Copyright 0 1989 by the Genetics Society of America
The Isolation and Sequence of Missense and Nonsense Mutations in the
Cloned Bacteriophage
P22
Tailspike Protein Gene
John J. Schwarz' and Peter B. Berget'
Department of Biochemistry and Molecular Biology, University of Texas Medical School and Graduate School of Biomedical Sciences, Houston, Texas 77025
Manuscript received August 5 , 1988 Accepted for publication December 19, 1988
ABSTRACT
Twenty-seven new mutations in the structural gene for the Salmonella typhimurium bacteriophage P22 tailspike protein have been isolated, mapped using a powerful plasmid-based genetic system and their DNA sequence changes determined. The mutations were generated by hydroxylamine treatment of the cloned gene on a plasmid expression vector. Assaying the activity of the tailspike protein produced from this plasmid and screening for plasmid mutants were accomplished by the in situ complementation of P22 capsids imbedded in soft agar to produce infectious phage. Deletion mutations in the cloned gene have been constructed by a two step procedure involving oligonucleotide linker insertion and in vitro deletion by restriction endonuclease digestion. The deletions, whose physical endpoints were determined by DNA sequencing, define 12 genetic and physical intervals into which the new mutations were mapped by marker rescue experiments. These deletions were transferred to phage P22 by recombination and used to map mutations carried on plasmids. Following mapping, the nucleotide change for each of the mutations was determined by DNA sequencing. The majority were absolute missense mutations although both amber and ochre nonsense mutations were also identified in the protein coding portion of the gene. T h e suppression pattern of the nonsense mutations was determined on several nonsense suppressors. Four of the mutations cause severely depressed levels of tailspike protein expression from both the cloned gene on the plasmid expression vector and from P22 phage carrying these mutations. These mutations were identified as nucleotide changes in what is probably the P22 late operon transcription terminator which immediately follows the tailspike protein coding sequence.
A
combined biochemical and genetic approach is providing valuable in the detailed structural and functional analysis of a number of proteins. Prob- lems of protein structure and function subjected to this approach include DNA-protein interactions(SCHMITZ, COULONDRE and MILLER 1978), catalytic mechanisms (KNOWLES 1987), protein stability (ALBER
and WOZNIAK 1985; SCHMITZ, COULONDRE and
MILLER 1978), and protein folding (HURLE, TWEEDY
and MATTHEWS 1986; JENNESS and SCHACHMAN
1983).
T h e tailspike protein of Salmonella typhimurium phage P22 has many interesting features which make it a useful model system for this type of study. It is a structural component of the phage. During the last step of the assembly pathway, 6 tailspike protein tri- mers attach to the phage capsid with strong noncova- lent bonds (KING, LENK and BOTSTEIN 1973). T h e capsid, which is the product of the penultimate step in P22 morphogenesis, and tailspike protein are both easily isolated and the assembly reaction has been
' Current address: Boyce Thompson Institute, Cornell University, Ithaca,
e Current address: Department of Biological Sciences, Carnegie Mellon New York 14853.
University, Pittsburgh, Pennsylvania 15213. Genetics 121: 635-649 (April, 1989)
studied in vitro under a wide range of conditions
(ISRAEL, ANDERSON and LEVINE 1967). T h e factors which control this strong, precisely timed attachment between proteins in the capsid and tailspike protein are not well understood but they should be at least partially amenable to investigation by genetic ap- proaches. A clearer understanding of this assembly reaction should also lead to a better understanding of less accessible assembly reactions in other supramo- lecular structures. T h e tailspike protein is also an endorhamnosidase which binds to and cleaves a-rham- nosyl-1,3-galactose linkages in the 0-antigen on the surface of the host bacterium (IWASHITA and KANE-
GASAKI 1973). As such, it is a member of a large, well- characterized group of sugar binding proteins, many members of which have known crystal structures
( QUIOCHO 1 9 8 6).
T h e tailspike protein is a trimeric molecule com- posed of the 666 amino acid product of P22 gene 9
(GOLDENBERG, BERGET and KING 1982; SAUER et al. 1982). This gene has been cloned into a plasmid expression vector and the DNA sequence determined
(SAUER et al. 1982; BERGET, POTEETE and SAUER
636 J. J. Schwarz and P. B. Berget
mal denaturation and to protease digestion. T h e ther- mal stability of the mature protein is in marked con- trast to its thermal lability during folding and subunit assembly. T h e mature protein experiences only a 10- 20% decrease in enzymatic activity after a 5-min in vitro incubation at 80" (IWASHITA and KANEGASAKI
1976), whereas the amount of polypeptide forming stable trimers in vivo drops from 90% at 27" to only 15% at 42" (GOLDENBERG, BERGET and KING 1982). In vivo folding and subunit assembly of this protein are coupled reactions in which the polypeptide chains assemble into a protrimer before maturing into the final trimer form. These two trimer forms can be distinguished by their different mobilities on poly- acrylamide gels and by their differential sensitivities to trypsin digestion and to denaturation by 1% SDS at room temperature. The mature trimer is resistant to trypsin digestion and to SDS denaturation while the protrimer is sensitive to both (GOLDENBERG and KING 1982).
An extensive collection of absolute lethal and con- ditional mutations have been isolated in gene 9 (SMITH, BERGET and KING 1980; FANE and KING
1987; BERGET and CHIDAMBARAM 1989). Because
gene 9 is an essential gene, the original genetic analysis of this protein utilized conditional mutants. T h e most thoroughly studied of these are the temperature-sen- sitive mutants. These mutants have nearly normal thermal stabilities when they mature at permissive temperature but fail to form trimers when maturation is carried out at the nonpermissive temperature. They are therefore defective in protein folding (GOLDEN- BERG, SMITH and KING 1983). T h e entire set of tem- perature sensitive mutants has recently been se- quenced and the mutations are all located within the middle third of the protein's primary structure be- tween residues 141 and 493 (VILLAFANE and KING, 1988). Many amber mutants also have a temperature sensitive phenotype when grown on at least one sup- pressing strain. These are apparently also defective in folding at the non-permissive temperature and they have been mapped genetically into the same region of the protein as the other temperature sensitive mutants (FANE and KING 1987). Although the analysis of mu- tants with a conditional lethal phenotype has been valuable, especially for the investigation of protein folding, they represent a limited class of mutations in which certain expected types of mutants such as those which affect attachment to the phage capsid or endor- hamnosidase activity have been absent or underrep- resented. In fact only one conditional mutant defec- tive in any property aside from folding has been isolated. This is an amber mutant which produces a endorhamnosidase defective protein when grow on a particular suppressor strain (BERGET and POTEETE
1980). In addition it is unlikely that all mutations
affecting a process as complex as folding of a large multisubunit protein would produce only a tempera- ture sensitive phenotype or all be located in one part of the protein. For these reasons a genetic analysis employing absolute lethal tailspike protein mutants isolated both from the phage (BERGET and CHIDAM-
BARAM 1989), and from the plasmid clone of the tailspike protein gene has been undertaken. This analysis is possible because of the ability to comple- ment absolute lethal phage tailspike protein mutants with purified tailspike protein in vitro. This allows one to propagate absolute lethal mutants by supplement- ing either solid or liquid media with purified tailspike protein.
