Acta Cryst.(2002). E58, i85±i87 DOI: 10.1107/S1600536802015659 Kissick and Keszler Rb2Al2B2O7
i85
inorganic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Rb2Al2B2O7
Judith L. Kissicka* and Douglas A. Keszlerb
aDepartment of Chemistry, University of
Reading, Whiteknights, Reading RG6 6AD, England, andbDepartment of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, OR 97331, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 293 K
Mean(O±B) = 0.005 AÊ Rfactor = 0.030 wRfactor = 0.075
Data-to-parameter ratio = 19.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
Rubidium aluminium borate, Rb2Al2B2O7, is characterized by
an association of AlO4 tetrahedra and BO3 triangles which
form a complete three-dimensional aluminium borate frame-work. Rb+cations occupy eight- and nine-coordinate positions
within the three-dimensional channel system created by the framework.
Comment
The phase Rb2Al2B2O7 is a new phase ®rst described here,
following a study of the systemM2O±Al2O3±B2O3, whereM=
Na, K, Rb. Rb2Al2B2O7 crystallizes in the monoclinic space
group P21/c and is characterized by a three-dimensional
framework built from corner-sharing AlO4 tetrahedra and
BO3 triangles surrounding a three-dimensional channel
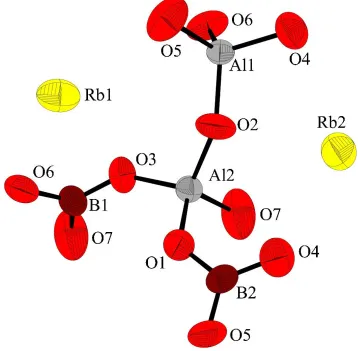
system in which the Rb atoms are located. Two crystal-lographically distinct Al atoms and two distinct B atoms are present in distorted tetrahedral AlO4and trigonal-planar BO3
groups (Fig. 1). Each AlO4group is connected to three BO3
groups and one AlO4group to form an Al2O7unit in which the
AlÐOÐAl bond angle is 146.9 (2).
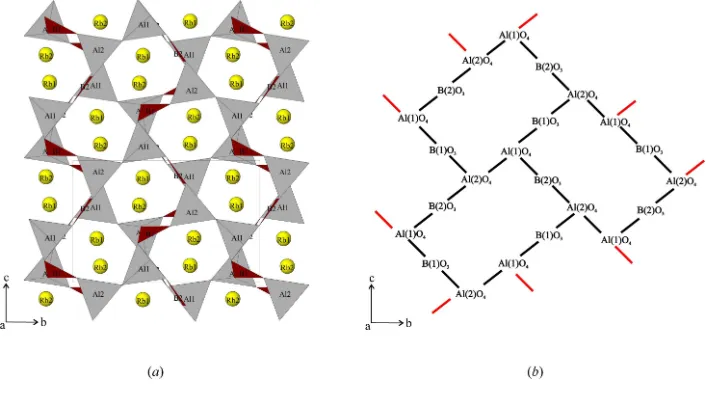
The structure can be considered to be built up from ten-membered Al6B4O10 rings, generated from corner-sharing
AlO4and BO3polyhedra. The rings are linked in herring-bone
fashion to form sheets in thebcplane (Fig. 2). Adjacent sheets are connected in a staggered formation through four-membered Al2B2O4rings and eight-membered Al4B4O8rings
perpendicular to thebandcaxes, respectively. Both crystal-lographically distinct Rb atoms have site symmetry 1. Rb1 is eight-coordinate within a coordination sphere of 3.5 AÊ and has a calculated bond valence of +1.01 (1). Rb2 is nine-coordinate within a 3.5 AÊ coordination sphere and has a calculated bond
Received 15 August 2002 Accepted 30 August 2002 Online 6 September 2002
Figure 1
inorganic papers
i86
Kissick and Keszler Rb2Al2B2O7 Acta Cryst.(2002). E58, i85±i87valence of +0.88 (1). Bond valences consistent with expected integral values are computed for each of the remaining atoms in the structure (Brese & O'Keeffe, 1991).
