organic papers
Acta Cryst.(2006). E62, o239–o240 doi:10.1107/S1600536805041334 Odabas¸og˘luet al. C
17H16O6
o239
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
Bis(3-formyl-4-hydroxy-5-methoxyphenyl)-methane
Mustafa Odabas¸og˘lu,a* C¸ig˘dem Albayrakaand Orhan Bu¨yu¨kgu¨ngo¨rb
aDepartment of Chemistry, Faculty of Arts and
Science, Ondokuz Mayıs University, TR-55139 Kurupelit Samsun, Turkey, andbDepartment of
Physics, Ondokuz Mayıs University, TR-55139 Kurupelit Samsun, Turkey
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 296 K
Mean(C–C) = 0.003 A˚ Rfactor = 0.046 wRfactor = 0.109
Data-to-parameter ratio = 13.4
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, C17H16O3, the asymmetric unit contains
one half-molecule; a twofold rotation axis bisects the
molecule. The structure is stabilized by O—H N
intra-molecular hydrogen bonds and C—H and –
inter-molecular interactions.
Comment
Hydroxy-substituted benzaldehyde reagents used for
condensation with primary amines, hydrazines, hydroxylamine and other primary amine derivatives afford imine derivatives which can function as ligands towards a number of metal cations (Loudon, 2002; Khandar & Nejati, 2000; Khandar & Rezvani, 1999).
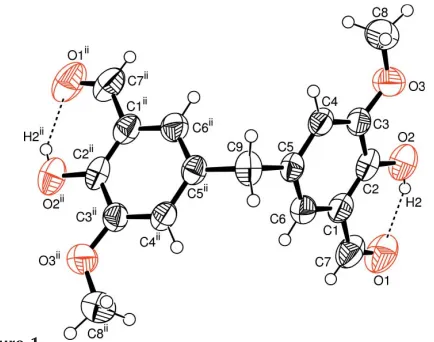
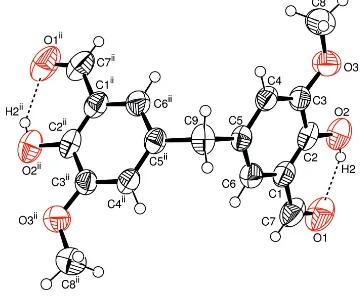
The asymmetric unit of the title compound, (I), contains one half-molecule; a twofold rotation axis passes through C9 (Fig. 1). The bond lengths and angles (Table 1) are in normal ranges (Allenet al., 1987).
In (I), molecules have strong intramolecular O—H O
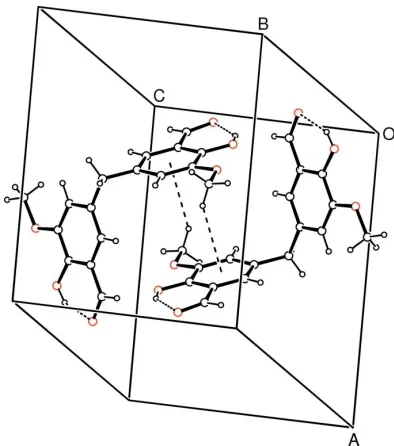
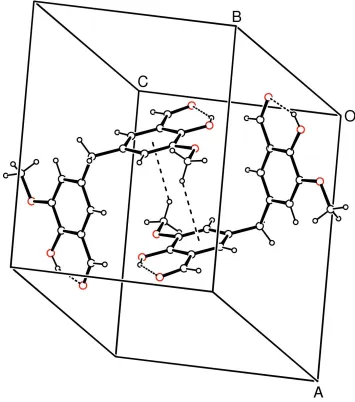
hydrogen bonds (Table 2) and they are linked through C8—
H8a Cg1 (Cg1 is the centroid of the C1–C6 ring) and
Cg1 Cg1 intermolecular interactions (Fig. 2). For the C8—
[image:1.610.219.436.536.707.2]Received 25 November 2005 Accepted 9 December 2005 Online 14 December 2005
Figure 1
The molecular structure of (I), with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probalitity level. The intramolecular hydrogen bonds are shown as dashed lines. [Symmetry code: (i) 1x,y,1
H8a Cg1 contact, the distance between atom H8aand the aromatic ring centroid is 3.01 (3) A˚ (symmetry code: 1 x,
1 y, z). There is also – stacking between adjacent
molecules at (x, y, z) and (x, y, 1
2 z), with distances of
3.608 (16) A˚ between the rings centroids and perpendicular distances of 2.481 (16) A˚ between the rings.