This paper describes the construction of a plasmid- based genetic system to facilitate the fine structure genetic analysis of this protein and its use to isolate and determine the nucleotide changes of 27 tailspike protein mutants. This system relies on a simple plate complementation assay to isolate mutations in the plasmid cloned version of gene 9. These mutations have been located to 12 deletion intervals using phage deletion mutants. T h e precise nucleotide change has been determined by DNA sequencing using a set of 10 oligonucleotide primers whose complementary se- quences are spaced at approximately 200 base inter- vals throughout gene 9. Significantly, the absolute lethal missense mutations which also prevent stable trimer formation
u.
J. SCHWARZ and P. B. BERGET, unpublished data) are preferentially located in the carboxy-terminal region of the protein which is devoid of mutations causing the temperature-sensitive phe- notype.MATERIALS A N D METHODS
Phage strains and procedures: T h e phage strains used and created in this study are shown in Table 1. General phage P22 procedures have been described (SUSSKIND, WRIGHT and BOTSTEIN 197 1; BOTSTEIN, CHAN and WAD DELL 1972). Lambda plates and soft agar are described in SIGNER and WEIL (1968). Phage strains that cannot make functional tailspike protein (tailspike dependent) were prop- agated in liquid culture by addition of purified tailspike protein at 10'' phage equivalents/ml and on plates by ad- dition of 10" phage equivalents of tailspike protein to the soft agar overlay. Phage equivalents of tailspike protein was determined as previously described (ISRAEL, ANDERSON and LEVINE 1967; BERGET and POTEETE 1980).
P22 capsids (9- particles) were made either by induction of a gene 9 deletion mutant prophage or by a single cycle of infection by a gene 9 deletion mutant in liquid culture. Induction was performed by growing a DB7000 lysogen of
637 P22 Tailspike Protein
TABLE 1
Phage P22 strains
Strain Genotype Reference
P22 Ap35 gene 9::Tnl(Ap35), sieA44, TR- WEINSTOCK (1 977) P22 Ap68tdpfrlO8 gene 9::Tnl(Ap68), sieA44, T R + YOUDERIAN a n d
SUSSKIND (1 980) P22 Dl P22 9A(-38 t o 69), sieA44 This study
P22 D2 P229A(72 t o 427), sieA44 This study
P22 D3 P22 9A(229 t o 510), sieA44 This study
P22 D4 P22 9A(430 t o 601), sieA44 This study
P22 D5 P22 9A(513 t o 724), sieA44 This study
P22 D6 P22 9A(604 t o 979), sieA44 This study
P22 D7 P22 9A(7 13 t o 999), sieA44 This study
P22 D8 P22 9A(982 t o 151 3), sieA44 This study
P22 D9 P22 9A(998 to 1434), sieA44 This study
P22 D l 0 P22 9A(1422 to 1513), sieA44 This study
P22 Dl 1 P22 9A( 15 19 to 1826), sieA44 This study
P22 D l 2 P22 9A(1752 to 2058), sieA44 This study
P22 hmH5 P22 9 - (Q59 amber), sieA44 This study
P22 hmH19 P22 9- (W640 amber), sieA44 This study
P22 hmH7, hmH30-3-3 P22 (G to A a t 201 7), sieA44 This study
P22 hmH3-5 P22 (G t o A a t 2019), sieA44 This study
P22 hmH3O-1-1 P22 (C t o T a t 2029), sieA44 This study
by infecting DB7000 with P22 Ap68tdpf1-108 at a multiplic- ity of 5 phage per cell. After 90 min of incubation with aeration the cells were lysed with a few drops of CHC13. Removal of cell debris and concentration of the capsids were by the same procedure as for the induced prophage. Induc- tion of a prophage produces around 2 X 10' capsids from 40 ml of culture, whereas infection produces around 5 X
10" capsids from 40 ml of culture.
Lysates of P22 Ap35, which has the transposon T n l inserted into gene 9, were prepared by inducing the pro- phage in the manner described above. Tailspike protein was attached to the phage capsid by adding tailspike protein to the phage lysate to a concentration of 10" phage equiva- lents/ml and incubating at 37" for at least 2 hr. The phage were then concentrated as described above. DNA was iso- lated from P22 by the method used for phage X (MANIATIS, FRITSCH and SAMBROOK 1982).
Bacterial strains and procedures: Bacterial stocks for plating P22 were made by growing DB7000 in LB broth to approximately 2 x 10' cells/ml, pelleting by centrifugation, and resuspending them in LB broth at 1/10 the original volume. Competent cells for transformation were prepared by the CaC& method (DAVIS, BOTSTEIN and ROTH 1980). T h e strain Escherichia coli RR1 was the recipient strain for most of the recombinant DNA work (Table 2).
Plate complementation assay of tailspike protein activ- ity: Colonies of bacteria containing plasmids which express gene 9 are assayed for functional tailspike protein by trans- ferring them with toothpicks to a lambda agar plate that has 2.5 ml of soft agar overlay containing 10' P22 capsids and 2 X 10' DB7000 cells. The plates are then incubated over- night at 30". Tailspike protein is supplied to the embedded capsids by the transferred colonies through release into the surrounding agar, presumably through cell death. When functional tailspike protein attaches to P22 heads they infect and lyse the surrounding bacteria to form a large plaque or
halo around the colony being tested. T h e efficiency at which this infectious process occurs depends on the amount of tailspike protein and its functional activity. If the amount of cells transferred to the indicator plate is kept roughly con-
stant between colonies, the size of the halo is a crude indicator of the quantity or specific activity of tailspike protein produced.
General recombinant DNA techniques: Small scale plas- mid preparations were made by the procedure of BIRNBOIM and DOLY (1 979). Large scale purification of plasmid DNA was by the CsCl/ethidium bromide density gradient method (DAVIS, BOTSTEIN and ROTH 1980). Purification of DNA from low melting point agarose gels was by the method in MANIATIS, FRITSCH and SAMBROOK (1982). T E is 10 mM Tris and 1 mM EDTA, pH 8.0. Restriction enzymes, Klenow fragment of DNA polymerase I , S1 nuclease a n d T 4 ligase were purchased from New England Biolabs, BRL Inc., and Boehringer Mannheim and used according to the manufac- turers' recommendations.
