Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Microsatellite Markers for Typing Aspergillus fumigatus Isolates
EMMANUELLE BART-DELABESSE,1JEAN-FRANC¸OIS HUMBERT,2E´RIC DELABESSE,3ANDSTE´PHANE BRETAGNE1*
Laboratoire de Parasitologie-Mycologie, Hoˆpital Henri Mondor, Cre´teil,1Institut National de Recherche Agronomique, Station d’Hydrobiologie Lacustre, Thonon-les-Bains,2and Laboratoire
d’He´matologie, CNRS URA 1461, Hoˆpital Necker, Paris,3France Received 16 March 1998/Returned for modification 22 April 1998/Accepted 1 June 1998
The use of microsatellites as highly polymorphic DNA markers for the typing of isolates of Aspergillus
fu-migatus was investigated. Four CA repeats were selected by screening an A. fufu-migatus DNA library with a (CA)10 oligonucleotide. Primers flanking these CA repeats were designed to amplify each locus. One primer of each pair was labeled with a fluorophore, and the PCR products were analyzed with an automatic sequencer and the GeneScan software. For each primer set and for a given isolate, one band was detected and was assigned to an allele because A. fumigatus is haploid. With 50 clinical isolates, 50 environmental isolates, and 2 reference strains we obtained 12, 11, 10, and 23 different alleles for the four CA microsatellites, respectively (discrim-inatory power, 0.994). The results were identical by whatever DNA extraction technique was used. Interestingly, no clustering between environmental and clinical isolates was observed, suggesting that every isolate is po-tentially pathogenic. Microsatellite markers appear suitable for use in large epidemiological studies of invasive aspergillosis.
Invasive aspergillosis due to Aspergillus fumigatus has be-come the leading cause of death in immunocompromised pa-tients such as allogeneic bone marrow transplant recipients. Invasive aspergillosis can be contracted during hospitalization, especially by patients during their first month after bone mar-row transplantation (26). Because inhalation of spores into the respiratory tract is the usual route of contamination, preven-tion can be achieved by using rooms with laminar air flow, but all patients at risk do not benefit from such conditions. There-fore, tracking the sources of contamination is an essential preventive measure. Molecular tools provide a means of com-paring isolates from patients and from their environment to determine the source of contamination.
DNA fingerprinting techniques include the direct detection of restriction fragment length polymorphisms (RFLPs) (6, 8) and the detection of RFLPs by Southern hybridization with the M13 phage (1), ribosomal probes (22), or a retrotransposon-like element (19). The last probe has proved to be highly dis-criminatory for A. fumigatus isolates (7). However, RFLP anal-ysis is a very time-consuming technique. Large amounts of DNA are needed, and DNA electrophoresis, blotting, and probing take more than 5 days for a limited number of isolates.
In contrast, the PCR-based methods are attractive because of their rapidity. Randomly amplified polymorphic DNA (RAPD) analysis has been applied to A. fumigatus. The prim-ers either were short and arbitrarily chosen (2, 13, 15, 21) or consisted of repetitive motifs known to detect variable DNA sequences in lower eukaryotes and prokaryotes (13, 25). How-ever, RAPD analysis exhibits a low level of reproducibility because of the low-stringency conditions used in the PCR, and these conditions lead to mismatched pairings (28). Therefore, the patterns may be complex and hardly comparable between laboratories. More recently, a high-stringency PCR technique with primers specific for RAPD products has been proposed
(16). This strategy requires the sequencing of RAPD products in a first step and overcomes the shortcomings of low-stringen-cy conditions, but several sets of primers are needed to obtain high discriminatory power. However, whatever RAPD tech-nique is used, the nature of RAPD polymorphisms is too poor-ly understood to be useful without limitations in phylogenetic studies (3).
