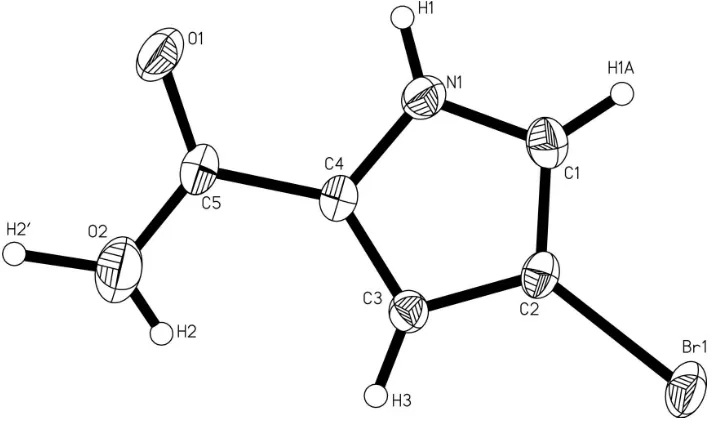
4 Bromo 1H pyrrole 2 carboxylic acid
7
0
0
Full text
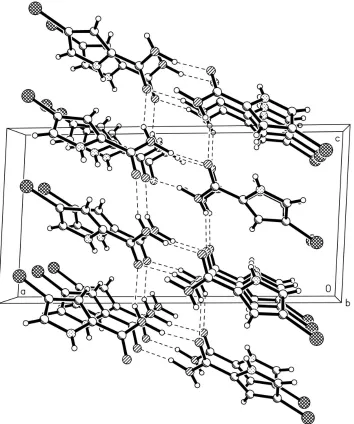
Figure

Related documents