Construction of pJS27 and pJS28: T h e tailspike protein gene, gene 9, was originally cloned into pPB13 and ex- pressed from the lacUV5 promoter (BERGET, POTEETE and SAUER 1983). This plasmid contains the origin of plasmid replication and the ampicillin resistance gene of pBR322 and approximately 4 kb of P22 DNA. T h e plasmid pJS27 was derived from this plasmid through removal of roughly 2 kb of P22 DNA from an AluI site near the 3'-end of gene 9 to the PuuII site which is the junction of pBR322 and P22 DNA (Figure 1). There are several AluI sites in pPBl3, so
this deletion was made by performing a partial digest with
AluI in the presence of ethidium bromide (PARKER, WATSON
638 J. J. Schwarz and P. B. Berget
TABLE 2
Bacterial strains
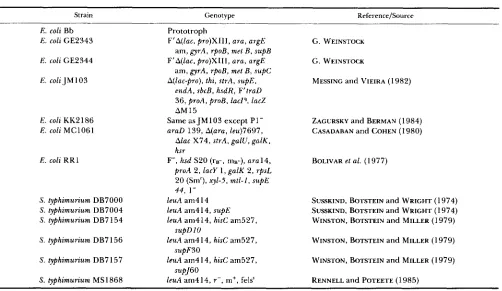
Strain Genotype Reference/Source
E. coli Bb
E. coli GE2343
E. coli GF.2344
E . coli J M 103
E. coli KK2 186
E . coli MC1061
E . coli R R l
S. typhimurium DB7000
S. typhimurium DB7004
S. typhimurium DB7 154
S. typhimurium DB7 156
S. typhimurium DB7 157
S. typhimurium MS 1868
Prototroph
F’A(lac, pro)XIII, ara, argE
F’A(lac, pro)XIII, ara, argE
A(1ac-pro), thi, strA, supE,
am, gyrA, rpoB, met B , supB
am, gyrA, rpoB, met B , supC
endA, sbcB, hsdR, F’traD
36, proA, proB, l a c P , lacZ
AM15
Same as JM103 except P1-
araD 139, A(ara, leu)7697,
Alac X74, strA, galU, galK, hsr
F-, hsd S20 ( r B - , mB-), a r a l 4 , proA 2, lacy 1 , galK 2, rpsL
20 (Smr), xyl-5, mtl-I, supE
44, 1-
leuA am4 14
leuA am4 14, supE leuA am414, hisC am527,
leuA am414, hisC am527,
leuA am414, hisC am527,
leuA am414, r - , m+, felss supD 10
supF3O
supJ60
G. WEINSTOCK
G. WEINSTOCK
MESSING and VIEIRA (1 982)
ZACURSKY and BERMAN (1 984) CASADABAN and COHEN (1 980)
BOLIVAR et al. (1 977)
SUSSKIND, BOTSTEIN and WRIGHT (1974) SUSSKIND, BOTSTEIN and WRIGHT (1 974) WINSTON, BOTSTEIN and MILLER (1 979)
WINSTON, BOTSTEIN and MILLER (1979)
WINSTON, BOTSTEIN and MILLER ( 1 979)
RENNELL and POTEETE (1985)
plasmid DNA for sequencing and oligonucleotide mutagen- esis by infecting with a filamentous helper phage and puri- fying the DNA from phage capsids (ZAGURSKY and BERMAN
1984).
Construction of gene 9 deletions in plasmids: Twelve gene 9 deletions were made for use in deletion mapping. These deletions were made in plasmids and then moved onto phage. T h e plasmid deletions were made by essentially the same two-step procedure as used by SHORTLE (1 983) for construction of deletions in the staphylococcal nuclease gene. In the first step an 8-base HindIII linker, 5’ CAAGCTTG 3‘ (Collaborative Research, Inc.) was inserted into gene 9 at various positions to make plasmids containing a single HindIII site. Insertion of the linker into gene 9 should in general inactivate gene 9 . Therefore plasmids with HindIII linker insertions in gene 9 were identified as trans- formants which had lost tailspike protein activity in the plate assay. The HindIII insert into the AluI site beginning at nucleotide 205 1 was isolated in the screen for transformants which had lost tailspike protein activity. This insertion is outside of gene 9 and in subsequent plate assays was found to not inactivate the gene.
The location of the HindIII linker insertion depends on the method used to linearize the plasmid. A number of methods for linearizing the plasmid were used in order to obtain insertions at useful positions throughout the gene. Most insertions were made semirandomly by partial diges- tion of plasmid DNA and either AluI, HaeII, or Hue111 in the presence of ethidium bromide (PARKER, WATSON and VINOGRAD 1977) followed by purification of full length linearized plasmids from low melting point agarose gels. T h e linear plasmid molecules were then circularized in the presence of the linker with T 4 DNA ligase. Two insertions were made by cleavage of the plasmid at random positions
by treatment with DNase I to make a single-stranded break in the phosphodiester backbone of the plasmid (GREEN- FIELD, SIMPSON and KAPLAN 1975) and linearizing the plas- mid at this break with S1 nuclease. A linker was also inserted into either the unique NcoI or BamHI restriction site in gene 9 by digesting the plasmid with the appropriate en- zyme, filling in single stranded ends with the Klenow frag- ment of DNA polymerase I, and ligation in the presence of the HindIII linker. A Hind111 site was created in P22 sequences 5‘ to the start of gene 9 at position -39 from the first codon of the mature protein by oligonucleotide di- rected mutagenesis using the oligonucleotide 5’
CCGTAGCCAAGCTTCGGCAATTCC 3‘. This and all
other oligonucleotides were made on an Applied Biosystems Model 380A DNA synthesizer unless specifically indicated and were purified by the procedure recommended by the manufacturer. The mutagenesis was done by the method of ECKSTEIN (TAYLOR, OTT and ECKSTEIN 1985) with reagents and procedures supplied by Amersham. The locations of the unique HindIII sites in all gene 9 containing plasmids are shown in Figure 2A.
P22 Tailspike Protein 639
A Eco R1
n G e n e9
Eco R1 Ecp R1
baGaeg
.-%:o
Nde I N d e I
Eco R1
N& I
FIGURE 1.-Construction of pJS27 and pJS28. The plasmid pJS27 was derived from pPBl3 by partial digestion with A h 1
followed by digestion to completion with PvuII, and ligation. The plasmid pJS28 was constructed by digestion of pJS27 and pZ152 with EcoRI and NdeI, mixing the digested plasmids together, and ligation. The M I 3 origin of DNA replication and single strand packaging is indicated by the anti-clockwise arrow on pZ152 and pJS28. The arrow indicates the 5' + 3' polarity of the packaged single strand. The smaller arrowhead indicates the location of the lacUV5 promoter found in plasmids pPB13, pJS27 and pJS28.
BamHI site in gene 9 of a plasmid which already carried a HindIII linker to make a plasmid with two HindIII sites. T h e region between the HindIII sites was then deleted by digesting with HindIII and ligating under dilute conditions which favor intramolecular ligation. T h e deletion D l 1 was made by digesting plasmid DNA with BamHI and HpaI, filling in the single stranded BamHI ends with Klenow fragment of polymerase I, and ligating the plasmid under dilute conditions in the presence of HindIII linkers.
Transfer of deletions from plasmids to phage: Deletions were transferred from plasmids to phage by recombination between P22 Ap35 and the desired plasmid. P22 Ap35 carries a T n l element in gene 9 and thus cannot form plaques even in the presence of tailspike protein because the phage genome is oversized and thus lacks terminal redun- dancy when packaged into particles. Replacement of the T n l element with the deletion by in vivo recombination restores terminal redundancy and allows the phage to form plaques on plates with tailspike protein. S. typhimurium MS1868 containing a plasmid with a gene 9 deletion was grown to approximately 2 X 10' cells/ml and infected with
I I I I I
1 500 1000 1500 2000
B
Eco R1
+
Eco R1
Hin
dm
/
E d R1FIGURE 2.-Construction of plasmid deletions. A , Location of HindIII sites created in gene 9 are indicated by vertical lines above the gene. These include the location of theHindIl1 linker insertions into both pPBI3 and pJS27; and the HindIII site in pJS28 created by oligonucleotide mutagenesis. B, Gene 9 deletions were con- structed by digesting two plasmids with HindIII sites at different locations in gene 9 with Hind111 and another enzyme that cuts only once in these plasmids, usually EcoRI. The fragments comprising the 3' and 5'junctions of the deletion were isolated from agarose gels and ligated together.
P22 Ap35 at a multiplicity of infection of 10. T h e infection was allowed to continue for 2 hr at 30" after which the cells were lysed with CHCls and the cell debris pelleted by centrifugation in a microfuge. T h e lysate was plated on DB7000 with tailspike protein. T h e plaques that arose were replica plated using toothpicks onto a plate with a lawn of DB7000 and a similar plate which also contained tailspike protein to identify those which were tailspike dependent. T h e tailspike dependent phage were presumed to carry the gene 9 deletion and were further tested. The endpoints of deletions Dl and D2 had been sequenced prior to being moved to phage, so the transfer of these deletions to phage was confirmed by digesting the phage DNA with HindIII and observing the presence of new restriction fragments of the expected size. The other plasmid deletion endpoints had not been sequenced prior to transferring to phage so they were sequenced after moving them to the plasmid pJS28 as described below.
