Abstract
Clinical presentation of osteoarthritis (OA) is dominated by pain during joint use and at rest. OA pain is caused by aberrant functioning of a pathologically altered nervous system with key mechanistic drivers from peripheral nerves and central pain pathways. This review focuses on symptomatic pain therapy exemplified by molecular targets that alter sensitization and hyperexcitability of the nervous system, for example, opioids and cannabinoids. We highlight opportunities for targeting inflam-matory mediators and their key receptors (for example, prosta-noids, kinins, cytokines and chemokines), ion channels (for example, NaV1.8, NaV1.7 and CaV2.2) and neurotrophins (for example, nerve growth factor), noting evidence that relates to their participation in OA etiology and treatment. Future neurological treatments of pain appear optimistic but will require the systematic evaluation of emerging opportunities.
Introduction
Osteoarthritis (OA) is recognized by degeneration of articular cartilage, synovitis, remodeling of subchondral bone and atrophy/weakness of joint muscles. Clinical presentation is dominated by pain during joint use and often at rest. There are circadian variations in pain severity in both knee and hand OA, with pain worsening in the evening [1,2]. Pain frequency and intensity has been related to obesity, helplessness and education as well as a significant co-morbid association with anxiety and depression [3].
There are major distinctions between physiological and pathophysiological (chronic) pain. Physiological pain is a
necessary defense mechanism, related directly to the degrees of existing or imminent tissue damage, and is essential for survival. On the other hand, chronic pain serves no defensive or helpful function, since neither the intensity nor quality of chronic pain is related to the degree of tissue damage and may persist long after the resolution of any initial insult. Chronic pain (nociceptive or neuropathic) is now recognized as a manifestation of an aberrant functioning of a pathologically altered nervous system. Pain therapy, and the emerging pharmacology, is seen in terms of symptomatic treatment (through modulation of aberrant function, that is, neural excitability) and disease modification (through neural restoration of physiological pain processing). This is the context in which we will develop new therapies and will be the focus of this review. However, this does not deny that disease modifying approaches, for example, to resolve joint or cartilage degeneration, may also impact on OA pain.
Pain in OA, like other chronic pain conditions, is a complex integration of sensory, affective and cognitive processes that involves a number of abnormal cellular mechanisms at both peripheral (joints) and central (spinal and supraspinal) levels of the nervous system. The relative contribution of these processes in the OA population appears to be strongly segmented. Intra-articular anesthetic studies in hip and knee OA support a peripheral drive to pain in approximately 60% to 80% of patients, depending on the affected joint [3,4]. In some individuals, however, central mechanisms, for example, dysfunction of descending inhibitory control [5] or altered
Review
Arthritis and pain
Future targets to control osteoarthritis pain
Andy Dray
1and Simon J Read
21AstraZeneca R&D Montreal, Frederick Banting St, Montreal H4S 1Z9, Canada 2AstraZeneca R&D, Mereside, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK
Corresponding author: Andy Dray, Andy.Dray@astrazeneca.com
Published: 30 May 2007 Arthritis Research & Therapy2007, 9:212 (doi:10.1186/ar2178) This article is online at http://arthritis-research.com/content/9/3/212
© 2007 BioMed Central Ltd
cortical processing of noxious information, may play a greater role [6].
With such patient heterogeneity, identifying pharmacological targets of the future is fraught with issues. Biomarker development and patient stratification will need to be progressed in parallel to ensure ‘tailor-made treatment’. More narrow titration of preclinical activities, for example, animal models, in vitroassays and so on, to specific patient subsets may also be required to improve predictability in humans. Nevertheless, rational mechanistic approaches can be taken. Alterations in the physiology of sensory pathways, such as sensitization (reduced threshold for stimulation), hyper-excitability (amplification or prolongation of nerve discharge) or spontaneous nerve activity, can be associated with specific molecular changes.
In this review we have selected examples of emerging pharmacology for the treatment of OA pain (Figure 1). Where appropriate, examples of inflammatory and neuropathic pain pharmacology have been highlighted, since there is
continuing discussion as to whether components of osteo-arthritic pain are also neuropathic (see [7] for a review). Ultimately, in any patient, multiple algogenic mechanisms may underpin the pain experience. Combinations of pharmaco-logical approaches may, therefore, be a requirement for effective pain management. However, ‘chasing’ efficacy with combinations will need to be balanced against the cumulative safety burden of treatments. Indeed, OA patients (particularly the elderly) may be willing to forgo efficacy in favor of lower adverse event risk [8].
