Fraction of Total SV
Actylated Amino Fractogel NIIVQR IATDGG SGKPVA
Figure 2.8 Injections of SV in PBS on Several Resins. Several resins were
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35
0 1 2 3 4 5
Column Volumes
Fraction of Total SV
Acyetylated Amino Fractogel NIIVQR SV 1 SKKPVA
Figure 2.9 Injections of SV in 50% HBP on Several Resins. Several resins were
challenged with injections of SV in 50% HBP. Fractions were collected and quantified using radiation.
Table 2.5 summarizes the best log clearances achieved of SV from PBS and 50%
HBP. Resins with a hexapeptide were able to clear between 1.4 and 3.0 logs. IATDGG
and SGKPVA cleared the most SV with 2.4 and 3.0 logs respectively. Positively and
negatively charged ion exchange resins from E-Merck Separations achieved 2.1 and 1.3
logs of SV clearance respectively. Toyopearl amino resin and acetylated amino resin
bound 0.6 and 0.1 logs of SV respectively.
In general, resins that made use of a ligand bound more SV than resins without a
ligand. The positive charge on the amino resin also allowed more SV to be adsorbed than
an uncharged resin. For both the amino resin and the FractoGel resins, the interaction
with SV may be a result of ionic interactions with charged patches on the surface of the