Transfer of deletions from phage to the plasmid pJS28:
640 J. J. Schwarz and P. B. Berget
multiplicity of 5 phage per cell. The transduction was done by incubating 0.2 ml of DB7000 plating bacteria with 0.1 ml of the lo-’ and 1 0-4 dilutions of the transducing lysate for 10 min at 37”. This mixture was then spread onto the selection plates. T h e cells which received the plasmid were selected for on green indicator plates (CHEN et al. 1972) containing 150 rg/ml ampicillin and 10 mM EGTA (WEIN-
STOCK, SUSSKIND and BOTSTEIN 1979) and incubated at 37 ”
.
Green indicator plates contain (per liter) 8 g tryptone, 1 g yeast extract, 5 g NaCI, 15 g agar, and 21 ml of 40% glucose, 25 ml of 2.5% (w/v) Alizarin Yellow G, GG (MATHESON, COLEMAN and BELL) and 3.3 ml of 2% (w/v) water-soluble Aniline Blue (BDH Chemicals). Stably lyso- genized cells produce large light green colonies on such plates while unstable lysogens or phage-infected colonies form small dark green colonies (SMITH and LEVINE 1967). T h e ampicillin-resistant colonies were pooled and used to make a rapid plasmid DNA preparation. This plasmid DNA was used to transform E. coli MC 106 1 or KK2 186 to ampi- cillin resistance. These colonies were then screened for tailspike protein production in the plate assay. Plasmids from cells that did not make functional tailspike protein were presumed to carry the gene 9 deletion. This was confirmed by demonstrating the presence of a Hind111 site in these plasmids and subsequent DNA sequencing. The extent of the missing DNA in these deletions has been determined by DNA sequencing.
Hydroxylamine mutagenesis of plasmih CsCl gradient purified plasmid DNA was mutagenized by a modification of the procedure described by HUMPHREYS et al. (1976). T h e only difference from that procedure was the DNA was incubated in the hydroxylamine solution for 30 min at 65” followed by dialysis against two changes of 1000-fold excess of T E before transformation into E. coli Bb, MC106 1, or JM 103.
In vivo deletion mapping: Deletion mapping of mutant plasmids was done by two procedures-a spot test in which the progeny of recombination were not isolated and a sen- sitive marker rescue test in which the progeny of the crosses were isolated and tested. T h e tailspike dependent phage which were used in these mapping procedures had tailspike protein added to them exogenously so that they could infect cells once. The spot test was done by transferring approxi- mately lo6 deletion phage in 10 rl onto a plate containing S. typhimurium MS1868 carrying the mutant plasmid in the soft agar. All of the different phage deletion strains were transferred at one time from a microtiter dish using a multiple prong applicator from West Coast Scientific. T h e plates were then incubated overnight at 30”. Because the phage deletion strains do not revert to tailspike independ- ence at a detectable rate, positive marker rescue was scored by the presence of any plaques in the spots. Mapping ambi- guities caused by failure to marker rescue with nonoverlap- ping deletion intervals were usually resolved by spotting a larger number of phage using a pipet. Marker rescue was only detected in the cross between Dl and hmH4-1 by plating 10-fold more phage than usual. Sensitive marker rescue was done in liquid culture. S. typhimurium MS1868 carrying the mutant plasmid was grown to mid-log phase and infected with phage carrying each of the gene 9 dele- tions at a multiplicity of 5 phage per cell. T o ensure that all of the progeny were infectious, tailspike protein was added to the culture to a final concentration of 5 X 10” phage equivalents/ml. After a 90-min incubation at 30” the cells were lysed with CHCI, and the cell debris removed by a 10- min centrifugation in a microfuge. The lysate was diluted and 0.1 ml of a 100-fold dilution was plated on DB7000
without the addition of tailspike protein to detect tailspike independent recombinant phage.
In vitro heteroduplex deletion mapping: The mutation hmH 10 has a dominant negative phenotype, so it is difficult to map using deletions carried on phage because mutant protein produced in large quantities from the plasmid can attach to the wild-type recombinant phage to make them noninfectious. We therefore mapped this mutation by the heteroduplex mapping protocol of SHORTLE ( 1 983) in which a heteroduplex is formed between a single strand from plasmid containing the mutation and a single strand from a plasmid containing a deletion. This heteroduplex is made in vitro by denaturing a mixture of the two plasmid mole- cules linearized at different restriction enzyme sites, allow- ing the linear molecules to renature to form both parental linear molecules and circular heteroduplexes. This plasmid DNA mixture is transformed into competent cells where the heteroduplexes are resolved in v i v o . The linear molecules transform with such a low frequency that generally only circular heteroduplexes are successfully introduced into the recipient cells. If the mutation is not within the deleted region then both mutant and wild-type plasmids will result from the heteroduplex depending on the strand choice during mismatch repair. In contrast, if the mutation is within the deleted interval only plasmids carrying the mutation will result.
In preparation for heteroduplex mapping the mutation hmH10 was moved from pPBl3 on which it was isolated to pJS28 in an EcoRI to HpaI fragment which contains about 90% of the tailspike protein gene. This was done by digest- ing pPBl3 hmHlO and pJS28 with EcoRI and HpaI, isolat- ing the fragments from a low melting point agarose gel, and ligating the fragments with T 4 ligase. T h e recombinant plasmid was tested for tailspike protein activity with the plate assay to determine if the mutation was transferred on the EcoRI to HpaI fragment. Because this plasmid failed to produce a halo in the plate test it was presumed to contain the hmH 10 mutation. Heteroduplexes were made between this plasmid and selected gene 9 deletions carried on pJS28 by the procedure of SHORTLE ( 1 983). After transformation of the heteroduplex reactions into MC1061 the ampicillin resistant colonies were assayed for functional tailspike pro- tein (wild-type recombinants) with the plate assay.
DNA sequencing: DNA sequencing was done by the dideoxy method (SANGER, NICKLEN and COULSON 1977) using the following 10 oligonucleotide primers: P1 ( 5 ‘
GCCGGTA 3’), P3 (5’ ATTGCAGGATGCAGC 3‘), P4
TAGGGGTCG 37, P6 (5’ CAGTAAGTAGCGCCCA 3’),
CACCCACGA 3’), P9 (5’ GGTAGACCCCTCTAGA 3’),
P10 (5’ GCACACCTGACGCTG 3’). These oligonucleo- tides are complementary to gene 9 DNA located at approx- imately 200 base intervals along the gene. DNA sequencing from these primers is in the same direction as gene 9
transcription.