Target classes
Opioids and their receptors
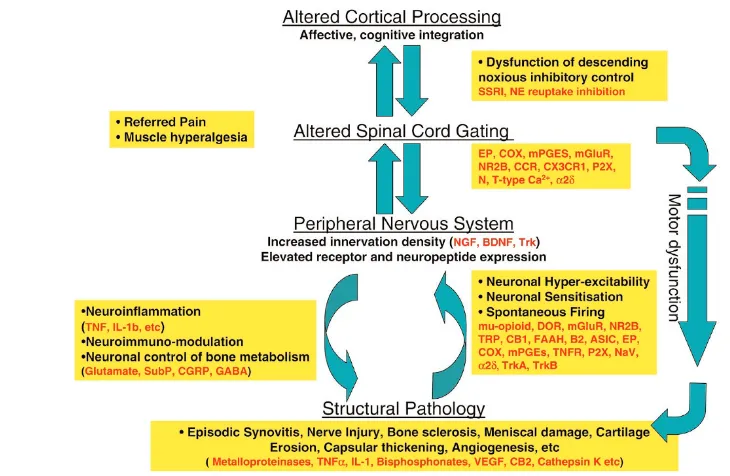
Opioids have been a mainstay of chronic pain therapy for many years. They act at peripheral, spinal, and supraspinal sites through a variety of opioid receptors (mu-, delta-, and kappa-opioid receptors) [9]. Opioids used in the clinic, such as morphine, act via mu-opioid receptors to cause a variety of well documented side effects, including sedation, dysphoria, respiratory depression and constipation. However, opioid receptor activation in the periphery, which directly hyper-Figure 1
Key elements of osteoarthritis (OA) pain pathophysiology and examples of pharmacological intervention points. Observations of pain resolution following intra-articular local anesthetic and following joint replacement would implicate a peripheral drive in the majority of OA patients. In the periphery, the interaction between structural pathology, and the immune and nervous systems perpetuate the pain experience. Over time, as structural pathology develops, the principle algogenic mechanisms and mediators will change. Furthermore, dysfunction in central processing of information at the spinal and cortical levels has also been observed in OA patients, affecting both sensory and motor systems. This, in combination with altered affective and cognitive functions, may underpin the pain experience in other patient subsets. ASIC, acid-sensing ion channel; BDNF, brain-derived neurotrophic factor; CB, cannabinoid receptor; CCR, chemokine receptor; CGRP, calcitonin gene-related peptide; COX, cyclo-oxygenase; DOR, delta opioid receptor; EP, E prostanoid receptor; FAAH, fatty acid amide hydrolysis; GABA, gamma-amino butyric acid; IL, interleukin; mGluR, metabotropic glutamate receptor; mPGES, membrane or microsomal PGE synthase; N-type Ca2+, neuronal-type calcium
channels; NE, noradrenaline; NGF, nerve growth factor; NR2B, -N-methyl-D-aspartate receptor 2B subunit; P2X, purinergic 2X ionotropic receptor; SSRI, selective serotonin reuptake inhibitor; SubP, substance P; T-type Ca2+, transient type Ca2+channels; TNF, tumor necrosis factor; TNFR,
[image:2.612.108.473.106.344.2]polarizes sensory neurones and attenuates nerve hyper-excitability caused by inflammation or injury [10,11], raises the possibility of therapy with minimal central nervous system (CNS) side effects. In keeping with this, limited clinical trials of intra-articular delivery of morphine in OA support the concept of peripherally restricted opiate analgesia [12]. Furthermore, novel mu-opioid ligands, such as [8-(3,3-diphenyl-propyl)-4-oxo-1-phenyl-1,3,8-triaza spiro [4.5]dec-3-yl]-acetic acid (DiPOA) and the antidiarrheal drug loperamide, which also do not penetrate the blood brain barrier, have shown efficacy in a number of post operative, inflammatory and bone cancer pain models [13,14].
Delta-opioid receptor (DOR) agonists have the potential for analgesic efficacy without the confounding side effects of other opioid receptor therapies (see [15] for a review). Thus, analgesia has been shown in primate and non-primate pain models with a number of DOR ligands, for example, [D-Pen2,D-Pen5]enkephalin, SNC80 and AM-390. However, DOR efficacy depends on the pain stimulus, the type of injury and the influence of the local neurochemical environment. Thus, delta ligands have low analgesic efficacy in acute pain models but show robust analgesia efficacy in a variety of chronic pain conditions accompanied by inflammation [16,17]. This can be explained by stimulus-dependent trafficking of DOR from the cytoplasm to nerve membranes in CNS neurons [16]. There is little clinical development of DOR agonists for analgesia, although ADL 5859 [17] is reported to be in clinical phase 1 for analgesia.
Kinins and their receptors
Bradykinin is an important mediator of inflammatory pain causing nociceptor activation and sensitization via B2 receptors [18]. The abundant metabolite of bradykinin, des-Arg9-bradykinin (kallidin), activates B1 receptors, which occur in low abundance, in the periphery and CNS [19-21].
B2 receptors undergo desensitization following prolonged kinin exposure, whereas B1 receptors do not desensitize rapidly and are dramatically up-regulated in many tissues following injury [22-25] or exposure to IL-1β or the neuro-trophin glial-derived neurotrophic factor (GDNF) [23,26]. Importantly, kinins cause a cascade of secondary changes, including prostanoid and nitric oxide production, phosphory-lation of signaling proteins such as PKC, and the sensitization of sensory transducers such as the transient receptor potential vanilloid (TRPV)1 receptor [27]. These events are linked with heat and mechanical hyperalgesia [28,29]. In keeping with this, B2 antagonists (for example, Icatibant and bradyzide) and a B1 antagonist (des-Arg10 HOE-140; SSR240612) produce robust anti-hyperalgesic effects in models of nerve injury-induced pain [30-33]. Importantly, intra-articular administration of Icatibant (HOE 140) in OA patients was shown to reduce pain intensity at rest and during activity [33].
Cannabinoids and their receptors
Two cannabinoid receptors, CB1 and CB2, are associated with pain modulation (reviewed in [35]). CB1 receptors are widely distributed in the CNS and peripheral sensory neurons while CB2 receptors have been found in peripheral tissues, including tissues of the immune system and keratinocytes, with limited expression in sensory and CNS cells [36]. More recently, constitutive expression of both CB1 and CB2 receptors have been isolated on chondrocytes and implica-ted in a potential disease modifying role in OA [37]. Several fatty acids, for example, anandamide, 2-arachidonylglycerol, and palmitoylethanolamide, have been identified as the endogenous ligands for these receptors while specific antagonists, such as SR141716A and SR147778 for CB1 and SR144428 for CB2, have been used to characterize receptor functions.
CB1 receptors attenuate pain by reducing peripheral nerve excitability and through inhibition of sensory transmitter release [38]. In the CNS, brain stem structures such as the periaqueductal grey appear to be important for stress-induced release of endocannabinoids, and CB1-stress-induced analgesia may involve activation of descending pathways that inhibit spinal excitability [39,40].
