Copyright 0 1990 by the Genetics Society of America
A Simple Method for Estimating Average Number of Nucleotide
Substitutions Within and Between Populations From Restriction
Data
Masatoshi Nei and Joyce
C.Miller’
Center f o r Demographic and Population Genetics, The University of Texas Health Science Center, Houston, Texas 77225
Manuscript received January 4, 1990 Accepted for publication May 5 , 1990
ABSTRACT
A simple method is proposed for estimating the average number of nucleotide substitutions per site within and between populations for the case where a large number of individuals are examined for many restriction enzymes. This method gives essentially the same results as those obtained by Nei and Li’s method but saves a large amount of computer time. The variances of the quantities estimated can be obtained by the jackknife method, and these variances are very similar to those obtained by Nei and Jin’s more sophisticated method. A similar simple method can also be applied to DNA sequence data.
T
HE extent of DNA polymorphism in a population is often measured by nucleotide diversity ( x ) , which is defined as the average number of either nucleotide differences or substitutions per site for a group of DNA sequences (alleles) sampled [NEI ( 1 987), chap. 101. When there is polymorphism within populations, the extent of nucleotide divergence be- tween two populations or species is also measured by sampling many DNA sequences from each population. A standard measure of this extent is the average number of net nucleotide substitutions per site (dA), where the effect of within-population polymorphism has been subtracted.NEI and LI (1 979) proposed a statistical method for estimating both x and dA from restriction-site or re- striction-fragment data, whereas NEI and JIN (1989) provided a method for computing the variances of these quantities. However, when the number of DNA sequences examined is large, these methods are quite cumbersome, because the number of nucleotide dif- ferences is first computed for each pair of DNA se- quences and then x or dA is estimated. Estimation of the variances is particularly computation-intensive.
When x is relatively small, however, there is a simpler method for estimating these quantities. While this method gives approximate estimates of x and dA, it can be shown that the estimates obtained are gen- erally sufficiently accurate. In the following we pres- ent this new method. In this paper, we will consider primarily restriction-site data, but the same method is applicable to restriction-fragment or DNA sequence data, as will be mentioned in the DISCUSSION.
Madison, Wisconsin 53706.
Genetics 125: 873-879 (August, 1990)
I Present address: Department of Agronomy, University of Wisconsin,
MATHEMATICAL FORMULATION
Nucleotide diversity: T h e nucleotide diversity in a population ( x ) is usually estimated by
7; = 2 dg/[n(n
-
l)], (1)i<j
where
d,
is an estimate of the number of nucleotide substitutions per site between DNA sequences i a n d j(dg) and n is the number of DNA sequences examined.
When DNA polymorphism is studied by using various restriction enzymes,
8,
can be computed either by NEI and LI’S (1979) method or by NEI and TAJIMA’S (1 983) maximum likelihood method (see also KAPLAN and LANGLEY 1979). In these methods it is convenient to construct a restriction-site map for each sequence, considering one restriction enzyme at a time or all enzymes simultaneously. Once the map is constructed for all sequences, one can compute the total number of restriction sites ( m i ) for each sequence and the number of shared restriction sites (mu) between se- quences i andj. These values are computed separately for each enzyme class. Here, restriction enzymes are classified according to the number of nucleotides in the recognition sequence ( r ) , which is usually 4, 6 , or16/3 [see NEI (1 987), chap. 51.
Once mi and mg are obtained, the following quantity is computed for each enzyme class.
2mg
s,
= mi + mj‘When only one class of restriction enzymes is used, one can estimate dg by
8,
= [-log,Sg]/r (3)874 M . Nei and J. C . Miller
TABLE 1
Number of restriction sites for the ith sequence (mi) and the number of shared sites (mij) when n DNA sequences are
compared
Sequence 1 2 3 n
1 ml
2 ~ I Y m y
3 m l . l my:! m :+
n m i , myn mYn m,
TAJIMA 1983). Here we use Equation 3 because we are considering relatively small values of do's and in this case the two methods give virtually the same estimate. When there are two or more different en- zyme classes, one can use NEI and TAJIMA'S (1983) maximum likelihood method for estimating x. How- ever, it is also possible to estimate d, by using the formula
fikrk&(k)
d.. A = h 11
fikrk ' (4)
k
where d o ( k ) is obtained by Equation 3 and m k is (mi(k)
+
m,(k))/2 (NEI and TAJIMA 1981). Here, subscript
k
refers to the kth class of restriction enzymes, and summation is taken over all different enzyme classes.