Mutations were isolated in two plasmids, pPB13 and pJS28. T h e plasmid pPB13 does not have the M13 origin,
so before mutations in this plasmid were sequenced they were either subcloned into pJS28 or the M13 origin was inserted into the plasmid carrying the mutation. T h e choice of procedure depended on the location of the mutation with respect to restriction enzyme sites that could be used for subcloning. Single stranded plasmid DNA was produced by infection of E. coli KK2186 containing the mutant plasmid with phage IR1 according to the method of DENTE, CB-
ARENI and CORTESE (1983). DNA sequencing was done
GCGGCAATTCCTTGC 3’) P2 (5’ ATCAACGCA-
(5’ CAAGCCTTGGACGGA 3’), P5 (5’ AATGTA-
P22 Tailspike Protein 64 1
D3
D4
D5 D6 D7 D8
D9
Dl 0 Dl 1 Dl2 pMC2 P M G pMC26 pMC26 pMC6 pMC12 pMC9 pMC13 pMCl0 pMCl5 pMC16 pMCl8 pMC25 pMC21 pMC23
Gene 9 Thr-1 Leu-666
Dl Ala-24
D2 LyS-23
-
Gly-143 Asn-75-
Ala-171Asp-142 pro-201 Lye170 Phe-242
Thr-200 Ser-327
Lys-241 Ala-334
Thr-326 Gly-505
ser-333 Asp479 Phe-472 Gly-505
Ile-506 HiS-601
Gly-582
7
-
Ala-90La-214 LyS-225
ne-245
lle-387 Asp399
Lvs-631 Lvs-651
FIGURE 3.-Deletion map of gene 9. The bars in this diagram represent the extent of gene Y DNA present in each of the deletions reported in this paper drawn to scale. In addition, the deletions previously reported (BERGET and CHIDAMBARAM 1989) are indi- cated for comparison. The first line represents the entire gene Y sequence. The complete codons which are near- est the endpoints of the deletions are indicated above each bar. The dele- tions D l through D l 2 exist on both plasmids and phage (Table 1).
using rea ents and procedures provided by BRL, Inc. The
32P and
k
S dATP were supplied by New England Nuclear.RESULTS
Gene 9 deletions: HindIII octomer linker insertions into gene 9 and flanking sequences were made in plasmids pPB13 and pJS27 and pJS28 as described in MATERIALS AND METHODS. Recombinant DNA tech- niques were used to combine fragments of these plas- mids to generate deletions in gene 9 and flanking sequences whose endpoints are marked by a HindIII site in the resulting plasmids. T h e location and extent of these deletions (Dl through Dl 2) is represented in Figure 3 along with the deletions previously reported
(BERGET and CHIDAMBARAM 1989). Each of the dele- tions was crossed onto P22 phage; and those deletions which did not originate on the plasmid pJS28 w e r e subsequently crossed from the resulting deletion
phage onto pJS28 in preparation for sequencing as described in MATERIALS AND METHODS. Each P22 phage which carries a deletion in gene 9 along with its genotype given as the extent of gene 9 DNA deleted is listed in Table 1. T h e deletions were pre-
cisely located by sequencing across the deletion end- points in the plasmid version of each deletion. T h e deletion D9 has 160 bp of pBR322 DNA inserted into gene 9 between the HindIII linker and the 3‘ region of gene 9 DNA. The inserted DNA is composed of 2 segments of pBR322: the segment nearest the 5’ end of gene 9 is composed of DNA from position 2580 to 2550 and the 3’ segment is from 3713 to 3841 in pBR322 (SUTCLIFFE 1979). T h e arrangement is such that pJS28 D l 0 contains an inverted repeat of the 5’ segment of pBR322 DNA and a direct repeat of the
3’ segment. This insertion of DNA occurred when the HindIII linker insertion that forms the 3’junction of Dl 0 was constructed. T h e plasmid was linearized by successive treatment with DNAse I and S1 nuclease which evidently created many small DNA fragments that were ligated into the linearized plasmid along with the HindIII linker. This insertion went unnoticed until the deletion junction was sequenced. Some of the surrounding DNA sequence and the HindIII linker which marks the position of the deletion is shown for each deletion in Table 3.
642 J. J. Schwarz and P. B. Berget
TABLE 3
Sequence of deletion junctions"
Dl
D2
D3
D4
D5
D6
D7
D8
D9
______
1-39 170
. . .
CGTAGCCAAGCTTGCTGTTGC...
171 1428
. . .
TTAAAGCAAGCTTGCCTTCTT. . .
1228 1511. . .
CAACGGCAAGCTTGCTAAATT.. .
1429 1602. . .
TGATGGCAAGCTTGCATGGGT. . .
1512 1725. . .
GTAAAGCAAGCTTGTCCCAGG. . .
1603 1980. . .
ACACCACAAGCTTGCTAT GGG...1712 11000
. . .
TAAGCGCAAGCTTGGCCCAGT.. .
1981 11514. . .
AACCAGCAAGCTTGGATCCGC.. .
1997 11435. . .
TAAGTACAAGCTTG***GACAC. . . *
11421 11514Dl0
. . .
TACCAACAAGCTTGGATCCGC. . .
I1518 11827Dl 1
. . .
TGGGATCAAGCTTGCCACAAA...
11751 12059Dl 2
. . .
GGGGGGCAAGCTTGCTAATTA. . .
a The numbering system is such that number 1 corresponds to
the first nucleotide of the first codon of the N-terminal amino acid (Thr) in the mature protein. The numbers above each sequence refer to the last 3' nucleotide and the first 5' nucleotide of the sequence which is identical to the phage P22 sequence flanking the deletion. The Hind111 linker sequence is underlined.
'
( * * * ) indicates an unexpected insertion of 160 bases of pBR322 DNA (see text for description).and pJS28 plasmid DNA was mutagenized with hy- droxylamine and then transformed into three differ- ent E. coli strains (Bb, JM103 and MC1061) as de- scribed in MATERIALS AND METHODS. T h e purpose of trying different strains was to find the strain in which tailspike protein mutants with low amounts of activity could most easily be distinguished from wild type in the plate assay. T h e strain MC 106 1 proved to be the most reliable indicator of tailspike protein activity simply because this strain produces the largest size halo with the wild-type gene. This is probably due to the fact that the lac repressor gene is deleted in this strain so expression of protein from the plasmid lac UV5 promoter may be higher than in strains which contain the lac repressor. T h e transformants were screened by the plate assay for halo production as described in MATERIALS AND METHODS. Twenty-seven independent mutants were isolated from a total of 13 10 colonies screened. Four of these, hmH7, hmH3- 5, hmH3O-1-1 and hmH30-3-3 generate smaller than normal size halos compared to wild type while the rest reproducibly generate no halos at all.
Deletion mapping of mutants: The mutant plas- mids were moved by transformation from the E . coli strains in which they were isolated into S. typhimurium MS 1868 in preparation for deletion mapping. For the majority of the mutants the rapid spot test described in MATERIALS AND METHODS was sufficient to locate the mutants to within a given deletion interval.
Seven of the mutants (hmH4, hmH7, hmH3-3,
hmH3-5, hmH3O-1-1, hmH30-2-3 and hmH30-3-3) could not be mapped with the spot test because cell lysis occurred in crosses with phage carrying each of the deletions. Because four of these mutants (hmH7, hmH3-5, hmH30-1-1 and hmH30-3-3) complement phage capsids in the plate complementation assay by making small halos as described above, it was thought that weak complementation was occurring in at least the case of these four mutants to obscure the marker rescue results. Therefore sensitive marker rescue ex- periments were done to determine the phenotype of the progeny under conditions where complementa- tion is not observed. Sensitive marker rescue was done by crossing these seven mutants in liquid culture with phage carrying deletions and testing the progeny for their ability to grow on plates containing S. typhimu- rium but no tailspike protein. Two of the mutants,
hmH3-3 and hmH30-2-3, produced phage which
were tailspike-independent for plaque formation when crossed with all deletions but Dl 1 where no tailspike-independent plaques were produced. These plasmid mutations must have produced tailspike pro- tein which could weakly complement deletion phage in the spot test but were unable to produce enough to be detected in the plate assay. Four mutations (hmH7, hmH3-5, hmH30-1-1 and hmH30-3-3) pro- duce progeny phage which make very small plaques when crossed with the deletion Dl 2. When crossed with all other deletions the progeny phage produce a mixture of very small and normal size plaques. So
these mutants have a small plaque phenotype when they are on phage and are located within the region of Dl 2 that does not overlap D l 1. All of the small plaque phenotype mutants, hmH7, hmH3-5, hmH30- 1-1 and hmH30-3-3 are able to complement phage capsids extracellularly in the plate assay to make small halos. Crosses between all of the deletions and the mutant hmH4 produced plaques of normal size. Be- cause the mutations which make small halos when on plasmids also make small plaques when on P22, it is unlikely that a mutant such as hmH4 which does not make a halo would make a normal size plaque when crossed onto P22. Therefore the most likely location of a plasmid mutation with a phenotype such as hmH4 is in the lac UV5 promoter region rather than in P22
DNA. DNA sequencing later showed that hmH4 is a
P22 Tailspike Protein 643
same site as the C A promoter down mutation L157 (REZNIKOFF and ABELSON 1978).