Several clinical studies have shown that many cannabinoids, such as delta(9)-tetrahydrocannabinol, that reduce pain by a CNS action also produce adverse effects, such as euphoria, dizziness and sedation [41]. Targeting peripheral cannabinoid receptors can reduce CNS side effects. Thus, localized administration of HU210 or oral administration of CB1 agonists with limited CNS availability, such as CT-3 (ajulemic acid), produced analgesia both in pain models [42,43] and in the clinic at a dose that causes minimal CNS side effects [44].
CB2 agonists (for example, HU-308, HU-210, CP55940, AM1241 and GW405833) also modulate acute and chronic pain [45-47] while JWH-133 also shows anti-inflammatory activity [48]. It is unclear how these effects are produced since few CB2 receptors are found in the CNS or on sensory neurons [49]. However, CB1 like side effects (sedation, catalepsy, motor impairments) have not been seen with CB2 selective compounds.
still unclear whether such synergy can be exploited in chronic pain treatment such as OA.
Prostanoids and receptors
A variety of prostanoid cyclo-oxygenase (COX) enzyme products (prostaglandin (PG)E2, PGD2, PGF2α, throm-boxane, PGI2) are made during inflammation, but PGE2 is considered to be the major contributor to inflammatory pain. Thus, blocking the major synthetic enzymes COX-1 and COX-2 or inhibition of prostanoid receptors continue to be important approaches for reducing inflammatory pain. PGE2 exerts its effects via a variety of E prostanoid (EP) receptors (EP1, EP2, EP3, EP4), which are present in both peripheral sensory neurones and the spinal cord. Activation of these receptors produces a complexity of effects, ranging from calcium influx to cAMP activation or inhibition. Sensitization of nociceptors by PGE2 is caused by the cAMP-mediated enhancement of sodium currents via ion channel phosphorylation [55,56]. However, in the spinal cord, prostaglandin-induced hyperexcitability was enhanced by EP1 receptors but reduced by an EP3α agonist (ONO-AE-248), suggesting further complexity in the prostanoid regulation of pain [57].
In addition to their important roles in the periphery, COXs are also present in the CNS. Important for pain is the increased spinal cord expression of COX-1 (glia) and COX-2 (ventral horn cells) caused by inflammation, peripheral nerve injury or cytokines. In keeping with this, several non-steroidal anti-inflammatory drugs (NSAIDs) have been shown to reduce inflammatory hyperalgesia via inhibition of spinal COX activity [58]. Several mechanisms have been proposed, including EP1 receptor activation and spinal release of glutamate as well as loss of spinal glycine receptor mediated inhibition [59]. Recently, COX-3 has been identified as a splice variant of COX-1 [60] and several NSAIDs (acetaminophen, diclofenac, phenacetin) show low efficacy but some degree of selectivity for COX-3. However, COX-3 has low enzymic capability and its distribution and low abundance in the CNS and periphery does not make this a compelling target for analgesia.
Since the 1990s, COX-2 selectivity has been associated with cardiovascular concerns following observations of reduction in anti-thrombotic prostacylin metabolites but not pro-thrombotic thromboxane A2 in urine. Large scale, controlled clinical trials for COX-2 inhibitors (VIGOR, CLASS, TARGET) comparing efficacy and safety of rofecoxib, celecoxib and lumiracoxib with traditional NSAIDs have confirmed an increased risk of serious cardiovascular events compared to placebo. Many key questions remain unanswered concerning the mechanism of cardiovascular risk of selective COX-2 inhibitors (see [61] for a review). Despite this uncertainty, development of COX-2 selective inhibitors still continues (for example, GW406381), reflecting the attraction of this path-way and the requirement for newer drugs with improved overall safety profiles.
An alternative route of PGE2 inhibition is via the blockade of PGE synthase (PGES), a major route of conversion of prostaglandin H2 to PGE2. Two iso-forms of the enzyme have been identified, membrane or microsomal associated (mPGES-1) and cytosolic (cPGES/p23), which are linked with COX-2 and COX-1 dependent PGE2 production, respectively [62,63]. Both isoforms are up-regulated by inflammatory mediators, and gene deletion studies in mice indicate an important role for mPGES in acute and chronic inflammation and inflammatory pain [64]. Additionally, inhibition of mPGES is thought to be associated with lower cardiovascular risk since PGI2 production would not be affected.
Cytokines, chemokines and their receptors
Inflammatory stimuli initiate a cascade of events, including the production of tumor necrosis factor (TNF)α, ILs, chemokines, nerve growth factor (NGF), sympathetic amines, leukotrienes and PGs, with a complex impact on pain production. Cyto-kines induce hyperalgesia by a number of direct and indirect actions. Thus, IL1β activates nociceptors directly via intra-cellular kinase activation, but it may also cause indirect nociceptor sensitization via the production of kinins and prostanoids [65]. TNFα also activates sensory neurones directly via the receptors TNFR1 and TNFR2 and initiates a cascade of inflammatory reactions through the production of IL1, IL6 and IL8 [66,67]. It is significant that direct TNFα application in the periphery induces neuropathic pain behavior that is blocked by ibuprofen and celecoxib [68], while nerve ligation causes increased TNFα in damaged as well as adjacent undamaged axons [69]. Interestingly, anti-TNFα treatment with the TNF antibody adalimumab produced a prolonged reduction of pain symptoms in OA [70]. These are encouraging preliminary data but will require further support.
sensory afferents, or desensitization of the endogenous opioid system.