Microsatellites represent another class of genetic markers that have not yet been used to differentiate A. fumigatus strains. Microsatellites are short tandem repeats of two to six nucleo-tides that are known to be highly polymorphic as well as nu-merous and spread equally throughout the human genome (27). Microsatellites have also been found in lower eukaryotic or-ganisms including fungi (4, 9). The polymorphism of microsat-ellites can be evaluated by PCR, and precise allele sizing can be achieved with fluorescent primers and an automatic sequencer. Therefore, we investigated whether microsatellites could be found in A. fumigatus and used as DNA markers to differen-tiate isolates of this fungal species.
MATERIALS AND METHODS
Screening of libraries, hybridization, and sequencing.Total DNA of A.
fu-migatus IP 2279.94 (Pasteur Institute, Paris, France) was partially digested with
either TaqI or MboI and was used to create two genomic libraries. The 150- to 1,000-bp fragments were inserted into the corresponding enzymatic site of bac-teriophage M13mp18 (Appligene Oncor, Illkirch, France) before transformation of Epicurian Coli XL1-Blue MRF9electroporation-competent cells (Stratagene, La Jolla, Calif.). Bacteriophage plaques were transferred onto Hybond-N1filters
(Amersham, Les Ulis, France) and were probed with a (CA)10oligonucleotide. The oligonucleotide was labeled with fluorescein-11-dUTP by using the ECL 39-oligolabelling system (Amersham) and was diluted in hybridization buffer to a final concentration of 10 ng per ml. After hybridization at 37°C for 2 h, the filters were washed twice at room temperature for 5 min each time in 53saline sodium citrate (SSC; 13SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% sodium dodecyl sulfate (SDS) and twice at 37°C for 15 min each time in 13SSC–0.1% SDS. Incubation with anti-fluorescein-horseradish peroxidase conjugate, detec-tion with the ECL detecdetec-tion system (Amersham), and exposure to Hyperfilm-ECL (Amersham) were carried out according to the manufacturer’s procedures. Clones showing hybridization signals were picked. Single-stranded DNAs from the bacteriophage were extracted, and automated sequencing was performed with the ABI dye primer kit (Perkin-Elmer, Courtaboeuf, France).
PCR amplification and analysis.For the clones containing a (CA)n ellite, primer pairs complementary to the flanking sequences of each microsat-* Corresponding author. Mailing address: Laboratoire de
Parasi-tologie-Mycologie, Hoˆpital Henri Mondor, 51 avenue du Ge´ne´ral De-Lattre de Tassigny, 94010, Cre´teil Cedex, France. Phone: 33 1 49 81 36 41. Fax: 33 1 49 81 36 01. E-mail: [email protected].
2413
on May 15, 2020 by guest
http://jcm.asm.org/
ellite were designed. One primer of each set was labeled with a fluorescent dye, either 6-carboxyfluorescein (6-FAM) or 4,7,29,49,59,79 -hexachloro-6-carboxy-fluorescein (HEX) (Oligo-Express, Paris, France), for detection with an auto-mated DNA sequencer. PCR amplification was performed in a 20-ml volume containing 1.5 mM MgCl2, 10 mM Tris-HCl (pH 9.0), and 50 mM KCl and with the forward and reverse primers at concentrations of 100 nM each, the de-oxynucleoside triphosphates at concentrations of 100mM each, 0.5 U of Taq DNA polymerase (Pharmacia Biotech, Orsay, France), 5% (vol/vol) dimethyl sulfoxide, and 50 ng of DNA template. Amplification was carried out in a Perkin-Elmer Cetus system 480 thermocycler, with denaturation for 5 min at 94°C, 30 times cycles of 30 s at 94°C, 30 s at 59°C, and 30 s at 72°C, and a final extension step at 72°C for 30 min. The PCR products were diluted 1/10 in water, and 1ml of each was run on a 36-cm-long acrylamide-urea gel (4.25% polyacryl-amide, 8.3 M urea, and 13Tris-borate-EDTA for 2 h at 3,000 V and 51°C). The
N,N,N9,N9-tetramethyl-6-carboxyrhodamine (TAMRA)-labeled GeneScan size standard (Perkin-Elmer) was loaded into each well along with the PCR products. Signals were read with an automatic sequencer (ABI 377; Applied BioSystems), and the data were stored and analyzed with GeneScan software (version 2.0.2; Perkin-Elmer) by the local Southern sizing method.