Experimental
A mixture of o-vanillin (0.1 mol) and formaldehyde (0.1 mol) was stirred at 393 K for 2 h. The solution was added to boiling ethyl alcohol and stirred at 393 K for 20 min and cooled to room temperature. The precipitate was filtered off and recrystallized from ethyl alcohol by slow evaporation (yield 1.58 g, 10%, m.p. 424– 426 K).
Crystal data
C17H16O6
Mr= 316.30
Monoclinic,C2=c a= 14.960 (3) A˚ b= 8.2889 (11) A˚ c= 13.249 (3) A˚
= 115.015 (15)
V= 1488.8 (5) A˚3
Z= 4
Dx= 1.411 Mg m
3
MoKradiation Cell parameters from 3016
reflections
= 2.9–27.5
= 0.11 mm1
T= 296 (2) K Plate, yellow
0.400.240.07 mm
Data collection
Stoe IPDS-II diffractometer
!scans
Absorption correction: integration (X-RED32; Stoe & Cie, 2002) Tmin= 0.963,Tmax= 0.993
6042 measured reflections 1470 independent reflections
828 reflections withI> 2(I) Rint= 0.065
max= 26.0
h=18!18 k=10!10 l=16!16
Refinement onF2 R[F2> 2(F2)] = 0.046
wR(F2) = 0.109 S= 1.00 1470 reflections 110 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.049P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.12 e A˚
3
min=0.11 e A˚
3
Table 1
Selected geometric parameters (A˚ ,). C1—C2 1.388 (3) C1—C6 1.399 (3) C2—O2 1.360 (2) C2—C3 1.395 (3)
C3—C4 1.381 (3) C4—C5 1.400 (3) C5—C6 1.368 (3) C7—O1 1.219 (3)
C2—C1—C7 120.9 (2) O2—C2—C3 118.0 (2)
O3—C3—C4 124.8 (2) C5—C9—C5i
113.6 (3)
C6—C1—C7—O1 176.1 (2) C6—C5—C9—C5i
72.25 (18)
C4—C5—C9—C5i
108.23 (19) C4—C3—O3—C8 1.6 (3) Symmetry code: (i)xþ1;y;zþ1
2.
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O2—H2 O1 0.82 1.94 2.648 (3) 145
Atom H9, attached to C9, was located in a difference map and refined isotropically [C—H = 0.98 (2) A˚ ]. The remaining H atoms were positioned geometrically [0.82 (OH), 0.93 (CH) and 0.96 A˚ (CH3)] and constrained to ride on their parent atoms withUiso(H) values of 1.5 (1.2 for methine) timesUeq(C,O).
Data collection: X-AREA (Stoe & Cie, 2002); cell refinement:
X-AREA; data reduction: X-RED32; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
ORTEP3 for Windows (Farrugia, 1997); software used to prepare material for publication:WinGX(Farrugia, 1999).
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G., & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp S1–19.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Khandar, A. A. & Nejati, K. (2000).Polyhedron,19, 607–613. Khandar, A. A. & Rezvani, Z. (1999).Polyhedron,18, 129–133.
Loudon, M. G. (2002).Organic Chemistry, , 4th ed., p. 837. Oxford University Press.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
[image:2.610.74.271.71.294.2]Stoe & Cie (2002).X-AREA(Version 1.18) andX-RED32(Version 1.04). Stoe & Cie, Darmstadt, Germany.