Theoretically,
2,
in Equation4
is less accurate than the estimate obtained by the maximum likelihood method, but the actual difference is negligibly small, as will be shown later. Therefore,ii
in Equation 1 can be computed by using2,
in either Equation 3 or4.
As mentioned above, however, this computation becomes laborious when n is large and many different enzymes are used. MILLER (1989) used 5 different restriction enzymes for each of 40 different nuclear DNA probes and examined a substantial number of individuals from each of several tomato species. In such a case as this, an enormous amount of computa- tion is required. A simpler method for this case is to compute a single S value for each enzyme class and then estimate x by using an equation equivalent to (4). That is, we first compute the following quantity
2
2
my-
i<jS = ( 5 )
C
mib)+
C
m(tb' 9 zCj
for each enzyme class, where r n , ( , ) and m(iu are the mi
and mj values, respectively, when seqyences i and j are compared (see Table 1). Note that S is an average of S , weighted with (mi
+
mj)/2. Therefore, if (mi+
mj)/ 2 is the same for all i ' s and j ' s ,s"
becomes the arith- metic mean of S,. T h e x value for the kth enzyme class can then be estimated byG k = [-lOgegk]/rk, (6)
Restriction Slte
1 2 3 4 5
Sequence
1
2 A A A A
3 n A A A
4 A A n
Y
A A A A A
"
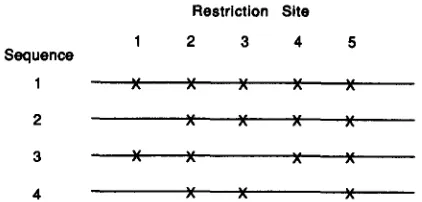
FIGURE 1 .-Hypothetical population of four sequences, used to illustrate the computation of L described in the text.
where subscript
k
refers to the kth enzyme class. This equation gives essentially the same estimate of x as that obtained by NEI and TAJIMA'S (1 98 1) method as long as it is small (see APPENDIX).Computation of the denominator of Equation 5 can be simplified in the following way if we note that mi is used n
-
1 times in the summation. That is, Mzicj
ml(])
+
m(i)j can also be written asM = (n
-
1) m, = n(n-
l)G(7)
where bi = mJn. Furthermore, when n is large, the following method facilitates the computation of L
ziv
mg in the numerator of Equation 5 . First consider the restriction-site maps given in Figure 1. In this case,m 1 2 = 4, m 1 3 = 4, m14 = 3, m23 = 3, mZ4 = 3, and mS4 =
2.
Therefore, L = 19. However, L can also be computed by counting the number of pairs of restric- tion sites( p )
that are possible for all DNA sequences at each restriction site. If the bth restriction site is shared by sb DNA sequences,p
is given by s b ( s b-
1)/2. L is then given by the sum of this value for all sites. That is,
i
1
L = sb(sb
-
1)/29 (8)b= 1
where t is the total number of restriction sites. There- fore
s"
is given by3
= 2 L / M . (9)In the example given in Figure 1, sb is 2, 4, 3, 3 and
4 for b = 1, 2, 3, 4 and 5 , respectively. Therefore, L
= 1
+
6+
3+
3+
6 = 19, which agrees with the value computed above. On the other hand, M = 3 X( 5
+
4+
4+
3) = 48. Thus,s"
= 38/48 = 0.792. Once the value ofs"
is obtained for the kth class of enzymes, i i k for this class is estimated by Equation 6.Therefore, x can be estimated by
mkrkxk x =
m h r k ' (10)
h
where 7jlk is the value of rii for the kth class of enzymes.