A. fumigatus isolates.One hundred isolates (50 environmental isolates and 50 clinical isolates) were studied. The isolates were identified on the basis of culture characteristics and the morphologies of the conidiophores and conidia. The environmental isolates were collected from three Parisian hospitals over a 2-year period and were epidemiologically unrelated to the clinical isolates (7). DNA was extracted from a single colony by a phenol-chloroform technique at the Pasteur Institute (10). Additionally, 20 of the same isolates were also sent to our labo-ratory as a culture on a 2% malt slant, and a new DNA extraction was performed by a cetyltrimethylammonium bromide technique (18).
Of the 50 clinical isolates, 39 were obtained from different patients with invasive aspergillosis followed at the Henri Mondor Hospital over a 6-year period, 3 were recovered from patients with invasive aspergillosis hospitalized in Germany, and 8 were from patients with cystic fibrosis followed in Parisian hospitals. Two reference strains originally isolated from two patients (strains IP 2279.94 and CBS 143.89) were also studied. DNA was extracted by a cetyltri-methylammonium bromide technique. Twenty of the clinical isolates were also tested without any specific DNA extraction. About 104to 105spores were suspended in 50ml of distilled water, frozen at280°C for 10 min, thawed, and spun, and 5ml of the supernatant was taken for PCR.
Discriminatory power and cluster analysis.The ability of microsatellite poly-morphism to discriminate between isolates was assessed by using Simpson’s index of diversity:
D512N~N121!
O
j51 s
xj~xj21!
where N is the number of isolates, and xjis the proportion of the isolates falling into jth group defined by the number of repeats obtained at each microsatellite locus (12). For each isolate, alleles were scored as 1 (present) or 0 (absent) in a contingency table. These data were analyzed by correspondence analysis, which is an ordination technique. This analysis was performed with the ADE-4 Soft-ware Package (24). For the graphic representations, the scatters module with ellipses option was chosen. The ellipses were centered on the means for each subgroup (environmental and patient isolates). The width and the height of these ellipses were given by the variances, and each covariance set the slope of the main axis of each ellipse.
RESULTS
Among 17,000 clones of the two A. fumigatus libraries screened, 9 clones hybridized with the (CA)10oligonucleotide probe and were sequenced. One of them was rejected as a TaqI cloning site and raised the possibility that recombination had occurred during the ligation process. Four other clones with less than eight CA repeats were also rejected because no poly-morphism was detected upon a preliminary PCR screening with 10 isolates. Therefore, only four microsatellites were re-tained for further analysis, and there are henceforth referred to as microsatellites A, B, C, and D (Table 1). A search of the sequences in the GenBank and EMBL databases with the Blastn search program did not yield any sequence homology between the microsatellite flanking sequences and prokaryotic or eukaryotic sequences.
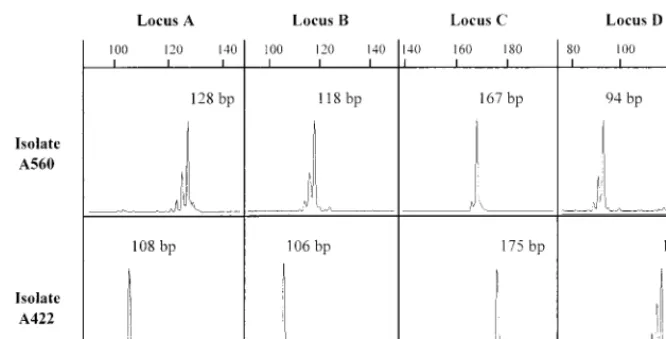
PCR products consisted mainly of a single band, but addi-tional bands shorter by 2 to 4 bp were seen for some alleles (Fig. 1). These extra bands were due to the slippage of the Taq DNA polymerase and were always less intense than the longest band. Only the latter band was used for scoring and was as-signed to an allele since A. fumigatus is thought to be haploid. For one isolate, we found two completely different peaks. We therefore performed a monospore culture and reamplified the DNA obtained for 10 different colonies. Each colony gave an unique peak. This isolate was actually a mixture of two A.
fu-migatus isolates and was therefore excluded from the analysis.