The structure of the materialM2Al2B2O7depends on the
nature of theMcation. Na, K and Rb analogues assume three different structures, even when synthesized under identical conditions. Both the Na and K analogues of M2Al2B2O7
crystallize in trigonal space groups [P31c,a= 4.8087 (6),c= 15.2734 (6) AÊ andZ= 2 (Chang, 1998; Heet al., 2001);P321,
a= 8.5657 (9), c= 8.463 (2) AÊ andZ = 3 (Hu et al., 1998)]. Their structures are characterized by six-membered Al3B3O6
rings, built from AlO4tetrahedra and BO3triangles, that are
linked together to form nearly planar sheets in theabplane. In the Na analogue, these sheets are connected in pairs through linear AlÐOÐAl bonds to form layers, which are linked through Na atoms to form a three-dimensional structure. In the K analogue, a three-dimensional AlÐBÐO framework is generated by AlÐOÐAl bonds between adjacent sheets and the K atoms are located in the space between these sheets.
We have found that up to 2.5% of the Rb atoms in Rb2Al2B2O7can be replaced by either Na or K and the
three-dimensional monoclinic structure is retained with essentially unchanged cell dimensions. Substitution of greater amounts of either Na or K causes the material to assume a structure more closely related to that of K2Al2B2O7.
Experimental
Single crystals of Rb2Al2B2O7were grown in a covered Pt crucible by melting a mixture of 42.0 wt% Rb2CO3 (99.8%, Alfa), 18.6 wt% Al2O3(99.997%, Alfa), 13.3 wt% B2O3(99.98%, Alfa) and 26.1 wt% LiBO2 (99.995%, Alfa), which acts as a ¯ux to ensure congruent melting. The melt was heated at 1373 K for 16 h to ensure homo-geneity, it was then cooled to room temperature at a rate of 0.07 K minÿ1. Numerous crystals formed in the crucible and a clear colourless block was physically separated from the matrix for analysis.
Crystal data Rb2Al2B2O7
Mr= 358.52
Monoclinic,P21/c
a= 8.901 (2) AÊ
b= 7.539 (1) AÊ
c= 11.905 (2) AÊ = 103.97 (1) V= 775.3 (2) AÊ3
Z= 4
Dx= 3.072 Mg mÿ3
MoKradiation Cell parameters from 21
re¯ections = 15±20
= 12.85 mmÿ1
T= 293 (2) K Block, colourless 0.200.150.10 mm Data collection
Rigaku AFC-6Rdiffractometer !/2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.113,Tmax= 0.277
4763 measured re¯ections 2281 independent re¯ections 1541 re¯ections withI> 2(I)
Rint= 0.058
max= 30.1
h=ÿ12!12
k=ÿ10!10
l=ÿ16!16 3 standard re¯ections
every 400 re¯ections intensity decay: 0.6% Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.030
wR(F2) = 0.075
S= 1.01 2281 re¯ections 119 parameters
w= 1/[2(F
o2) + (0.0237P)2
+ 0.6214P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001 max= 0.65 e AÊÿ3 min=ÿ0.57 e AÊÿ3
Extinction correction:SHELXL97 Extinction coef®cient: 0.0097 (5)
Table 1
Selected geometric parameters (AÊ,).