Estimating Nucleotide Substitutions 875
TABLE 2
The mi and mg values for 13 mtDNAs from chimpanzees
- 1 2
3
4
5 6 7 8
9
10 1 1 12 13
____
~ ~ _ _ _ ~~
1 2 3 4 5 6 7 8 9 10 11 12 13
35, 1 1 , 6
3 3 , I O , 6 36, I O , 6
3 3 , 10, 6 36, 10, 6 36, 10, 6
3 2 , 1 0 , 6 34, 9, 6 34,9, 6 38, 12, 7
32, 10.6 34, 9, 6 34,9, 6 38, 12, 7 38, 12, 7 3 5 . 1 1 . 6 3 3 , 1 0 , 6 3 3 , 1 0 , 6 3 2 , 1 0 , 6 3 2 , 1 0 , 6 3 5 , l l , 6
32, 11,6 34, 10, 6 34, 10, 6 3 2 , 10, 6 32, 1 0 , 6 32, 11.6 35, 11, 6
32, 10, 6 34, 9, 6 34, 9 , 6 38, 12, 7 38, 12, 7 33, 1 0 , 6 32, 10, 6 38, 12, 7 3 4 , 1 0 , 6 3 3 . 9 . 6 3 3 , 9 , 6 3 3 , 9 , 6 3 3 , 9 , 6 3 4 , 1 0 , 6 3 2 , 1 0 , 6 3 3 , 9 , 6 3 5 , 1 0 , 6
33, 1 0 , 6 36, 10.6 36, 10.6 34,9, 6 34, 9 , 6 33, 10, 6 34, 10, 6 3 4 , 9 , 6 33, 9 , 6 36, 1 0 , 6 3 0 , 8 , 6 3 0 . 7 . 6 3 0 , 7 , 6 3 0 , 8 , 6 3 0 , 8 , 6 3 0 , 8 , 6 2 8 , 8 , 6 3 0 , 8 , 6 3 1 , 8 , 6 3 0 , 7 , 6 3 7 , 8 , 9
2 9 , 8 , 6 3 1 , 7 , 6 3 1 , 7 , 6 3 1 , 8 , 6 3 1 , 8 , 6 2 9 , 8 , 6 2 9 , 8 , 6 3 1 , 8 , 6 3 0 , 8 , 6 3 1 , 7 , 6 3 3 , 8 , 8 3 6 , 8 , 8 3 0 , 8 , 6 3 0 , 7 , 6 3 0 , 7 , 6 3 0 , 8 , 6 3 0 , 8 , 6 3 0 , 8 , 6 2 8 , 8 , 6 3 0 , 8 , 6 3 1 , 8 , 6 3 0 , 7 , 6 3 7 , 8 , 9 3 3 , 8 , 8 3 7 , 8 , 9
t11tDNAs 1 through 10 are from the common chimpanzee Pan troglodytes, and 11 through 13 are from the pygmy chimpanzee P. paniscus.
Values on and below the diagonal are the m,’s and my’s. respectively. T h e first, second and third numbers within each comparison show the m , and m,,’s for 6-base, 16/3-base and 4-base enzymes, respectively.
When the number of restriction-sites examined is large, Equation 1 is expected to give a more reliable estimate. However, when this number is small, the new method seems to be better, because the averaging of m, and mli would reduce the extent of sampling error. The computational process in the latter method is also much simpler than that in the former, as mentioned earlier.
Nucleotide divergence: Suppose that nx and ny
DNA sequences are sampled from population X and
Y , respectively. T h e extent of nucleotide divergence (average number of net nucleotide substitutions per site) between the two populations can be measured by the following quantity (NEI and LI 1979).
d , = dxy
-
(dx+
d y ) / 2 , (1 1)where dx and dy are the r values in populations X and
Y , respectively, whereas dxy is the average number of nucleotide substitutions per site between X and Y .
NEI and LI’S method for estimating dxy is to estimate
d , for each sequence from population X and each
sequence of Y , and to take the average of d, for all combinations. Here, we propose the following simple method for estimating this quantity. We first compute
s x y defined by
2
e
mx,y,c
mxsy,, +c
m(x,)Y,’-
i<JS X Y = ( 1 2 )
‘ Y i-q
where mx,y, is the number of restriction sites shared by the ith sequence from population X and the jth se- quence from Y , whereas mX,(%) and are the num-
bers of restriction sites for the ith sequence from X
and the jth sequence from Y , respectively. Here M x
=
Et<j mx,(v,). MY E i < j m(x,)Y,, and LxY
E,<j
mx,% can beTABLE 3
Estimates of T , dm, and dA and their standard errors ( ~ 1 0 0 0 )
obtained by method I and method XI (present method)
Method chimpanzee chimpanzee
Common Pygmy
Ci,, a,
I“ 13.4& 3.3 9.4, 3.4 35.8 f 7.5 24.4 f 6.7 11 13.3 f 2.9 9.2 k 3.3 35.6 & 7.8 24.4& 6.5 T h e results involving common chimpanzees are slightly differ- ent from those of NEI and JIN ( 1 989), because there was an error in counting the number of restriction sites in the latter.
written in the following way.