For the 102 isolates tested in the study, each PCR amplifica-tion was repeated at least twice with the same DNA template, and identical profiles were obtained. In addition, identical re-sults were obtained regardless of the DNA extraction tech-nique used. Besides, the amplifications were specific to A.
fu-migatus because no band was observed upon amplification of
other Aspergillus species: A. flavus IP 597.69, A. niger IP 1431.83,
A. terreus IP 1136.76, and A. nidulans IP 17.60.
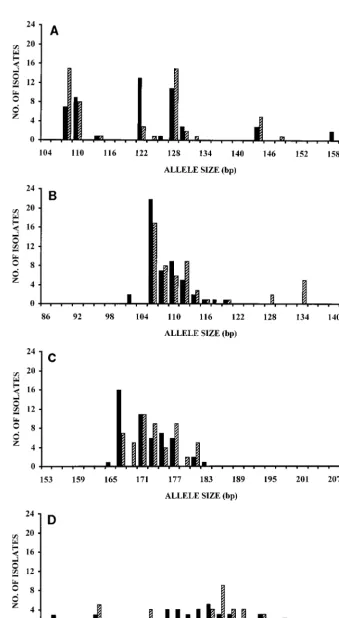
Upon the analysis of the 2 reference strains and the 100 un-related isolates, 12, 11, 10, and 23 alleles were detected for mi-crosatellites A, B, C, and D, respectively (Fig. 2). The combina-tion of the four markers led to 80 different allelic types, which corresponded to a discriminatory power of 0.994 (Table 2).
[image:2.612.50.550.82.210.2]To check that the differences in the lengths of the PCR products were due to differences in the number of CA repeats at each microsatellite locus, PCR products of strain IP 2279.94 and an isolate (A.224) with distinct alleles were inserted into pGEM-T Easy vector (Promega, Madison, Wis.). After trans-formation of competent XL1-Blue cells (Stratagene), two white colonies were selected from each PCR product and the inserts TABLE 1. Features of the four polymorphic microsatellite sequences of A. fumigatus upon analysis of 100 isolates and 2 reference strains
Microsatellite Primer sequences (59to 39) Microsatellitesequencesa PCR products (bp)Length range of Range of no.of repeats Total no.of alleles
A GCCTACGATGACCGAAATGA (CA)9(GA)25 108–158 16–41 12
CTGTTTTGAGAAGCGGATGG
B TTGCCATCGCTTGTCATAGA (CA)2C(CA)23 102–134 9–25 11
GCAGGTGGTTCAATAGGACAG
C CGAAGCTCTCCCCTGCAAATC (CA)8 165–183 7–16 10
GATGCCGCTGGTGGTGTTGT
D AGGGATACGGCTACGGACAA (CA)21 76–134 7–36 23
AAAGCGTCTGTCAGCGTGTCT
aBased on the original DNA sequences of strain IP 2279.94.
on May 15, 2020 by guest
http://jcm.asm.org/
were sequenced. The sequences obtained showed the different numbers of CA repeats which were responsible for the differ-ent sizes of alleles: (CA)9(GA)25and (CA)9(GA)8for micro-satellite A, (CA)2C(CA)23and (CA)2C(CA)10for microsatel-lite B, (CA)8and (CA)13for microsatellite C, and (CA)21and (CA)32for microsatellite D for strain IP 2279.94 and isolate A.224, respectively. Subsequently, we decided to systematically run reference strain IP 2279.94 on each gel to check the correct automatic sizing with the GeneScan software.