Rb1ÐO6 2.808 (2) Rb1ÐO1i 2.946 (3)
Rb1ÐO3 2.968 (3) Rb1ÐO3ii 3.048 (3)
Rb1ÐO1 3.056 (3) Rb1ÐO5 3.105 (3) Rb1ÐO5i 3.126 (3)
Rb1ÐO7iii 3.403 (3)
Rb2ÐO7iv 2.924 (3)
Rb2ÐO2v 3.009 (3)
Rb2ÐO2vi 3.014 (3)
Rb2ÐO6 3.043 (3) Rb2ÐO4vii 3.086 (3)
Rb2ÐO4 3.200 (3) Rb2ÐO1i 3.297 (3)
Rb2ÐO7v 3.405 (3)
Rb2ÐO4viii 3.455 (3)
Al1ÐO2 1.716 (3) Al1ÐO6v 1.746 (3)
Al1ÐO4ix 1.749 (3)
Al1ÐO5i 1.762 (3)
Al2ÐO2 1.725 (3) Al2ÐO3 1.747 (3) Al2ÐO7x 1.755 (3)
Al2ÐO1x 1.764 (3)
O3ÐB1 1.360 (5) O6ÐB1 1.380 (5) O7ÐB1ii 1.359 (5)
O1ÐB2i 1.370 (5)
O4ÐB2 1.366 (5) O5ÐB2 1.360 (5)
O2ÐAl1ÐO6v 112.30 (13)
O2ÐAl1ÐO4ix 109.32 (14)
O6vÐAl1ÐO4ix 105.08 (14)
O2ÐAl1ÐO5i 111.00 (15)
O6vÐAl1ÐO5i 105.17 (14)
O4ixÐAl1ÐO5i 113.82 (14)
O2ÐAl2ÐO3 109.87 (14) O2ÐAl2ÐO7x 109.16 (15)
O3ÐAl2ÐO7x 109.03 (14)
O2ÐAl2ÐO1x 112.30 (13)
O3ÐAl2ÐO1x 108.52 (13)
O7xÐAl2ÐO1x 107.89 (14)
O7vÐB1ÐO3 122.8 (4)
O7vÐB1ÐO6 118.6 (3)
O3ÐB1ÐO6 118.5 (3) O5ÐB2ÐO4 123.0 (4) O5ÐB2ÐO1i 116.9 (3)
O4ÐB2ÐO1i 120.0 (3)
Al1ÐO2ÐAl2 146.85 (18)
Symmetry codes: (i) ÿx;ÿy;1ÿz; (ii) ÿx;1
2y;32ÿz; (iii) ÿx;1ÿy;1ÿz; (iv)
1x;1
2ÿy;12z; (v) ÿx;yÿ12;32ÿz; (vi) 1x;y;z; (vii) x;ÿ12ÿy;12z; (viii)
1ÿx;1
2y;32ÿz; (ix)xÿ1;y;z; (x)x;12ÿy;12z.
Data collection: MSC/AFC Diffractometer Control Software (Molecular Structure Corporation, 1999); cell re®nement:MSC/AFC Diffractometer Control Software; data reduction: TEXSAN for Windows(Molecular Structure Corporation,1997); program(s) used to solve structure:SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics: ATOMS(Shape Software, 1998); software used to prepare material for publication:WinGX(Farrugia, 1999).
Figure 2
This work was supported by the Solid State Chemistry Program of the National Science Foundation. JLK would like to thank Dr A. M. Chippindale for helpful discussion about this work.
References
Brese, N. E. & O'Keeffe, M. (1991).Acta Cryst.B47, 192±197. Chang, K.-S. (1998). PhD dissertation, Oregon State University, USA. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837±838.
He, M., Chen, X. L., Zhou, T., Hu, B. Q., Xu, Y. P. & Xu, T. (2001).J. Alloys Compds,327, 210±214.
Hu, Z. G., Higashiyama, T., Yoshimura, M., Yap, Y. K., Mori, Y. & Sasaki, T. (1998).Jpn. J. Appl. Phys. Part2Lett.32, L1093±1094.
Molecular Structure Corporation (1997).TEXSAN for Windows. Version 1.0. MSC, 9009 New Trails Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation (1999).MSC/AFC Diffractometer Control Software. MSC, 9009 New Trails Drive, The Woodlands, TX 77381 USA. North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351±
359.