M X = n y mx, = nxny mx, (1 3)
i
MY = nx m? = n x n y my, (14)
I
t
LXY =
c
SX*SY*’ ( 1 5 )b= 1
where mx = mx,/nx, m =
zj
myz/ny, and sxb and sybare the numbers of restriction sites for all sequences at the bth site in populations X and Y , respectively.
We can then estimate dxy for the Kth enzyme class by
and the estimate of dxy for all enzyme classes is given by
where
Gk
=(mX
+
G y ) / 2 . Therefore, d,., is estimated by876 M. Nei and J. C. Miller
TABLE 4
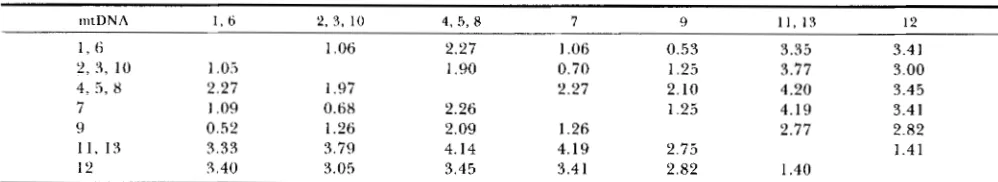
Estimates of the numbers of nucleotide substitutions per site ( d q ) between mtDNAs from different individuals of chimpanzees (X100)
m t D N A I , ti 2 , 3 , 10 4, 5, 8 7 9 1 1 , 1 3 12
1 , (5 1.06 2.27 1.06 0.53 3.35 3.41
2 , 3 , 10 1.05 1.90 0.70 1.25 3.77 3.00
4, 5,
x
2.27 1.97 2.27 2.10 4.20 3.457 1 .09 0.68 2.26 1.25 4.19 3.4 1
9 0.52 1.26 2.09 1.26 2.77 2.82
11.13 3.33 3.79 4.14 4.19 2.75 1.41
1 2 5.40 3.05 3.45 3.41 2.82 1.40
M;lxilntllll-li~elihood estimates are on the upper right half of the matrix, whereas the estimates from Equation 4 are on the lower left half.
This method is also known to give essentially the same results as those obtained by NEI and TAJIMA’S (1983) method (see the numerical example given later).
Variances of
?;,
dxr, and dA: NEI and TAJIMA (1 98 1) have studied the variances of?r
andd,
that are gen- erated at the time of allelic sampling, whereas NEI and JIN (1 989) have presented a method for comput- ing the variance due to estimation errors of nucleotide substitutions, including some effects of stochastic changes of allele frequencies in the past. Since the latter variance is generally much larger than the for- mer, we will consider only the latter variance here. NEI and JIN’S method w?s developed for computing the variances of;r
and d, obtained by NEI and LI’S(1 979) estimation method (method I). Unfortunately, their method cannot be used inAthe present case. However, the variances of
k
and d A can be obtained by the jackknife or bootstrap method (EFRON 1982). I n practice, the jackknife method is easier to apply for the present case than the bootstrap method and seems to give quite accurate values of the variances.T o compute the variance of 7; by the jackknife method, we first compute
;r
by using Equation 10. We then eliminate data for the first restriction enzyme and again computeG
by Equation 10. We denote thisG
value byGI.
We do the same computation for the second, third a.
, mth enzymes and denote them byp 2 , T : ~ ,
. .
.,
Gm, respectively, where m is the numberof restriction enzymes used. (When m enzymes are used for each of
p
DNA probes from nuclear DNA, this computation is done for each enzyme/probe, SOthat mp G,’s are computed.) Once these values are computed, the variance of ?; is given by
A I
m - 1
V ( G ) =
-
(Gi
-
G)?
m I = ]
The variance of
dxy
or d A can be computed in thesame way.