To check the stability of the microsatellite repeats over time, DNAs from four subcultures of strain CBS 143.89 obtained in 1971, 1986, 1989, and 1995 (provided by J.-P. Latge´) were test-ed. This strain has been regularly subcultured since 1989 at the Pasteur Institute. Identical microsatellite profiles were observed between the four subcultures tested. The same re-sults were obtained with strain IP 2279.94, which has been regularly subcultured in our laboratory since 1994.
Of the 100 isolates and 2 reference strains studied, 64 had a unique type and 38 were grouped into 16 types (12 types of two isolates each, 3 types of 3 isolates each, and 1 type of 5 iso-lates). Some isolates had a common type, although they were undoubtedly independent because they were collected at com-pletely different places. For instance, one environmental iso-late from a Parisian hospital had the same type as a clinical isolate from a German patient. For other isolates with a com-mon type, their independence was questionable because they were collected at the same place, although at different times. For instance, the same type was recovered from four patients hospitalized in the same hospital but in different wards over a 22-month period.
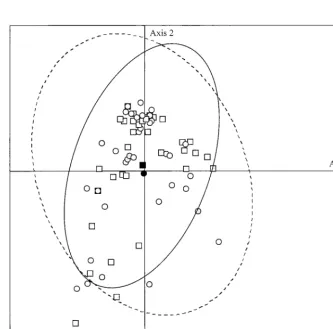
The isolates were analyzed by correspondence analysis ac-cording to their clinical or environmental origin. Correspon-dence analysis produced a multidimensional representation of the data, which were reduced in plane to be more easily visu-alized. The first three planes were defined by the three major axes which explained most of the variance (relative inertia, 19%). The two ellipses which contained 90% of the individuals of each subgroup (patient and environmental isolates) widely overlapped and their centers were very close (Fig. 3). The same results were obtained for the other projections. Therefore, the diversity of patient isolates was of the same order as that of the environmental isolates.
DISCUSSION
For the first time, the characterization of microsatellites of
A. fumigatus is reported and the use of microsatellites as a
typing system was investigated. These microsatellites are poly-morphic, and reproducible typing can be obtained by analysis with an automatic sequencer. Our results are consistent with those obtained with microsatellites of other species. In the human genome, the higher the number of uninterrupted CA repeats, the more polymorphic the microsatellite (27). Indeed, no polymorphism was detected with three microsatellites with CA repeat numbers of less than 8; 10 alleles were found with microsatellite C (CA repeat range, 7 to 16), and 23 alleles were found with microsatellite D (CA repeat range, 7 to 36). The reproducibility of this technique is high because (i) the PCR amplifications are performed under high-stringency conditions, (ii) the results do not depend on the DNA isolation technique, (iii) and the sizing is automatic, with an internal size standard systematically loaded along with the amplified products. Thus, the coding of alleles for further analysis is simple. Moreover, these microsatellite markers appear stable upon subculturing. Another advantage is that there is no need for monospore culture. Indeed, a single peak is expected for a given locus because A. fumigatus is haploid. If several peaks are observed, the isolate tested is actually a mixture of several isolates and can be rejected.
The combined discriminatory power of the four microsatel-lites described is 0.994 and outscores the discriminatory power obtained with other PCR-based markers (14, 16). Therefore, these markers appear promising for large epidemiological stud-ies. From a technical point of view, the throughput of A.
fu-migatus isolates could be increased if several fluorescent dyes
were used to label the primers in multiplex PCRs.