It was difficult to map conclusively the mutation hmHlO using the phage deletions because the fre- quency of wild type phage produced in both the spot test and the sensitive marker rescue test was very low. As will be more fully discussed in a later paper, the mutation hmH 10 produces a tailspike protein which attaches to phage capsids but is defective in the en- dorhamnosidase activity which is required for infec- tion. It is likely that the protein produced by the mutant hmH 10, which is overproduced from the plas- mid, attaches to the progeny phage capsids generated in the mapping crosses to make them noninfectious regardless of their genotype (a dominant negative phenotype). This mutation was therefore mapped us- ing the plasmid-based heteroduplex deletion mapping procedure of SHORTLE (1983) as described in MATE- RIALS AND METHODS. T h e recombination products of this mapping protocol are clonally isolated plasmids residing in individual colonies. These plasmids are assayed independently for production of functional tailspike protein in the plate assay. Therefore there should be no competition between mutant and wild type proteins for phage capsids to complicate the interpretation of the mapping results. T h e results from the heteroduplex mapping of hmH10 indicate that hmH 10 maps to deletion intervals D8 and D 10. With the exception of hmH4, each of the mutations isolated on the cloned gene 9 could be unambiguously located genetically in one of the deletion intervals described above. T h e results of the deletion mapping experiments are summarized in Table 4.
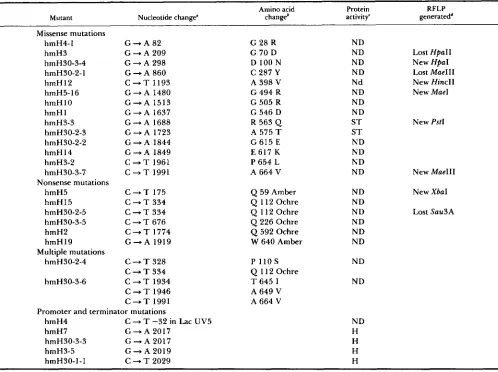
DNA sequence of hmH mutants: Once the muta- tions had been located within a deletion interval of known physical size, the exact nucleotide change for each mutation was determined by DNA sequencing using the appropriate primers described in MATERIALS A N D METHODS. For each mutation the entire interval in which the mutation was genetically located was sequenced to insure that every nucleotide change within that region was found. T h e nucleotide and inferred amino acid changes of all the mutants are listed in Table 5. All of the changes are the expected outcome of the deamination of cytosine by hydroxyl- amine. Even though the mutations hmH15 and
hmH30-2-5, and hmH7 and hmH30-3-3 have the same nucleotide changes they are independent iso- lates.
Suppression of nonsense mutations: Several of the hmH mutants contain nonsense mutations. Suppres- sion of these mutations by different suppressor strains provides information on the suitability of different amino acids at a particular position. T h e ochre mu- tations were suppressed by moving the plasmids with these mutations into the two suppressor strains
GE2343 and GE2344 which insert Gln and Tyr, re- spectively, at the ochre codon. T h e tailspike protein activity was tested by the plate assay. T h e results for ochre suppression were fairly uniform (Table 6). Al- though the halo size was small, there was no distin- guishable difference between the wild-type glutamine and the tyrosine substitution at codons 11 2, 226 and 592. T h e nonpermissive temperature for tempera- ture-sensitive tailspike protein mutants is 39". How- ever, the halo size of suppressed ochre mutants was almost imperceptibly small at 39", even when the correct amino acid was being inserted, so it was not possible to compare the effect of different amino acid insertions at the nonpermissive temperature. T h e for- tuitous occurrence of a proline to serine change at codon 1 10 in combination with the ochre mutation at codon 112 in hmH30-2-4 made it possible to test the effect of Pro 1 1 OSer both by itself and in combination with Glnl 1 2Tyr. Neither of the changes affects the protein to an extent that can be detected by this assay. Because there is an extensive set of amber suppressors in S. typhimurium and the level of suppression of amber mutations on phage is easier to determine than on plasmids, hmH5 and hmH19 were crossed onto P22 and tested on these suppressor strains. T h e transfer of the amber mutations to P22 was confirmed by crosses between the presumed P22 amber mutants and the identical allele on the plasmid, as well as with nearby hmH mutations. Backcrosses to plasmids car- rying the identical allele failed to produce tailspike independent phage above the background reversion level, whereas recombination to produce wild type was detected for plasmids with nearby mutations. In addition, because hmH5 creates a new XbaI site, its transfer was confirmed by restriction enzyme diges- tion of isolated phage DNA. T h e phage carrying the amber mutations were then plated on the different suppressor strains and incubated at either 30" or 39" to assess the effect of different amino acid substitu- tions. T h e results for amber suppression are more diverse than for ochre suppression. T h e requirement for T r p at codon 640 is stringent because four other amino acids at this site failed to produce infectious phage particles above the level of reversion of the amber mutation (Table 6). In contrast, insertion of the amino acids Ser, Leu and Tyr at codon 59 pro- duced functional infectious phage at about the same level as insertion of the wild-type amino acid, Gln, at both 30" and 39".
644 J. J. Schwarz and P. B. Berget
TABLE 4
Deletion mapping of gene 9 mutations'
Deletion intervals
D l D2 D3 D4 D5 D6 D7 D8 D9 D l D l 0 1 D l 2
Spot testb Mutant
hmH4-1
+
-+
+
+
+
+
+
+
+
+
hmH5
+
+
+
+
+
+
+
+
+
+
+
+
hmH3
+
+
+
+
+
+
+
+
+
+
hmH30-3-4
+
-
-+
+
+
+
+
+
+
+
+
hmH30-2-4
+
- -+
+
+
+
+
+
+
+
+
hmH 15
+
-
-
+
+
+
+
+
+
+
+
+
hmH30-2-5
+
--
+
+
+
+
+
+
+
+
+
hmH30-3-5
+
+
+
+
- -+
+
+
+
+
+
hmH30-2-1
+
+
+
+
+
--
+
+
+
+
+
hmH12
+
+
+
+
+
+
+
-
-+
+
+
hmH5-16
+
+
+
+
+
+
+
-
+
-
+
+
hmH 1
+
+
+
+
+
+
+
+
+
+
-
+
hmH2
+
+
+
+
+
+
+
+
+
+
hmH30-2-2
+
+
+
+
+
+
+
+
+
+
+
hmH14
+
+
+
+
+
+
+
+
+
+
+
hmH 19
+
+
+
+
+
+
+
+
+
+
+
hmH30-3-6
+
+
+
+
+
+
+
+
+
+
+
hmH3-2
+
+
+
+
+
+
+
+
+
+
+
hmH30-3-7
+
+
+
+
+
+
+
+
+
+
+
Mutant
hmH4
+
+
+
+
+
+
+
+
+
+
+
+
hmH3-3
+
+
+
+
+
+
+
+
+
+
-
+
hmH30-2-3
+
+
+
+
+
+
+
+
+
+
-+
hmH7
+
+
+
+
+
+
+
+
+
+
+
ShmH30-3-3
+
+
+
+
+
+
+
+
+
+
+
ShmH3-5
+
+
+
+
+
+
+
+
+
+
+
ShmH3O-1-1
+
+
+
+
+
+
+
+
+
+
+
SMutant
hmH 10
+
+
+
+
+
+
nd-
nd-
+
+
-
+
--
-
-
-
- - -Sensitive marker rescue'
-
Heteroduplex deletion mappingd
a (+) indicates marker rescue, (-) indicates lack of observable marker rescue, (s) indicates small plaques were observed when the progeny
The spot test was done by spotting P22 gene 9 deletion phage on a lawn of S. typhimurium containing plasmids with the mutant gene 9 .