NUMERICAL EXAMPLE
Computation of ;i and
d
and their variances by the present method (method 11) is different from theprevious one (method I) in three ways, i.e., (1) com- putation of
;r
when there are several enzyme classes, (2) computation ofS
values and (3) computation of the variances. T o see the difference between the two methods, let us consider a numerical example. FERRIS,WILSON and BROWN (1 98 1) and FERRIS et al. (1 98 1) studied the restriction-site polymorphism among 10 mtDNAs from common chimpanzees and 3 mtDNAs from pygmy chimpanzees. They used 13 enzymes with
r = 6, two enzymes with r = 16/3, and one enzyme with r = 4. T h e mi and m,, values for the 13 mtDNAs examined are presented in Table 2. Chimpanzee mtDNAs are highly polymorphic. Therefore, if the two methods give similar ;i and
2,
values for these two species, method I1 can be used for most species (see APPENDIX).Let us first compute 7; for pygmy chimpanzees using Equation 10. To obtain this 7;, we must first compute
3
separately for three different enzyme classes. For the enzyme class with r = 6, M = ( n-
1)E,
m, = 2 X(37
+
36 + 37) = 220, whereas 2L = 2 X m,, = 2 x(33 + 37
+
33) = 206. Therefore, = 0.936. Simi- larly, we obtainS2
= 1 .OOO for r = 16/3 and 3 3 =0.962 for r = 4. From these values, one can obtain ; i l
= 0.01 1, G2 = 0.000, and G 3 = 0.0098 by using
Equation 6. To obtain
G
from Equation 10, one has to know&.
From Table2,
we obtain r i i l = 36.67,&
= 8.00, and f i 3 = 8.67. Therefore,
G
is 0.009. T h e Gfor common chimpanzees can be obtained in the same way. I t becomes 0.0133. T h e variance or standard error of
G
can be obtained by the jackknife method described above. T h e results obtained are presented in Table 3.To obtain
dxy
and d A between common and pygmychimpanzees, we must first compute $xy for the three classes of enzymes. It becomes 0.824,0.815 and 0.962 for r = 6, r = 16/3 and r = 4, respectivelylTherefore, usingAEquations 16, 17 and 18, we obtain d ~ 0.0356 =
and d A = 0.0244. T h e standard errors of these quan- tities are given in Table 3.
Estimating Nucleotide Substitutions 877
in Table 3. Comparison of these values and those obtained by the present method (method 11) indicates that the differences between them are very small. Particularly, the
k ,
dxy, and2,
values are virtually identical, though method I1 tends to give a little smaller value than method I as expected from the theoretical study given in the APPENDIX. T h e largest difference is observed in the standard error of i; for common chimpanzees.Table 4 is presented to show the differences be- tween the maximum likelihood estimates obtained by NEI and TAJIMA’S method and the estimates obtained by Equation 4. It is interesting to see that the simple Equation 4 gives essentially the same values as those obtained by the maximum likelihood method.
DISCUSSION
As mentioned earlier, the present method depends o n a number of approximations but has some advan- tage over method I when the number of restriction enzymes used is relatively small. Therefore, the esti- mates of a, d x y , and dA obtained by method I1 are not necessarily less accurate than those obtained by method I. However, the theoretical study in the AP-
PENDIX and the numerical example given above show that the differences in the estimates obtained by the two methods are generally very small as long as k is not extremely large. Note that k is generally smaller than 0.02 for most nuclear and mitochondrial DNA and that the k for chimpanzee mtDNA is exceptionally high (NEI 1987; CLARK 1990). Therefore, the simple method proposed here seems to be widely applicable. T h e simplicity of the present method also makes it less prone to computational errors than method I.
T h e computional time saved by method I1 is also substantial when the data set is as large as that of MILLER (1 989).
As is well known, the jackknife is a nonparametric statistical method. Therefore, if the pattern of nucleo- tide substitution varies during evolutionary time, this method may give a more reliable variance than the parametric method proposed by NEI and JIN (1989).
In practice, however, both methods seem to give very similar results.
Restriction fragment data: In the present paper we have been primarily concerned with restriction site data. However, the same method is applicable to re- striction fragment data. In this case, the number of nucleotide substitutions per site for a pair of DNA sequences is estimated by solving for G in the following equation
G = [F(3
-
2G)’’4, (20)where F is the fraction of shared fragments between two sequences (NEI and LI 1979). When the number of fragments is mi for the ith sequence and mj for the
j t h sequence and the number of shared fragments is mq, F is given by
F = 2mjj/(m,
+
mj). (2 1)Equation 20 may be solved by an iteration metho$ (see NEI 1987). Once G is obtained,
2,
is given by d ,When many DNA sequences are examined in a population, a can be estimated by the same method as method I. However, it can also be estimated by method 11, if we consider a single F, i.e.,
= -(2/r)log,G.