Shape Software (1998).ATOMS. Shape Software, 521 Hidden Valley Rd, Kingsport, TN 37663, USA.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Acta Cryst.(2002). E58, i85±i87 Kissick and Keszler Rb2Al2B2O7
i87
supporting information
sup-1
Acta Cryst. (2002). E58, i85–i87
supporting information
Acta Cryst. (2002). E58, i85–i87 [doi:10.1107/S1600536802015659]
Rb
2Al
2B
2O
7Judith L. Kissick and Douglas A. Keszler
S1. Comment
The phase Rb2Al2B2O7 is a new phase first described here following a study of the system M2O—Al2O3—B2O3, where M
= Na, K, Rb. Rb2Al2B2O7 crystallizes in the monoclinic space group P21/c and is characterized by a three-dimensional
framework built from corner-sharing AlO4 tetrahedra and BO3 triangles surrounding a three-dimensional channel system
in which the Rb atoms are located. Two crystallographically distinct Al atoms and two distinct B atoms are present in
distorted tetrahedral AlO4 and trigonal-planar BO3 groups (Fig. 1). Each AlO4 group is connected to three BO3 groups and
one AlO4 group to form an Al2O7 unit in which the Al–O–Al bond angle is 146.9 (2)°.
The structure can be considered to be built up from ten-membered Al6B4O10 rings, generated from corner-sharing AlO4
and BO3 polyhedra. The rings are linked in herring-bone fashion to form sheets in the bc plane (Fig. 2). Adjacent sheets
are connected in a staggered formation through four-membered Al2B2O4 rings and eight-membered Al4B4O8 rings
perpendicular to the b and c axes, respectively. Both crystallographically distinct Rb atoms have site symmetry 1. Rb1 is
eight-coordinate within a coordination sphere of 3.5 Å and has a calculated bond valence of +1.01 (1). Rb2 is
nine-coordinate within a 3.5 Å coordination sphere and has a calculated bond valence of +0.88 (1). Bond valences consistent
with expected integral values are computed for each of the remaining atoms in the structure (Brese & O′Keeffe, 1991).
The structure of the material M2Al2B2O7 depends on the nature of the M cation. Na, K and Rb analogues assume three
different structures, even when synthesized under identical conditions. Both the Na and K analogues of M2Al2B2O7
crystallize in trigonal space groups [P31c, a = 4.8087 (6), c = 15.2734 (6) Å and Z = 2 (Chang, 1998; He et al., 2001);
P321, a = 8.5657 (9), c = 8.463 (2) Å and Z = 3 (Hu et al., 1998)]. Their structures are characterized by six-membered
Al3B3O6 rings, built from AlO4 tetrahedra and BO3 triangles, that are linked together to form nearly planar sheets in the ab
plane. In the Na analogue, these sheets are connected in pairs through linear Al–O–Al bonds to form layers which are
linked through Na atoms to form a three-dimensional structure. In the K analogue, a three-dimensional Al–B–O
framework is generated by Al–O–Al bonds between adjacent sheets and the K atoms are located in the space between
these sheets.
We have found that up to 2.5% of the Rb atoms in Rb2Al2B2O7 can be replaced by either Na or K and the
three-dimensional monoclinic structure is retained with essentially unchanged cell dimensions. Substitution of greater amounts
of either Na or K cause the material to assume a structure more closely related to that of K2Al2B2O7.
S2. Experimental
Single crystals of Rb2Al2B2O7 were grown in a covered Pt crucible by melting a mixture of 42.0 wt% Rb2CO3 (99.8%,
Alfa), 18.6 wt% Al2O3 (99.997%, Alfa), 13.3 wt% B2O3 (99.98%, Alfa) and 26.1 wt% LiBO2 (99.995%, Alfa), which acts
as a flux to ensure congruent melting. The melt was heated at 1373 K for 16 h to ensure homogeneity, it was then cooled
to room temperature at a rate or 0.07 K min−1. Numerous crystals formed in the crucible and a clear colourless block was
supporting information
sup-2
Acta Cryst. (2002). E58, i85–i87
S3. Refinement
The Rb and Al atoms were located using the direct methods program SHELXS97 (Sheldrick, 1997) and the remaining
[image:5.610.125.483.125.476.2]atoms were placed following analysis of difference electron-density maps.