Adrenergic receptors
Several chronic pain disorders termed ‘sympathetically main-tained pain’ have highlighted the importance of the release of sympathetic transmitters (epinephrine or norepinephrine) from sympathetic varicosities and the involvement of adrenergic receptors in pain etiology. The joint capsule, synovium and bone are richly innervated by sympathetic postganglionic neurons [74]. These regulate vascular tone and permeability, bone homeostasis and, during inflammation, sensitizing of afferent sensory pathways. In rheumatoid arthritis, sympathetic innervation is reduced, probably by increased release of sympathetic nerve repellents such as semaphorins, although no such denervation is observed in OA [75]. Interactions between sympathetic and afferent peripheral neurons may take place at several sites. NGF may play an important role in linking sympathetic and C-fibre inner-vation as sympathetic actiinner-vation stimulates NGF secretion from vascular smooth muscle [76]. Other pain conditions have demonstrated sympathetic/sensory coupling at the level of the dorsal root ganglion [77] and at the peripheral sites of injury (for example, neuroma) [78].
Studies have also shown the expression of α-1 and α-2 adrenergic receptors on sensory neurons or on post-ganglionic sympathetic terminals after nerve injuries [79,80]. Under these conditions sensory neurones can be directly activated by the endogenous release of sympathetic trans-mitters (via α-1 receptors) or in the clinic by intradermal injection of norepinephrine [81].
Clonidine and other α-2 agonists such as dexmedetomidine have also been used systemically to inhibit sensory trans-mission in the spinal cord by block of pre- and postsynaptic membrane excitability and intra-articularly following joint replacement. Unfortunately, sedation and hypotension are major target-related systemic side effects of these compounds. Great efforts have been made to identify ligands with improved α-2 receptor subtype selectivity, to avoid side effects, but thus far this has not been particularly successful.
Glutamate regulation and glutamate receptors
In OA, synovial fluid levels of glutamate and aspartate are significantly elevated above controls [82]. Glutamate acts through a variety of receptor-coupled, ligand-gated ion channels, including α -amino-3-hydroxy-5-methylisoxazole-4-proprionate (AMPA)/kinate receptors, ionotropic glutamate receptors (iGluRs) and G-protein coupled metabotropic glutamate receptors (mGluRs). Injections of glutamate or metabolically stable receptor-selective agonists such as NMDA, AMPA, and kainate cause a pro-nociceptive response upon thermal and mechanical stimulation, while application of iGluR and mGluR antagonists attenuate pain in acute models (see [83,84] for reviews). Glutamate may also have a
disease-modifying role, with receptors found on non-neuronal cells, that is osteoblasts, osteoclasts, and chondrocytes, mediating bone remodeling and cartilage mechano-trans-duction, respectively [85,86].
NMDA antagonists show robust attenuation of pain behaviors but also induce a number of side effects (sedation, confusion, motor incoordination) and thus have insufficient therapeutic margin. There has been a refocus on more specific NMDA-receptor subtype blockers (NR1 and NR2) directed towards the strychnine-insensitive glycineBmodulatory site to avoid side effects. This site modulates the NMDA channel only during the sustained stimulation of the receptor, which is considered to occur during chronic pain. Selective NR1-Gly antagonists have been claimed to reduce pain with reduced side effects [87,88]. However, clinical experience has not confirmed this. GV196771 did not show efficacy against clinical pain, possible due to inadequate penetration into the CNS [89].
Alternative initiatives have targeted other NMDA receptor subtypes, such as the NR2B receptor, which has a specific distribution in sensory pathways. Blockade of this receptor has also been claimed to produce anti-nociception (ifen-prodil, traxoprodil (CP-101,606)) with reduced side effects [90]. To date, traxoprodil has advanced into phase I safety and efficacy study for acute ischemic stroke.
The mGluRs, particularly mGluR1 and mGluR5, have been reported to play a key role in sustaining heightened central excitability in chronic pain with minimal involvement in acute nociception. Thus, spinal administration of selective agonists such as dihydroxy phenyl glycine produced allodynia, while mGluR5 was shown to be significantly over-expressed in some, but not all, chronic pain models [91]. Peripheral mGluR5 receptors have also been claimed to modulate pain. Thus, local administrations of mGluR5 antagonists 2-methyl-6[phenylethynyl]-pyridine (MPEP) and SIB1757 have been effective in reducing pain behavior, suggesting a potential use in pain therapy [92,93].
Metabotropic group II receptors (mGluR2 and mGluR3) also modulate pain transmission. mGluR2 is located in sensory neurones and presynaptic nerve terminals whereas mGluR3 is found all over the brain. mGluR3 can be selectively increased in the spinal dorsal horn neurones after peripheral UV injury [94]. mGluR2/3 receptor activation appears necessary to reduce nerve terminal excitability and to modulate pain transmission since treatment with the agonist L-acetyl carnitine reduced inflammatory hyperalgesia and mechanical allodynia and increased the expression of mGluR2/3. The effects of L-acetyl carnitine were attenuated by LY379268, an mGluR2/3 antagonist [95].
Ion channels
targeted for pain control. The mammalian TRP channel represents a large receptor family, subdivided into six subfamilies: TRPA, TRPC, TRPM, TRPP, TRPV, and mucolipin. Many TRP channels are localized to sensory neurones and play a major role in temperature and mechanical transduction.