Figure 2
supporting information
sup-1
Acta Cryst. (2006). E62, o239–o240
supporting information
Acta Cryst. (2006). E62, o239–o240 [doi:10.1107/S1600536805041334]
Bis(3-formyl-4-hydroxy-5-methoxyphenyl)methane
Mustafa Odabaşoğlu, Çiğdem Albayrak and Orhan Büyükgüngör
S1. Comment
Hydroxy-substituted benzaldehyde reagents used for condensation with primary amines, hydrazines, hydroxylamine and
other primary amine derivatives afford imine derivatives which can function as ligands towards a number of metal cations
(Khandar & Nejati, 2000; Khandar & Rezvani, 1999).
The asymmetric unit of the title compound, (I), contains one-half molecule (Fig. 1). The bond lengths and angles (Table
1) are in normal ranges (Allen et al.,1987).
In (I), molecules have strong intramolecular O—H···O hydrogen bonds (Table 2) and they are linked through C8—
H8a···Cg1 (Cg1 is the centroid of the C1–C6 ring) and Cg1···Cg1 intermolecular interactions (Fig. 2). For the C8—
H8a···Cg1 contact, the distance between atom H8a and the aromatic ring centroid is 3.01 (3) Å (symmetry code: 1 − x, 1
− y, −z). There is also π–π stacking between adjacent molecules at (−x, y, 1/2 − z), with distances of 3.608 (16) between
the rings centroids and a and perpendicular distance of 2.481 (16) Å between the rings.
S2. Experimental
A mixture of o-vanilline (0.1 mol) and formaldehyde (0.1 mol) was stirred at 393 K for 2 h. The solution was added to the
boiling ethyl alcohol and stirred at 393 K for 20 min and cooled to room temperature. The precipitate was filtered off and
recrystallized from ethyl alcohol by slow evaporation (yield 1.58 g, 10%, m.p. 424–426 K).
S3. Refinement
Atom H9 was located in a difference map and refined isotropically [C—H = 0.98 (2) Å]. The remaining H atoms were
positioned geometrically [0.82 (OH), 0.93 (CH) and 0.96 Å (CH3)] and constrained to ride on their parent atoms with
Figure 1
The molecular structure of (I), with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50%
supporting information
sup-3
[image:5.610.126.481.72.474.2]Acta Cryst. (2006). E62, o239–o240 Figure 2
A packing diagram of (I) with the C—H···π and π–π intermolecular interactions shown as dashed lines.
Bis(3-formyl-4-hydroxy-5-methoxyphenyl)methane
Crystal data
C17H16O6 Mr = 316.30
Monoclinic, C2/c Hall symbol: -C 2yc a = 14.960 (3) Å b = 8.2889 (11) Å c = 13.249 (3) Å β = 115.015 (15)° V = 1488.8 (5) Å3 Z = 4
F(000) = 664 Dx = 1.411 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 3016 reflections θ = 2.9–27.5°
µ = 0.11 mm−1 T = 296 K Plate, yellow
Stoe IPDS-II diffractometer
Radiation source: fine-focus sealed tube Plane graphite monochromator
Detector resolution: 6.67 pixels mm-1 ω scans
Absorption correction: integration (X-RED; Stoe & Cie, 2002) Tmin = 0.963, Tmax = 0.993