Figure 1
supporting information
sup-3
[image:6.610.129.482.73.270.2]Acta Cryst. (2002). E58, i85–i87
Figure 2
View of Rb2Al2B2O7 along [100] showing the three-dimensional Al–B–O framework and the Rb atoms in the channels.
Yellow spheres = Rb atoms, grey tetrahedra = AlO4 and brown triangles = BO3 (ATOMS; Shape Software, 2002).
(I)
Crystal data
Rb2Al2B2O7
Mr = 358.52
Monoclinic, P21/c
a = 8.901 (2) Å
b = 7.539 (1) Å
c = 11.905 (2) Å
β = 103.97 (1)°
V = 775.3 (2) Å3
Z = 4
F(000) = 664
Dx = 3.072 Mg m−3
Mo Kα radiation, λ = 0.71069 Å Cell parameters from 21 reflections
θ = 15–20°
µ = 12.85 mm−1
T = 293 K Block, colourless 0.20 × 0.15 × 0.10 mm
Data collection
Rigaku AFC-6R diffractometer
Radiation source: Rigaku rotating anode Graphite monochromator
ω/2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.113, Tmax = 0.277 4763 measured reflections
2281 independent reflections 1541 reflections with I > 2σ(I)
Rint = 0.058
θmax = 30.1°, θmin = 3.2°
h = −12→12
k = −10→10
l = −16→16
3 standard reflections every 400 reflections intensity decay: 0.6%
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.030
wR(F2) = 0.075
S = 1.01 2281 reflections 119 parameters 0 restraints
w = 1/[σ2(F
o2) + (0.0237P)2 + 0.6214P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.65 e Å−3 Δρmin = −0.57 e Å−3
supporting information
sup-4
Acta Cryst. (2002). E58, i85–i87
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Rb1 0.03371 (4) 0.14003 (5) 0.62650 (3) 0.02092 (11)
Rb2 0.45632 (4) −0.13338 (6) 0.85435 (3) 0.02143 (11)
Al1 −0.34247 (12) −0.12434 (14) 0.62207 (8) 0.0101 (2)
Al2 −0.18525 (12) 0.08342 (14) 0.86588 (8) 0.0113 (2)
O1 −0.2027 (3) 0.1892 (3) 0.3953 (2) 0.0176 (5)
O2 −0.3084 (3) 0.0199 (4) 0.7370 (2) 0.0191 (6)
O3 0.0064 (3) 0.0407 (4) 0.8620 (2) 0.0180 (6)
O4 0.4631 (3) −0.1894 (4) 0.5892 (2) 0.0195 (6)
O5 0.2828 (3) 0.0324 (4) 0.4962 (2) 0.0226 (6)
O6 0.2373 (3) 0.1790 (3) 0.8455 (2) 0.0167 (5)
O7 −0.2294 (3) 0.5381 (4) 0.4795 (2) 0.0249 (7)
B1 0.1552 (5) 0.0873 (5) 0.9114 (3) 0.0134 (8)
B2 0.3191 (5) −0.1147 (5) 0.5634 (3) 0.0129 (7)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Rb1 0.0239 (2) 0.02130 (18) 0.01522 (17) 0.00202 (16) 0.00023 (14) −0.00159 (15)