TRPV1 is a non-selective cation channel, gated by capsaicin, noxious heat (>45°C), acidic pH (<5.3), and regulated by a variety of inflammatory agents, including protons, bradykinin, ATP, PGE2, 12-lipoxygenase products, protease-activated receptor-2, anandamide, CCL3 and NGF. Sensitization of TRPV1 involves a variety of pathways that regulate receptor phosphorylation [96]. Analgesia approaches in OA have used capsaicin preparations or capsaicin-like agonists to induce TRPV1 desensitization or reversible sensory nerve terminal degeneration caused by prolonged cation influx into the nerve, osmotic damage and metabolic collapse [97]. In a randomized study of intra-articular injections of placebo or capsaicin (ALGRX 4975) prior to knee replacement, ALGRX 4975 was found to decrease visual analogue scales (VAS) scores without effecting proprioreception or joint histopathology [98]. Currently, there is a focus on TRPV1 channel blockers or selective TRPV1 receptor antagonists [99]. Supporting these approaches, competitive (AMG-9810) [100] and non-competitive (DD161515) [99] TRPV1 antagonists block chemical and thermal pain sensitivity, heralding the emergence of a novel therapy. Indeed, recent studies in volunteers have shown that oral SB705498 attenuated capsaicin and ultra-violet (UV)-induced pain and hyperalgesia [101]. Other TRP channels (TRPV3, TRPV4, TRPA1) have also been suggested to be involved in pain transduction. Thus, TRPA1 (ANKTM1) is co-localized with TRPV1 and is activated by capsaicin and mustard oil but can also be sensitized by inflammatory mediators, including bradykinin, known to be significantly elevated in osteoarthritic synovial fluid, to produce cold-induced burning pain [102]. In addition, TRPV1 can oligomerize with other TRP family members, including TRPV3. The latter is found in keratinocytes and appears to be upregulated in inflammatory pain conditions. So far there are few reliable chemical tools to help characterize the functions of these TRP receptors and support their value as analgesia targets.
Purinergic receptor-regulated channels
The unique localization of the purinergic 2X ionotropic (P2X)3 receptor to small sensory fibres has highlighted its importance in pain. Large amounts of the endogenous ligand ATP are released after tissue injury and during inflammatory injuries while both ATP and a stable analogue, α,β-methyl ATP, induce pain and are pronociceptive when administered intradermally in volunteers [103].
In chronic inflammatory pain, P2X3-mediated excitability is enhanced while reduction of P2X3 receptors by antisense oligonucleotide administration reduces inflammatory hyper-algesia as well as that evoked by α,β-methyl ATP [104]. In
keeping with this, several antagonists, including 2′,3′ -O-(2,4,6-trinitrophenyl)-adenosine triphosphate (TNP-ATP), pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid, and suramin, reduce pain behavior. More selective, and drug like, antagonists, such as A-3174919, reduced pain in a number of acute and chronic pain models, supporting the possibility for future analgesia therapy of nociceptive pain such as OA [105].
It should be noted that several other purinergic receptor sub-types, including P2X4 and P2X7, have also been suggested to modulate pain through altered central excitability and the release of neuroglial-cell products [106-108]. Thus, activated microglia, astrocytes and satellite cells release a variety of inflammatory mediators, including IL1β, TNFα, prostanoids and nitric oxide upon ATP stimulation. Indeed, increased expression of P2X4 has been shown to occur in spinal microglia after peripheral nerve lesions and this was related to painful mechanical allodynia. This behavior was blocked by spinal administrations of the selective P2X4 antagonist TNP-ATP [106]. Remarkably, spinal administration of activated microglia reproduced TNP-ATP sensitive mechanical allodynia in naïve animals.
Increased P2X7 expression has been found in peripheral macrophages following inflammation but this receptor is also expressed in spinal neurones and microglia following peripheral nerve injury [107]. In keeping with an important role in chronic pain, both microglia and P2X7 receptors are up-regulated in human chronic pain patients [108] while deletion of the P2X7 receptor gene produced a complete absence of mechanical and thermal pain in mice [108].
It is worth noting that other nucleotide-gated ion channels have also been shown to be important for regulating periph-eral excitability. Thus, the Na/K re-polarizing ‘pacemaker current’, Ih, which is activated during membrane hyper-polarization, is important for generation of rhythmic and spontaneous action potentials in sensory neurons. Ih currents are controlled by cyclic nucleotides (cAMP and cGMP) via a family of hyperpolarization-activated, cyclic nucleotide-gated (HCN1-4) ion channels. These have been found to be differentially expressed and redistributed after inflammatory nerve injuries [109,110].
Acid sensing ion channels
emerging, but data indicate specificity differences for both species and nerve fibre subtypes (Isolectin B4-/+) [112].
A novel blocker (A-317567) of peripheral ASIC 1, 2 and 3 channels has been described [113]. This reduces hyper-algesia in models of inflammatory and post-operative pain, but there have been no reports of therapeutic advances with ASIC inhibitors.
Sodium channels
Voltage-gated sodium channels are characterized by their primary structure and sensitivity to tetrodotoxin (TTX). A variety of TTX sensitive (NaV1.3, Nav1.7) and TTX insensitive (NaV 1.8, NaV1.9) channels are involved in regulating sensory neural excitability [114,115]. Changes in the expression, trafficking and redistribution of NaVs following inflammation or nerve injury are considered to account for the abnormal firing and the generation of ectopic activity in afferent nerves [116]. Mutations of NaV1.7 have been identified as the cause of burning pain in erythromelalgia [117], while inflammation causes the over-expression of NaV 1.7 in animal models and in inflamed human tooth pulp [118]. Interestingly, NaV1.7 over-expression could be prevented by pre-treatment with COX-1 and COX- 2 inhibitors (ibuprofen, NS-398).
The clinical utility of non-selective Na channel blockade in OA pain has been well established with the experimental use of local anesthetics such as intra-articular levobupivacaine, the active enantiomer of bupivacaine. It is noteworthy that the OA population is stratified in response to intra-articular local anesthetic, indicating a significant central component to the pain in some patients [3]. Systemic and central exposure to local anesthetics has been attempted in other pain paradigms. Intravenous administration has been reported to produce long lasting pain relief in both animal models [119] and intractable neuropathic pain [120]. The major dis-advantages of the systemic use of non-selective Na channel blockers are cardiotoxicity and CNS sedation and confusion, considered to be produced by NaV1.5 and NaV1.2 channel blocking, respectively. Considerable activity is currently focused on discovering novel, selective Na channel blockers.