[image:3.612.135.468.71.240.2]However, some technical problems can be encountered with this technique. Amplification of dinucleotide repeats can lead to PCR artifacts with several peaks separated by intervals of two nucleotides. These extra bands are mainly due to the slippage of Taq DNA polymerase during amplification (11). In our experience, this trouble was overcome by performing the amplification with dimethyl sulfoxide. Thus, the longest band was always the most intense and was the only one retained for analysis. Another PCR artifact is the addition of an extra A to
FIG. 1. GeneScan analysis of PCR profiles obtained with two independent isolates (isolates A560 and A422). PCRs were performed with a 6-FAM-labeled primer (loci A, B, and C) or a HEX-labeled primer (locus D), and the PCR products for each microsatellite locus (A, B, C, and D) were run in an acrylamide-urea gel. Bands produced fluorescent peaks, and their molecular sizes were automatically determined by comparison to the TAMRA-labeled GeneScan internal size standards loaded in each well (data not shown). The numbers refer to the sizes (in base pairs) of the PCR products by considering the fluorescent peak with the maximum height.
on May 15, 2020 by guest
http://jcm.asm.org/
FIG. 2. Allele size distributions of A. fumigatus isolates at microsatellite loci A (A), B (B), C (C), and D (D) upon analysis of 50 environmental isolates (solid bars) and 52 clinical isolates including 2 reference strains (striped bars).
on May 15, 2020 by guest
http://jcm.asm.org/
the 39 end of product strands, more specifically, when the 59
end of the reverse primer is a G, which is the preferential template for the addition of extra nucleotides by the Taq poly-merase (5). The final extension at 72°C for 30 min in our PCRs was aimed at ensuring the systematic addition of an extra A to
make the PCR products homogeneous. Nevertheless, a refer-ence strain must be systematically included as an internal con-trol in all amplification runs and analyses. Because the results expected for the reference strain are known, it is possible to detect an artifact of the PCR or a sizing error.
Another question related to the microsatellite markers is whether the variations in the sizes of the PCR products are due to variations in the number of CA repeats. It has recently been reported (20) that the addition or deletion of nucleotides in the flanking regions of the dinucleotide repeat instead of in the microsatellite sequence itself could account for length varia-tions. For the microsatellites tested in this work, the primers for amplification were chosen close to the CA repeat in order to generate PCR products shorter than 200 bp. Thus, the probability that mutations outside the microsatellite account for the length polymorphism is low. Therefore, in contrast to RAPD techniques, the nature of the differences is known. Currently, this information is not usable because it is impossi-ble to assess the mutation rate at a given microsatellite in the absence of sexual reproduction for A. fumigatus. Nevertheless, it is reasonable to think that knowledge obtained with a model species with a sexuality, such as Emericella nidulans, could be transposable to A. fumigatus.
By RFLP, Debeaupuis et al. (7) have shown that no partic-ular genotype was associated with virulence. We were also unable to cluster isolates depending on their clinical or
[image:5.612.52.289.90.254.2]envi-FIG. 3. Correspondence analysis performed for all alleles showing the dispersion observed for the 102 A. fumigatus isolates. The projection in the plane defined by the two most informative axes is shown. Squares, environmental isolates; circles, clinical isolates; empty symbols, unique profiles; shaded symbols, profiles shared by two or more isolates. The two ellipses contain 90% of the individuals of each subgroup (dotted line for patient isolates; continuous line for environmental isolates). The centers of the ellipses are indicated, with a black circle for the patient isolate ellipse and a black square for the environmental isolate ellipse.
TABLE 2. Discriminatory power of microsatellite markers for the 100 A. fumigatus isolates and the 2 reference strains tested
Marker
association(s) No. oftypes No. of types representedby a single isolate Discriminatorypower
A 12 4 0.835
B 11 1 0.793
C 10 2 0.869
D 23 4 0.948
A1C 31 14 0.935
B1C 33 16 0.944
A1B 35 17 0.950
A1D 52 26 0.981
C1D 55 30 0.980
B1D 60 36 0.986
A1B1C 49 30 0.968
A1C1D 67 45 0.987
A1B1D 73 52 0.992
B1C1D 77 58 0.992
A1B1C1D 80 64 0.994
on May 15, 2020 by guest
http://jcm.asm.org/
[image:5.612.132.465.361.689.2]ronmental origin. This finding may indicate that every A.
fumi-gatus isolate is potentially pathogenic and that the risk for the
patient is inhalation of the conidia of any A. fumigatus isolate. This hypothesis is in contrast to the hypothesis that certain isolates are more pathogenic than others (17, 23).