2
C
m,-F =
i<j(22)
C
mib) +C
m(:)j’i c j i<j
Once
F
is obtained, a for the kth enzyme class is estimated by?;k = -(2/r)10&dj
where
d
is the value of G given by Equation 20 whenF
is used. One can then computek
by using Equation 10. In this case, however,6k
represents the average number of restriction fragmFnts per sequence for the kth class of enzymes. The dxy and dA values can be obtained in the same way.However, note that
F
declines to 0 more rapidly than as a or d x y increases. Therefore, this method should be applied only to conspecific populations or to very closely related species.DNA sequence data: T h e method of estimating a and d A presented here can easily be extended to the case of DNA sequence data. In this case, the total number of nucleotide sites to be compared ( m ) is the same for all sequences, and a site may have one to four different kinds of nucleotides (A, T , C and G). (Deletions/insertions are neglected). When n se- quences are sampled from a population and a is small (say a
<
0.02), a can be estimated bym
i p = h,lm, (23)
i= 1
where h, is an estimate of nucleotide diversity at the ith nucleotide site. Let x A , xT, XC and XG be the relative
frequencies of nucleotides A , T , C and G at the ith site among n sequences. An estimate of the nucleotide diversity at this site is then given by
h, =
-
nn - 1 (1
-
X:-
X;-
X:-
X:). (24)Therefore, a can be estimated by using this h,. Note that h, = 0 for monomorphic sites and thus one needs to compute hi only for polymorphic sites. This method gives essentially the same result as that obtained by Equation 1 as long as a is small (<0.02). When ?r is
878 M. Nei and J. C. Miller
correction for multiple hits may give a better estimate. That is,
;r
=- -
3 loge( 1-
3
4.).
4The dA and dxy values can be estimated in the same way. In this case the average proportion of different nucleotides between nx sequences from population X and ny sequences from population Y can be computed by
m
-
PXY
= hxY,/m, (26)I= 1
where h x , is the average proportion of different nu- cleotides at the ith site, i,e.,
h x , = 1
-
XAYA-
X T Y T-
xc:yc-
XCYC. (27)Here, x A , xT, etc., and yA, yT, etc., refer to the relative
frequencies of A , T , etc., in populations X and Y ,
respectively. An approximate estimate of d x y is then given by
d,,
= " log, 1-
-
p,,
,4
(
; - )
and
2,
= d x y-
( d x+
d y ) , (29)where
dx
andd y
are the values of & in populations X and Y , as before.Equation 28 should, however, be used only when
d x y is relatively small, say d x y
<
0.1, since it may give an underestimate when d x y is large.Computer program: A phylogenetic analysis pro- gram package (RESTSITE) that includes the method
11 in this paper (both for restriction-site and restric- tion-fragment data) is available from the authors (pref- erably from J.C.M.). T o receive this package, send a 5'A-inch high density diskette or three double-density diskettes.
This study was supported by research grants from the National Institutes of Health and the National Science Foundation.
LITERATURE CITED
BROWN, W. M., 1980 Polymorphism in mitochondrial DNA of humans as revealed by restriction endonuclease analysis. Proc. Natl. Acad. Sci. USA 77: 3605-3609.
BROWN, W. M . , and H. M. GOODMAN, 1979 Quantitation of intra- population variation by restriction endonuclease analysis of h ~ c n ~ t n n1itochondrial DNA, pp. 485-500, in Extrachromosomal
DNA, ICN-UCLA Symposia, Vol. 15, edited by D. CUMMINGS, P. BORST, 1. DAVID, S. WEISSMAN, and C. F. FOX. Academic Press, New York.
CLARK, A,, 1990 Inference of haplotypes from PCR-amplified sample of diploid populations. Mol. Biol. Evol. 7: 11 1-122. F.FRON, B., 1982 The Jackknije, the Bootstrap, and Other Resampling
Plans. Society for Industrial and Applied Mathematics, Phila-
delphia.
FERRIS, S. D., A. C. WILSON and W. M. BROWN, 1981 Evolu- tionary tree for apes and humans based on cleavage maps of mitochondrial DNA. Proc. Natl. Acad. Sci. USA 78: 2432- 2436.