An alternative approach to regulate ion channels is to block the trafficking of channels to the nerve membrane. For example, the functioning of NaV1.8 may be reduced by preventing an association with p-11, an annexin II related protein that tethers the channel to the nerve membrane [121]. In addition, channel-associated cell surface glycoproteins such as contactin may be involved in concentrating specific channel subtypes, for example, NaV1.8 and NaV1.9 (Isolectin B4+) but not NaV1.6 and NaV1.7 (Isolectin B4-) in DRG nerve membranes, with an associated increased in ionic current density [122]. Although these approaches are attractive, they have not been explored significantly and it is unclear whether they will impact on nerve excitability associated with specific pain etiology.
Calcium channels
Voltage-gated calcium channels are subdivided into two major categories, low voltage-activated calcium channels (T-type channels) and high activated. High voltage-activated channels are further subdivided, based on pharma-cology and biophysical characteristics, into L-, N-, R-, P-, and Q-types. Several have been shown to be prominently involved in pain regulation [123]. The N-type calcium channel is an important regulator of nerve terminal excitability and neuro-transmitter release. N-type channels can be regulated, particularly through GPCR signaling by analgesic drugs such as opioids, with a resultant modulation of sensory transmitter release, for example, substance P, calcitonin gene-related peptide (CGRP) and glutamate, at both spinal and peripheral sensory nerve terminals. Channel trafficking may also be affected; for example, activation of the opioid receptor-like receptor by nociceptin causes channel internalization and downregulation of calcium entry [124].
Gene deletion of the α2δ subunit of the N-type channel reduces inflammatory and neuropathic pain [125,126]. Moreover, selective blockers such as Ziconotide (SNX-111, Prialt; a synthetic form of omega-conotoxin) and verapamil have been used to characterize channel activity while Ziconitide has been used experimentally and clinically by spinal intrathecal administration for pain relief [127,128]. Building on this concept, small molecule channel blockers, with oral availability, are now reported to be undergoing clinical evaluation for analgesia, for example, NMED-160 [128].
Low voltage-activated T channels also appear important for pain transmission and as targets for pain therapy. Thus, they are expressed in superficial laminae of the spinal cord and in dorsal root ganglion neurones [123]. T-channels play a prominent role in regulating spinal excitability and spinal sensitization following repetitive C-fibre stimulation [129]. Moreover, nerve injury-induced hyper-responsiveness was blocked by the T-channel blocker ethosuximide [130], which also attenuated mechanical allodynia in animal models of vincristine and paclitaxel-induced neuropathic pain [131].
was observed in patients with knee OA but patients with hip OA experienced improvement in sleep quality and improve-ments in the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) pain subscale [134].
Neurotrophins and their receptors
Neurotrophins and their receptors represent an important family of regulatory proteins essential for sensory nerve development, survival and the determination of neurochemical phenotype important for the regulation of excitability [135,136]. Several neurotrophins (NTs) have been identified, including NGF, brain derived growth factor (BDNF) and NT3 and NT4/5. Each NT binds with high affinity to a receptor tyrosine kinase (Trk): NGF to TrkA, BDNF and NT4/5 to TrkB and NT3 to TrkC. NT3 also binds with TrkA and TrkB. Mature NTs also bind to a structurally distinct receptor, p75, which affects neuronal development through downstream signaling. NTs arise from pro-NT precursors following extracellular cleavage by metalloproteinases and plasmin. It is notable that pro-NTs may signal through the p75 receptor in a manner that opposes the effects of NTs, for example, to produce apoptosis rather than cell survival [137].
NGF has been most studied with respect to inflammatory hyperalgesia as its production is unregulated by inflammation in macrophages, fibroblasts and Schwann cells. NGF has emerged as a key regulator of sensory neurone excitability and as an important mediator of injury-induced nociceptive and neuropathic pain [138-140]. Thus, NGF acts via TrkA and p75 to activate a number of other kinase pathways, for example, that of p38 kinase, leading to altered gene transcription and increased synthesis of sensory neuropeptides (substance P, CGRP), ion channels (TRPV1, NaV1.8, ASIC3) [141-143], membrane receptors such as bradykinin and P2X3 [144,145], and structural molecules, including neurofilament and channel anchoring proteins such as the annexin light chain p11 [121].
Increased expression and release of NGF have been demonstrated in several painful conditions in animal models (for example, UV injury, surgical injury) [146,147] and in human conditions, including arthritis, cystitis, prostitis and headache [148-150]. Administration of exogenous NGF induces thermal and mechanical hyperalgesia in animals and humans [151,152], which is considered to be due, in part, to mast cell degranulation and by directly increasing sensory neuronal excitability [153].
Only a few small molecule NGF antagonists are available, but ALE0540, which inhibits the binding of NGF to TrkA and p75, and PD90780, which inhibits NGF binding to p75, have been proposed to have efficacy in chronic pain models [154,155]. The importance of NGF has also received clinical confirmation since RN624, a humanized ant-NGF monoclonal antibody, has been reported to be efficacious in reducing pain and improved mobility in OA [156]. Anti-NGF mono-clonal antibody therapy appears to be an attractive
thera-peutic approach with the potential for long lasting pain treat-ment, similar in efficacy to morphine, without compromising physiological nociception.
NGF also induces the synthesis and accumulation of BDNF in peptide-containing sensory neurones following painful nerve injury [135]. Release of BDNF in the spinal dorsal horn increases spinal excitability and pain sensitization via TrkB receptors. This initiates a variety of effects, including direct neural excitation, activation of a signaling cascade via the phophorylation of NMDA receptors, and altered regulation of the neural chloride-ion transporter that contributes to pain hypersensitivity [157]. In addition, spinal BDNF administration induces thermal and mechanical allodynia whereas anti-BNDF neutralization or TrkB IgG administration reduces inflammation or nerve injury hypersensitivity in a number of animal models [139,158,159].