The microsatellites described in this work represent a new class of highly polymorphic markers for A. fumigatus. Epide-miological studies by automated procedures with large num-bers of isolates can be designed. Moreover, microsatellites offer perspectives for studying the genetic relatedness among
A. fumigatus isolates and for genome mapping, as has already
been done for the genomes of other species.
ACKNOWLEDGMENTS
We thank J.-P. Latge´ from the Pasteur Institute for providing A.
fu-migatus isolates and R. Calderone from Georgetown University,
Wash-ington, D.C., for critical reading of the manuscript.
This work was supported by grant BQR 9014 R1 from the Paris XII University.
REFERENCES
1. Anderson, M. J., K. Gull, and D. W. Denning. 1996. Molecular typing by random amplification of polymorphic DNA and M13 Southern hybridization of related paired isolates of Aspergillus fumigatus. J. Clin. Microbiol. 34:87– 93.
2. Aufauvre-Brown, A., J. Cohen, and D. W. Holden. 1992. Use of randomly amplified polymorphic DNA markers to distinguish isolates of Aspergillus
fumigatus. J. Clin. Microbiol. 30:2991–2993.
3. Backeljau, T., L. De Bruyn, H. De Wolf, K. Jordaens, S. Van Dongen, R. Verhagen, and B. Winnepenninckx.1995. Random amplified polymorphic DNA (RAPD) and parsimony methods. Cladistics 11:119–130.
4. Bretagne, S., J. M. Costa, C. Besmond, R. Carsique, and R. Calderone. 1997. Microsatellite polymorphism in the promoter sequence of the elongation factor 3 gene of Candida albicans as the basis for a typing system. J. Clin. Microbiol. 35:1777–1780.
5. Brownstein, M. J., J. D. Carpten, and J. R. Smith. 1996. Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modi-fications that facilitate genotyping. BioTechniques 20:1004–1010. 6. Burnie, J. P., A. P. Coke, and R. C. Matthews. 1992. Restriction
endonucle-ase analysis of Aspergillus fumigatus DNA. J. Clin. Pathol. 45:324–327. 7. Debeaupuis, J. P., J. Sarfati, V. Chazalet, and J. P. Latge´. 1997. Genetic
diversity among clinical and environmental isolates of Aspergillus fumigatus. Infect. Immun. 65:3080–3085.
8. Denning, D. W., K. V. Clemons, L. H. Hanson, and D. A. Stevens. 1990. Restriction endonuclease analysis of total cellular DNA of Aspergillus
fu-migatus isolates of geographically and epidemiologically diverse origin. J.
In-fect. Dis. 162:1151–1158.
9. Field, D., and C. Wills. 1996. Long, polymorphic microsatellites in simple organisms. Proc. R. Soc. Lond. Ser. B Biol. Sci. 263:209–215.
10. Girardin, H., J. P. Latge´, T. Srikantha, B. Morrow, and D. R. Soll. 1993. Development of DNA probes for fingerprinting Aspergillus fumigatus. J. Clin. Microbiol. 31:1547–1554.
11. Hauge, X. Y., and M. Litt. 1993. A study of the origin of ’shadow bands’ seen when typing dinucleotide repeat polymorphisms by the PCR. Hum. Mol. Genet. 2:411–415.
12. Hunter, P. R. 1991. A critical review of typing methods for Candida albicans and their applications. Crit. Rev. Microbiol. 17:417–434.