Rb2 0.02018 (19) 0.0257 (2) 0.01822 (17) −0.00420 (16) 0.00437 (13) −0.00026 (15)
Al1 0.0111 (5) 0.0101 (5) 0.0085 (4) 0.0004 (4) 0.0016 (4) 0.0002 (4)
Al2 0.0115 (5) 0.0122 (5) 0.0101 (5) −0.0013 (4) 0.0025 (4) −0.0005 (4)
O1 0.0185 (13) 0.0151 (11) 0.0212 (13) 0.0038 (11) 0.0086 (11) 0.0047 (10)
O2 0.0177 (13) 0.0190 (13) 0.0185 (12) 0.0021 (11) −0.0001 (10) −0.0093 (11)
O3 0.0118 (12) 0.0217 (13) 0.0209 (13) −0.0033 (11) 0.0048 (10) −0.0019 (11)
O4 0.0141 (12) 0.0216 (13) 0.0214 (13) −0.0024 (11) 0.0018 (10) −0.0014 (11)
O5 0.0311 (16) 0.0193 (13) 0.0214 (13) 0.0061 (12) 0.0140 (12) 0.0098 (11)
O6 0.0228 (13) 0.0163 (13) 0.0109 (11) −0.0081 (11) 0.0037 (10) −0.0016 (9)
O7 0.0190 (14) 0.0343 (16) 0.0197 (14) 0.0047 (13) 0.0012 (11) −0.0138 (13)
B1 0.0122 (18) 0.0109 (17) 0.0171 (18) −0.0008 (14) 0.0035 (15) −0.0013 (14)
B2 0.0177 (19) 0.0136 (18) 0.0080 (14) −0.0002 (16) 0.0042 (14) −0.0028 (14)
Geometric parameters (Å, º)
Rb1—O6 2.808 (2) Rb2—B1 3.361 (4)
Rb1—O1i 2.946 (3) Rb2—B2 3.387 (4)
supporting information
sup-5
Acta Cryst. (2002). E58, i85–i87
Rb1—O3ii 3.048 (3) Rb2—O4viii 3.455 (3)
Rb1—O1 3.056 (3) Rb2—Al2vi 3.5577 (13)
Rb1—O5 3.105 (3) Al1—O2 1.716 (3)
Rb1—O5i 3.126 (3) Al1—O6v 1.746 (3)
Rb1—B1 3.326 (4) Al1—O4ix 1.749 (3)
Rb1—O7iii 3.403 (3) Al1—O5i 1.762 (3)
Rb1—B2i 3.403 (4) Al2—O2 1.725 (3)
Rb1—B2 3.410 (4) Al2—O3 1.747 (3)
Rb1—Al2ii 3.5976 (12) Al2—O7x 1.755 (3)
Rb2—O7iv 2.924 (3) Al2—O1x 1.764 (3)
Rb2—O2v 3.009 (3) O3—B1 1.360 (5)
Rb2—O2vi 3.014 (3) O6—B1 1.380 (5)
Rb2—O6 3.043 (3) O7—B1ii 1.359 (5)
Rb2—O4vii 3.086 (3) O1—B2i 1.370 (5)
Rb2—O4 3.200 (3) O4—B2 1.366 (5)
Rb2—O1i 3.297 (3) O5—B2 1.360 (5)
O2—Al1—O6v 112.30 (13) O3—Al2—O1x 108.52 (13)
O2—Al1—O4ix 109.32 (14) O7x—Al2—O1x 107.89 (14)
O6v—Al1—O4ix 105.08 (14) O7v—B1—O3 122.8 (4)
O2—Al1—O5i 111.00 (15) O7v—B1—O6 118.6 (3)
O6v—Al1—O5i 105.17 (14) O3—B1—O6 118.5 (3)
O4ix—Al1—O5i 113.82 (14) O5—B2—O4 123.0 (4)
O2—Al2—O3 109.87 (14) O5—B2—O1i 116.9 (3)
O2—Al2—O7x 109.16 (15) O4—B2—O1i 120.0 (3)
O3—Al2—O7x 109.03 (14) Al1—O2—Al2 146.85 (18)
O2—Al2—O1x 112.30 (13)