Finally, GDNF represents an extensive family of ligands and membrane receptor complexes that have an important role in regulating peripheral and central neural phenotypes. GDNF related ligands include neurturin and artemin, which act via the complex c-Ret proto-oncogene receptor tyrosine kinase and co-receptors glial cell line-derived neurotrophic factor receptor (GFR)α1, α2, α3 and α4. Although there appears not to be a specific role in inflammation, GDNF has been shown to have neuroprotective and restorative properties in a number of neurodegenerative and neuropathic pain states [135]. Specifically, GDNF treatment has been shown to restore peripheral sensory neurone function, including peptide and ion channel expression patterns, following painful peripheral nerve injury accompanied by an attenuation of pain behaviors. Unfortunately, clinical observations using GDNF have shown unacceptable side effects, such as weight loss and allodynia, which has discouraged therapeutic developments [160].
Botulinum toxin
Another approach to pain modulation has been the use of botulinum toxins (BoTNs). The mechanism of action of BoTN is related to inhibition of transmitter release from motor fibers through proteolytic cleavage of a number of synaptosomal regulatory proteins (soluble N-ethyl maleimide-sensitive fusion protein attachment protein receptors (SNAREs), syntaxin, synaptosome-associated protein of 25 kDa (SNAP-25) and synaptobrevin). More recent studies have also indicated potential for inhibition of neuropeptide transmitter release from small afferent nerves [161,162]. In keeping with this, BoNT has been shown to provide long lasting pain relief following administration into human OA joints [163] and improve bladder dysfunction in overactive bladder patients. This was correlated with loss of both P2X3 and VR1 receptors in the urinary bladder [164].
development. Animal models of cutaneous inflammatory pain were developed initially as pharmacodynamic assays of anti-inflammatory drug activity, particularly for NSAIDs. Typically, primary endpoints were reduction in hindpaw swelling, induced by Freund’s adjuvant or carrageenan, and reflex limb withdrawal to a mechanical stimulation. At this time, the lack of activity of NSAIDs in models of acute nociceptive pain, such as the tail-flick [165] and hot plate assays [166], raised an awareness that clinical pain pathophysiology and pharma-cology, in which a sensitized state is induced in the presence of inflammation (or nerve damage), differ significantly from normal physiological pain observed in healthy animals. From that time a major emphasis on models that reproduce specific elements of chronic pain have allowed the systematic mecha-nistic exploration of excitability changes in pain pathways [167]. This has also provided the building blocks for rational translation of findings in animal models, for example, pharmacodynamic/pharmacokinetic measures of the reduction of neuro-excitability and pain behavior to reduction of clinical pain.
However, there is concern that current models still lack the tissue and disease specificity of some key patient popula-tions. OA pain is an example where an improved clinical understanding of joint pathology and its relationship to pain can focus disease specific approaches. Magnetic resonance imaging studies have reported significant association of specific tissue pathologies such as subchondral bone lesions, synovial thickening and knee effusion with pain [168-170]. These clinical observations, along with histo-pathology samples from joint arthroplasty, synovial fluid collections and so on, allow an investigation of specific elements of structural pathology, the potential mediators involved and the presence/absence of pain. It is clear that while no single animal model replicates human OA, specific elements can be modeled in animals. The choice of model, interpretation of endpoints and translation to the clinic are critical future challenges in therapeutic development.
While a comprehensive analysis of OA models is beyond the scope of this review, recent developments have focused on intra-articular injection of monoiodoacetate into rodent femorotibial joint or surgical destabilization of the joint in rats and guinea pigs. These models seek to emulate aspects of OA pathology. For example in the monoiodoacetate model following chondrocytic cell death and cartilage fragmentation, a subchondral bone lesion develops with active resorption and remodeling of cancellous bone typically by day 21. Inflammation is observed as mononuclear cell infiltrates and hyperplastic synovium but this is transient and resolves [171-173]. In addition, mechanical allodynia (weight bearing) [173,174] and mechanical hyperalgesia (von Frey hair stimulation) [175] are exhibited. Further characterization shows that, in the early stages, there is sensitivity to NSAIDs [173,174] whereas later stages appear to demonstrate evidence of nerve damage with elevated activating
transcription factor-3 (ATF-3) immunoreactivity in innervating cell bodies of lumber-DRG and sensitivity to morphine, amitriptyline and gabapentin [173,176]. The correlation of bone lesion with onset of ATF-3 immunoreactivity makes osteoclast-induced injury or mechanical compression of bone Aδ and C-fibres candidate mechanisms for nerve damage. These observations indicate the importance of relating animal model histopathology with clinical samples to gain understanding of putative analgesic targets and to propose clearer hypotheses for testing. Detailed translation of this kind may also be applied to the analysis of OA heterogeneity and the evaluation of personalized approaches to OA treatments.
Summary and conclusions
Clinical presentation of OA is dominated by pain during joint use and often at rest. Effective pain therapy has been a key therapeutic challenge not only in OA but in a variety of chronic pain disorders. OA represents a complexity of pain conditions, including manifestations of both nociceptive and neuropathic mechanisms driven by joint pathophysiology and abnormal excitability in peripheral and central pain pathways. A mechanisms-based focus on the key molecular drivers of neural excitability offers a multiplicity of possible intervention points. Indeed, a rich diversity of molecular events has been identified in the pathophysiology of chronic pain, representing most families of regulatory proteins. Many molecules are inflammatory mediators and their key receptors (kinins, mPGES) while others, such as ion channels (TRPV1, NaV1.7) and NTs (NGF), are key regulators of membrane excitability and cellular phenotype. We have highlighted these and a number of other important targets for future pain therapy, noting in particular evidence that relates to their participation in animal model systems of OA, translatability to humans as well as efficacy in the clinical setting. The future treatment of pain appears optimistic but will require the systematic evaluation of emerging opportunities.
Competing interests
Both authors are employees and shareholders of AstraZeneca Pharmaceuticals.