13. Leenders, A., A. van Belkum, S. Janssen, S. de Marie, J. Kluytmans, J. Wielenga, B. Lowenberg, and H. Verbrugh.1996. Molecular epidemiology of apparent outbreak of invasive aspergillosis in a hematology ward. J. Clin. Microbiol. 34:345–351.
14. Lin, D., P. F. Lehmann, B. H. Hamory, A. A. Padhye, E. Durry, R. W. Pinner, and B. A. Lasker.1995. Comparison of three typing methods for clinical and environmental isolates of Aspergillus fumigatus. J. Clin. Microbiol. 33:1596– 1601.
15. Loudon, K. W., J. P. Burnie, A. P. Coke, and R. C. Matthews. 1993. Appli-cation of polymerase chain reaction to fingerprinting Aspergillus fumigatus by random amplification of polymorphic DNA. J. Clin. Microbiol. 31:1117– 1121.
16. Mondon, P., M. P. Brenier, E. Coursange, B. Lebeau, P. Ambroise-Thomas, and R. Grillot.1997. Molecular typing of Aspergillus fumigatus strains by sequence-specific DNA primer (SSDP) analysis. FEMS Immunol. Med. Mi-crobiol. 17:95–102.
17. Mondon, P., C. De Champs, A. Donadille, P. Ambroise-Thomas, and R. Grillot. 1996. Variation in virulence of Aspergillus fumigatus strains in a murine model of invasive pulmonary aspergillosis. J. Med. Microbiol. 45: 186–191.
18. Murray, M. G., and W. F. Thompson. 1980. Rapid isolation of high molec-ular weight plant DNA. Nucleic Acids Res. 8:4321–4325.
19. Neuve´glise, C., J. Sarfati, J. P. Latge´, and S. Paris. 1996. Afut1, a retro-transposon-like element from Aspergillus fumigatus. Nucleic Acids Res. 24: 1428–1434.
20. Orti, G., D. E. Pearse, and J. C. Avise. 1997. Phylogenetic assessment of length variation at a microsatellite locus. Proc. Natl. Acad. Sci. USA 94: 10745–10749.
21. Rinyu, E., J. Varga, and L. Ferenczy. 1995. Phenotypic and genotypic analysis of variability in Aspergillus fumigatus. J. Clin. Microbiol. 33:2567–2575. 22. Spreadbury, C. L., B. W. Bainbridge, and J. Cohen. 1990. Restriction
frag-ment length polymorphisms in isolates of Aspergillus fumigatus probed with part of the intergenic spacer region from the ribosomal RNA gene complex of Aspergillus nidulans. J. Gen. Microbiol. 136:1991–1994.
23. Tang, C. M., J. Cohen, A. J. Rees, and D. W. Holden. 1994. Molecular epidemiology study of invasive pulmonary aspergillosis in a renal transplan-tation unit. Eur. J. Clin. Microbiol. Infect. Dis. 13:318–321.
24. Thioulouse, J., D. Chessel, S. Doledec, and J.-M. Olivier. 1997. ADE-4: a multivariate analysis graphical display software. Stat. Comput. 7:75–83. 25. van Belkum, A., W. G. Quint, B. E. de Pauw, W. J. Melchers, and J. F. Meis.
1993. Typing of Aspergillus species and Aspergillus fumigatus isolates by interrepeat polymerase chain reaction. J. Clin. Microbiol. 31:2502–2505. 26. Wald, A., W. Leisenring, J. A. van Burik, and R. A. Bowden. 1997.
Epide-miology of Aspergillus infections in a large cohort of patients undergoing bone marrow transplantation. J. Infect. Dis. 175:1459–1466.
27. Weber, J. L. 1990. Informativeness of human (dC-dA)n-(dG-dT)n polymor-phisms. Genomics 7:524–530.
28. Weising, K., R. G. Atkinson, and R. C. Gardner. 1995. Genomic fingerprint-ing by microsatellite-primed PCR: a critical evaluation. PCR Methods Appl. 4:249–255.