FERRIS, S. D., W. M. BROWN, W. S. DAVIDSON and A. C. WILSON, 198 1 Extensive polymorphism in the mitochondrial DNA of apes. Proc. Natl. Acad. Sci. USA 78: 6319-6323.
JUKES, T. H., and C. R. CANTOR, 1969 Evolution of protein molecules, pp. 21-132, in Mammalian Protein Metabolism, ed- ited by H. N. MUNRO. Academic Press, New York.
KAPLAN, N., and C. H. LANGLEY, 1979 A new method of sequence divergence of mitochondrial DNA using endonuclease map- ping. J. Mol. Evol. 13: 295-304.
MILLER, J. C., 1989 RFLP analysis of phylogenetic relationships, genetic variation, and factors affecting the detection of poly- morphism in the genus Lycopersicon. Ph.D. thesis, Cornell Uni- versity, Ithaca, N.Y.
NEI, M., 1987 Molecular Euolutionary Genetics. Columbia Univer- sity Press, New York.
NEI, M., and L. JIN, 1989 Variances of the average numbers of nucleotide substitutions within and between populations. Mol. Biol. Evol. 6: 240-300.
NEI, M., and W.-H. LI, 1979 Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76: 5269-5273.
NEI, M., and F. TAJIMA, 1981 DNA polymorphism detectable by restriction endonucleases. Genetics 97: 145-163.
NEI, M . , and F. TAJIMA, 1983 Maximum likelihood estimation of
the number of nucleotide substitutions for restriction sites data. Genetics 105: 207-216.
Conlmunicating editor: A. G. CLARK
APPENDIX. ACCURACY O F EQUATIONS 6 A N D 16
Let us first compare Equation 6 with the estimate of 7r obtainable from Equations 1, 2, and 3. We call
NEI and LI'S (1979) method method
Z
and the new method methodZZ.
In method I, Sg = mg/mg is com- puted for all pairs of DNA sequences, where rfig = (mi+
m,)/2. Slj can be written as 1-
ag, so thatSince aq is generally small (<O. 15 in most cases) when DNA sequences from the same population are compared, we ignore the third and higher order
terms. T h e estimate of .rr is therefore approximately
1
=
G
[$
2 l i<j lalj
+
-
a;where ci and a; are the mean and respectively, and nT = n(n
-
1)/2.(A2)
Estimating Nucleotide Substitutions 879
In method 11, Equation 5 can be written in the following way, since m,/mij = 1
-
a,.If we note that
m
,
and a, are not correlated andm
,
= n+z, (A3) is approximately written as
?ilay
n7ln
s =
1-~
= 1
- a
(A4)Therefore, the estimate of 7r becomes
1
7rlI =
;
[- log,(l-
Z)]Comparison of (A2) and (A5) shows that the differ-
ence between
;,
and&,,
is approximately u,2/(2r). a, (or a,) is generally much smaller than c i 2 (or 5). In the case of common chimpanzees discussed earlier we have ua = 0.004 and 6 = 0.1 16. These values can be computed for pygmy chimpanzees as well, but the number of mtDNAs used for this species is only 3, sothat the estimates of u, and ci are not reliable. We therefore computed the values from BROWN and
GOODMAN’S (1979) and BROWN’S (1980) data for 2 1
human mtDNAs. T h e ua and ci values obtained for these data were 0.00004 and 0.017, respectively. Therefore, the difference between T I and ~ 1 seems 1 to be generally very small.
T h e accuracy of ~ X Y in equation ( 1 6) can be evalu-
ated by examining ~ X Y in ( 1 2) in the same way. Here
mx8yl can again be written as ? i t x y ( 1
-
a&, where ?itxy= (mx,
+
m,,)/2. In this case, U G can be large, particu-larly when X and Y represent different species. There- fore, the third and higher order terms in ( A l ) may not be neglected. However, ay’s are expected to be similar to each other between different qpecies. This can be seen from the near-equality of dq’s between common and pygmy chimpanzees in Table 4. T h e variation among aq’s is mainly due to stochastic errors. Therefore, Equation 16 and NEI and LI’S (1979)
previous method are expected to give essentially thesame result, as long as 2y.s between different spe- cies are large relative to 7r.
2