6042 measured reflections 1470 independent reflections 828 reflections with I > 2σ(I) Rint = 0.065
θmax = 26.0°, θmin = 2.9° h = −18→18
k = −10→10 l = −16→16
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.046 wR(F2) = 0.109 S = 1.00 1470 reflections 110 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.049P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max < 0.001
Δρmax = 0.12 e Å−3 Δρmin = −0.11 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full
covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-5
Acta Cryst. (2006). E62, o239–o240
O1 0.85964 (13) 0.6153 (2) 0.47781 (18) 0.0999 (7) O2 0.77382 (12) 0.5455 (2) 0.61102 (14) 0.0774 (6) H2 0.8151 0.5887 0.5944 0.116* O3 0.63516 (12) 0.3852 (2) 0.63842 (13) 0.0755 (5) H9 0.5368 (17) 0.111 (3) 0.221 (2) 0.081 (8)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0436 (11) 0.0583 (14) 0.0675 (15) 0.0026 (10) 0.0168 (10) 0.0136 (12) C2 0.0383 (10) 0.0496 (12) 0.0657 (15) 0.0016 (9) 0.0052 (10) −0.0012 (11) C3 0.0463 (11) 0.0598 (13) 0.0477 (12) 0.0058 (10) 0.0140 (9) 0.0005 (10) C4 0.0390 (10) 0.0568 (13) 0.0565 (13) −0.0011 (9) 0.0147 (10) 0.0047 (11) C5 0.0427 (10) 0.0498 (12) 0.0502 (13) 0.0056 (9) 0.0119 (9) 0.0070 (10) C6 0.0516 (12) 0.0657 (15) 0.0553 (13) 0.0051 (11) 0.0199 (10) 0.0092 (11) C7 0.0499 (14) 0.0795 (18) 0.098 (2) −0.0012 (12) 0.0242 (14) 0.0246 (16) C8 0.0811 (16) 0.0969 (19) 0.0712 (17) −0.0058 (15) 0.0389 (14) 0.0012 (15) C9 0.0597 (19) 0.053 (2) 0.0543 (19) 0.000 0.0114 (16) 0.000 O1 0.0593 (10) 0.0937 (14) 0.1302 (17) −0.0221 (10) 0.0239 (11) 0.0181 (13) O2 0.0574 (9) 0.0699 (11) 0.0835 (11) −0.0114 (8) 0.0090 (8) −0.0117 (9) O3 0.0685 (10) 0.0971 (13) 0.0632 (11) −0.0187 (9) 0.0299 (8) −0.0153 (9)
Geometric parameters (Å, º)
C1—C2 1.388 (3) C6—H6 0.9300 C1—C6 1.399 (3) C7—O1 1.219 (3) C1—C7 1.454 (3) C7—H7 0.9300 C2—O2 1.360 (2) C8—O3 1.417 (3) C2—C3 1.395 (3) C8—H8A 0.9600 C3—O3 1.365 (3) C8—H8B 0.9600 C3—C4 1.381 (3) C8—H8C 0.9600 C4—C5 1.400 (3) C9—C5i 1.514 (3) C4—H4 0.9300 C9—H9 0.98 (2) C5—C6 1.368 (3) O2—H2 0.8200 C5—C9 1.514 (3)
C6—C5—C9 121.26 (19) C5i—C9—H9 109.2 (14) C4—C5—C9 120.44 (17) C2—O2—H2 109.5 C5—C6—C1 121.6 (2) C3—O3—C8 117.75 (18)
C6—C1—C2—O2 178.34 (18) C3—C4—C5—C9 178.3 (2) C7—C1—C2—O2 0.6 (3) C4—C5—C6—C1 0.0 (3) C6—C1—C2—C3 −0.7 (3) C9—C5—C6—C1 −179.6 (2) C7—C1—C2—C3 −178.5 (2) C2—C1—C6—C5 1.0 (3) O2—C2—C3—O3 −1.5 (3) C7—C1—C6—C5 178.8 (2) C1—C2—C3—O3 177.60 (19) C2—C1—C7—O1 1.6 (4) O2—C2—C3—C4 −179.63 (19) C6—C1—C7—O1 −176.1 (2) C1—C2—C3—C4 −0.5 (3) C6—C5—C9—C5i −72.25 (18) O3—C3—C4—C5 −176.39 (19) C4—C5—C9—C5i 108.23 (19) C2—C3—C4—C5 1.5 (3) C4—C3—O3—C8 −1.6 (3) C3—C4—C5—C6 −1.2 (3) C2—C3—O3—C8 −179.6 (2)
Symmetry code: (i) −x+1, y, −z+1/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A