Heteroduplexes were made between plasmids with deletions and with the hmH10 mutation, transformed into E . coli and tested for of the cross were plated, and (nd) indicates the heteroduplex was not made.
' Sensitive marker rescue was done by crossing the deletion phage with the mutant plasmids in liquid culture and plating the progeny.
marker rescue in the plate complementation assay.
plaque mutants can also arise from mutations in other HmH mutants which create RFLPs: Missense mu- genes. Four small halo mutants were isolated. All of tations which cause the creation or loss of restriction these were moved to phage during the process of enzyme sites lead to restriction fragment length poly- deletion mapping by the sensitive marker rescue tech- morphisms (RFLPs). RFLPs are useful markers for nique and found to make very small plaques. Unex- the presence of alleles during the transfer of mutations pectedly none of these mutations were identified by between Plasmids and Phage and during reversion DNA sequencing to be in the protein coding sequence analysis. Such was the case for hmH5 as well as others of the gene but rather in the sequence 3' to the stop listed in 5.
codon which has been suggested to be the transcrip- tion terminator of the P22 late operon (SAUER et al.
1982). As shown in Figure 4, these mutations individ- ually change three of the four GC pairs in the stem of the proposed stem and loop structure to either AC or GU pairs which decrease the calculated stability of the structure from the wild-type AG of -21.4 kcal/mole to -13.55 kcal/mole (hmH7, 3-5, and 30-3-3) or -1 1.7 kcal/mole (hmH30.1.1) (SALSER 1977).
DISCUSSION
There is currently an impressive collection of mu- tagenic techniques applicable to genes which are cloned in plasmids and single stranded vectors. Some of these techniques even permit isolation of mutations without a phenotypic screen (MYERS, LERMAN and
P22 Tailspike Protein 645
TABLE 5
Nucleotide and amino acid substitutions of gene 9 mutants
Amino acid Protein RFLP
Mutant Nucleotide change” changeb activity’ generatedd
Missense mutations hmH4-1 hmH3 hmH30-3-4 hmH30-2-1 hmH 12 hmH5-16 hmH10 h m H l hmH3-3 hmH30-2-3 hmH30-2-2 hmH14 hmH3-2 hmH30-3-7
hmH5 hmH 15 hmH30-2-5 hmH30-3-5 hmH2 hmH 19 Nonsense mutations
Multiple mutations hmH30-2-4
hmH30-3-6
G - A 8 2
G + A 209 G + A 298 G - A 860 C + T 1193
G + A 1480 G - A 1 5 1 3
G + A 1637 G + A 1688 G + A 1723
G + A 1844
G + A 1849 C + T 1961 C + T 1991
C + T 175 C + T 334 C + T 3 3 4 C + T 6 7 6 C + T 1774
G + A 1919
C + T 3 2 8 C + T 3 3 4 C + T 1934 C + T 1946 C + T 1991
G 28 R G 70 D D 100 N C 287 Y
A 398 V
G 494 R
G 505 R G 546 D
R 563 Q
A 575 T
G 615 E E 617 K P 654 L A 664 V
Q 59 Amber
Q 112 Ochre Q 11 2 Ochre Q 226 Ochre
Q 592 Ochre
W 640 Amber
P 110
s
Q 112 Ochre T 645 I A 649 V A 664 V
ND ND ND ND Nd ND ND ND S T S T ND ND ND ND
ND ND ND ND ND ND
ND
ND
Lost HPaII New HpaI Lost Mae111
New HincII New Mae1
New PstI
New Mae111
New Xbal
Lost Snu3A
Promoter and terminator mutations h m H 4 C + T -32 in Lac UV5 ND hmH7 G + A 2017 H
hmH30-3-3 G + A 2017 H
hmH3-5 G
-
A 2019 HhmH3O-1-1 C + T 2029 H
* Nucleotide numbering starts from the first base of the first codon of the mature protein.
‘ The protein activity is indicated by (ND) for none detected, (ST) for detected only in the spot test, and (H) for those which make smaller
RFLPs a r e restriction fragment length polymorphisms produced by the loss or creation of restriction enzyme recognition sites by the Amino acid numbering starts with the first amino acid of the mature protein (Thr).
than normal size halos in the plate complementation assay.
mutations.
assay of its product’s function, the ability to rapidly screen for the loss or restoration of a particular func- tion is certainly advantageous. It was possible to de- velop such a screen for the tailspike protein because its attachment to P22 capsids to make infectious phage occurs under a wide variety of conditions. T h e ability of the P22 tailspike protein to assemble onto capsids in liquid culture during the growth of tailspike de- pendent phage mutants allows the propagation of these defective mutants. In the plate assay which was developed for these studies the level of production or
functionality of the tailspike protein expressed from a plasmid carrying gene 9 is measured by the ability of the protein produced to complement P22 capsids in soft agar to form a halo of lysis around the colonies making tailspike protein. T h e size of the halo was a reliable enough indicator of overall tailspike protein
activity to isolate mutations which make smaller than normal size halos when on plasmids and smaller than normal size plaques when on phage.
A small number of mutants were isolated in order to test the feasibility of mutant isolation using the plate assay and of deletion mapping of tailspike pro- tein mutants. One of the goals of isolating mutants made in the plasmid clone of the tailspike protein gene was to find mutants which would be difficult or
impossible to isolate on phage such as those with a marginal decrease in activity or those which prevent the propagation of mutant phage by the addition of exogenous tailspike protein. Examples of both of these classes of mutants were found. Four of the mutants produce small plaques when they are on phage and one mutant has a dominant negative phenotype.
646 J. J. Schwarz and P. B. Berget
TABLE 6
Suppression patterns of gene 9 nonsense mutants
Mutant Suppressed 30" 39'
Amber" hmH5
hmH19
Ochreb hmH30-2-4
hmH30-2-5
hmH30-3-5
hmH2
Q 59 amber-Q
Q 59 amber-L Q 59 amber-S
Q 59 amber-Y
W 640 amber-+Q W 640 amber-L W 640 amber-S W 640 amber-Y
P 110 S, Q 112 ochre+ P 110 S, Q 112 ochre-Y Q 1 12 0chr-Q
Q 1 12 ochre-Y
Q 226 ochre-Q Q 226 ochre-Y Q 592 ochre+Q
Q 592 ochre-Y
+
+
+
+
-
- - -+
+
+
+
+
+
+
+
T h e a m b e r mutants were transferred to phage P22 by recom- bination and titered on various amber Salmonella suppressor strains which insert Gln, Leu, Ser or Tyr at both 30" which is the permis- sive temperature for temperature-sensitive tailspike protein mutants and 39" which is the nonpermissive temperature. For the amber mutants a (+) indicates that the titer is decreased less than one order of magnitude from that on a suppressor strain which inserts the wild type codon. A (-1 indicates that the titer on these strains is no higher than the background reversion frequency.
* Plasmids carrying the ochre mutations were transformed into ochre suppressing E. coli strains which insert Gln or Tyr and the tailspike protein activity tested by the plate complementation assay. T h e ochre mutants all had about the same small halo on both suppressor strains at 30" and no halo at 39", even when the wild- type amino acid was inserted.
which mutations on plasmids could be easily mapped using deletions carried on phage. Twelve deletions were made on plasmids and moved onto phage by recombination. This system worked extremely well for all but one mutant which required the use of heteroduplex deletion mapping. Deletion mapping with phage deletions was by two methods: a spot test where the cross is done on a plate and sensitive marker rescue where the cross is done in liquid media. Most mutations required only a rapid spot test. T h e ability of the spot test to discriminate between mutants lo- cated within a deletion interval and those which are nearby is quite good. T h e result of the cross between
D l 1 and hmH30-2-2 show that spotting lo6 deletion phage onto a lawn containing a mutation which is 17 bases removed from the deletion endpoint is sufficient to detect marker rescue. Under these same conditions however, the cross between D l and hmH4-1 which are separated by 12 bases failed to produce marker rescue. The problem of producing wild type phage by recombination is compounded in this case by there being only 64 bases of homology 5' to gene 9 between the plasmid and the phage. In recombination between
A A
U A
G-C
-A
hmh3.5 A + G"-C
AG-13.55 kcal/mole +C +U hmh30.1.1
AG-13.55 kcal/mole hmh 30.3.3 A
+
-c AG-11.7 kcal/moleF A
A-U A-U A-U A-U A-U
F A
A-U A-U
5". AUGU UUAUG..J'
FIGURE 4.-Mutations in the presumptive transcription termi- nator. Mutations were isolated on plasmids based on their small halo size in the plate complementation assay. They were moved onto phage P22 by in vivo recombination where they produced very
small plaques. All of these mutations disrupt a potential G-C base pair in the stem of the presumptive transcription terminator. l ' t w free energies shown are the calculated stabilities of the proposed mutant stem and loop structure by the method of SALSER (1977) and should be compared to the calculated free energy of the wild type sequence of -2 1.4 kcal/mole.
phage h and a plasmid the threshold for efficient recombination was determined to be at approximately 74 bp even though recombination can be detected with as little as 20 bp (WATT et al. 1985). T h e spot test for this cross was repeated using larger amounts of phage. Spotting 10' phage onto a lawn was suffi- cient to detect marker rescue in this cross. So the spot test can correctly map a mutation which is at least 12 nucleotides from a deletion endpoint. For the majority of mutants the spot test is a facile and accurate means of mapping mutations. However for a subset of mu- tants a slightly more elaborate but still simple proce- dure was necessary.
T h e spot test was unable to map 8 of the 27 mutants tested. Seven of these formed large zones of lysis indicative of positive marker rescue with all of the deletions. This apparent marker rescue in crosses with all deletions could have three causes: the mutation is not in the region corresponding to any of the dele- tions, phage carrying the mutation make plaques SO
P22 Tailspike Protein 647
make small plaques when on phage. Therefore, for these mutants the apparent positive marker rescue in the spot test is the result of both complementation and their phage phenotype. T h e mutants hmH3-3 and HmH30-2-3 were neither able to make plaques when on the phage nor were they able to complement capsids extracellularly in the plate assay, yet they formed large areas of lysis when crossed in the spot test with phage carrying the deletions in which they are located. It is possible these mutants possess an extremely small amount of activity which, although insufficient for extracellular complementation in the plate assay, is nevertheless sufficient to complement the capsid under the conditions of the spot test. T h e critical difference between the conditions of the spot test and the plate assay is the phage used in the spot test have had tailspike protein added in vitro to allow them to infect cells once whereas those used in the plate assay are devoid of tailspike protein and require the cells to supply tailspike protein for infection. So during the spot test cells producing the mutant protein from a plasmid are infected and lysed. Complemen- tation under these circumstances could be the result of both intracellular complementation and of the large amounts of mutant protein released by lysis of in- fected, mutant protein producing cells. T h e hmH4 mutation is located in the lacUV5 promoter which is not included in the gene 9 deletions carried on phage, so this mutation produces true positive marker rescue with all deletions in sensitive marker rescue. It was therefore mapped by inference and later confirmed by DNA sequencing.
The other mutant that was not mapped in the spot test, hmH10, appeared to produce an extremely low level of positive marker rescue in the spot test with some deletions and none with others. This same be- havior was observed with sensitive marker rescue. From the analysis of protein defects of these mutants
(J.
J. SCHWARZ and P. B. BERGET, unpublished data) this behavior appears to be the result of enzymatically defective tailspike protein interfering with attachment of functional tailspike protein to capsids rather than multiple mutations in the gene. Therefore it was mapped with the plasmid heteroduplex deletion map- ping procedure (SHORTLE 1983) in which there can be no interference between mutant and functional tailspike protein. T h e ability of heteroduplex deletion mapping to discriminate whether or not a mutant is in a deletion interval is impressive. T h e hmH10 mu- tation is separated from the deletion D l 1 by only 5 nucleotides but the products of the heteroduplex be- tween D l l and hmH IO produced functional tailspike protein in 1 out of the 50 colonies tested.A crude indication of tailspike protein activity is provided by the results of the plate assay and the spot deletion mapping technique. T h e plate assay appears
to be as sensitive an indicator of tailspike protein activity as the plaque-forming ability of phage because plasmid gene 9 mutations that show up as small halo mutants in the plate assay form extremely small plaques when crossed onto phage. T h e spot deletion mapping technique seems to be a more sensitive indi- cator because it detected activity in two mutants (hmH3-3 and hmH30-2-3) which was not detected in the plate assay or by phage phenotype. Therefore the mutants can be separated into three categories of increasing tailspike protein activity based on the re- sults of the plate assay and the spot deletion mapping technique: 1) Those which have no detectable activity (ND). 2) Those whose activity is only detected in the spot test (ST). 3) Those whose activity is detected by the plate assay (H). These categories are indicated in Table 5 . T h e two mutants hmH3-3 and hmH30-2-3 are the only ones whose mutations are in the protein coding region of the gene and have any detectable activity. They are also located very close to each other, codons 563 and 575 respectively. It is remarkable that the four mutants which have enough activity to make small halos and small plaques are all in the presump- tive transcription terminator and not in protein cod- ing sequence. It is possible that mutations in the protein coding sequence which cause marginal activity decreases commensurate with small halos are ex- tremely rare.
Mutations in the putative transcription terminator were entirely unexpected. It has been suggested in several reports that a RNA stem and loop structure is a barrier to mRNA digestion by exonuclease (WONG and CHANG 1986; BELASCO et al. 1985; GUARNEROS et al. 1982). T h e mutations all decrease the calculated stability of the stem and loop structure of the mRNA, so it is possible this structure is a less effective barrier to mRNA degradation in these mutants because of their lower stability. This could lead to lower expres- sion of the gene by decreasing the lifetime of its message.