References
1. Bellamy N, Sothern RB, Campbell J: Rhythmic variations in pain perception in osteoarthritis of the knee. J Rheumatol1990, 17: 364-372.
2. Bellamy N, Sothern RB, Campbell J, Buchanan WW: Rhythmic variations in pain, stiffness and manual dexterity in hand osteoarthritis. Ann Rheum Dis2002, 61:1075-1080.
This review is part of a series on
Arthritis and pain
edited by Jason McDougall.
Other articles in this series can be found at http://arthritis-research.com/articles/
3. Creamer P, Hunt M, Dieppe P: Pain mechanisms in osteoarthri-tis of the knee: Effect of intra-articular anaesthetic. J Rheuma-tol1996, 23:1031-1036.
4. Crawford RW, Gie GA, Ling RS, Murray DW: Diagnostic value of intra-articular anaesthetic in primary osteoarthritis of the hip. J Bone Joint Surg1998, 80:279-281.
5. Kosek E, Ordeberg G: Lack of pressure pain modulation by heterotopic noxious conditioning stimulation in patients with painful osteoarthritis before, but not following, surgical pain relief. Pain2000, 88:69-78.
6. Buffington ALH, Hanlon CA, McKeown MJ: Acute and persistent pain modulation of attention-related anterior cingulated fMRI activations. Pain2005, 113:172-184.
7. Rowbotham MC, Kidd BL, Porreca F: Role of central sensitiza-tion in chronic pain: Osteoarthritis and rheumatoid arthritis compared to neuropathic pain. In Proceedings of the 11th World Congress on Pain. August 21 - 26th 2005, Sydney, Aus-tralia. Edited by Flor H, Kalso E, Dostrovsky J. Seattle; IASP Press: 2006.
8. Fraenkel M, Bogardus ST, Concato J, Wittink DR: Treatment options in knee OA. Arch Intern Med2004, 164:1299-1304. 9. Yaksh TL: Pharmacology and mechanisms of opioid analgesic
activity. Acta Anaesthes Scand1997, 41:94-111.
10. Hurley RW, Hammond DL: The analgesic effects of supraspinal µµand δδopioid receptor agonists are potentiated during per-sistent inflammation. J Neurosci2000, 20:1249-1259. 11. Sawynok J: Topical and peripherally acting analgesics.
Phar-macol Rev2003, 55:1-20.
12. Likar R. Schafer M. Paulak F. Sittl R. Pipam W. Schalk H. Geissler D. Bernatzky G: Intraarticular morphine analgesia in chronic pain patients with osteoarthritis. Anesth Analg1997, 84: 1313-1317.
13. Menendez L, Lastra A, Meana A, Hidalgo A, Baamonde A: Anal-gesic effects of loperamide in bone cancer pain in mice. Phar-macol Biochem Behav2005, 81:114-121.
14. Whiteside GT, Boulet JM, Walker K: The role of central and peripheral mu opioid receptors in inflammatory pain and edema: a study using morphine and DiPOA ([8-(3,3-diphenyl-propyl)-4-oxo-1-phenyl-1,3,8-triaza-spiro[4.5]dec-3-yl]-acetic acid). J Pharmacol Exp Ther2005, 314:1234-1240.
15. Dray A: Alternatives to mu-opioid analgesics: delta-opioid and galanin-receptor selective compounds. In Progress in Pain Research and Management. Edited by Kalso E, McQuay HJ, Wiesenfeld-Hallin Z. Seattle: IASP Press; 1999:269-280. 16. Cahill CM, Morinville A, Lee M-C, Vincent J-P, Collier B, Beaudet
A: Prolongedmorphine treatment targets δδopioid receptors to neuronal plasma membranes and enhances δδ-mediated antinociception. J Neurosci2001, 21:7598-7607.
17. Koblish M, LaBuda CJ, Ajello CW, Gu M, Zhou QJ, Tuthill PA, Chu G, Le Bourdonnec B, Dolle RE, Little PJ:Anti- hyperalgesic activity of a novel delta-opioid receptor agonist. Soc Neurosci Abst2005, 490.14.
18. Dray A: Kinins and their receptors in hyperalgesia. Can J Phar-macol1997, 75:704-712.
19. Dray A, Perkins MN: Bradykinin and inflammatory pain. Trends Neurosci1993, 16:99-104.
20. Wotherspoon G, Winter J: Bradykinin B1 receptor is constitu-tively expressed in the rat sensory nervous system. Neurosci Letts2000, 294:175-178.
21. Shughrue PJ, Ky B, Austin CP: Localization of B1 bradykinin receptors mRNA in the primate brain and spinal cord: an in situhybridization study. J Comp Neurol2003, 46:372-384. 22. Levy D, Zochodne DW: Increased mRNA expression of the B1
and B2 bradykinin receptors and antinociceptive effects of their antagonists in an animal model of neuropathic pain. Pain 2000, 86:265-271.
23. Fox A, Wotherspoon G, McNair K, Hudson L, Patel S, Gentry C, Winter L: Regulation and function of spinal and peripheral neuronal B(1) bradykinin receptors in inflammatory mechani-cal hyperalgesia. Pain2003, 104:683-691.
24. Ferreira, J, Beirith A, Mori MAS, Araujo RC, Bader M, Pesqero JB, Calixto JB: Reduced nerve injury induced neuropathic pain in kinin B1 receptor knock-out mice. J Neurosci2005, 25: 2405-2412.
25. Eisenbarth H, Rukwied R, Petersen M, Schmelz M: Sensitization to bradykinin B1 and B2 receptor activation in UV-B irradiated human skin. Pain2004, 110:197-204.
26. Vellani V, Zachrisson O, McNaughton PA: Functional bradykinin B1 receptors are expressed in nociceptive neurons and are upregulated by the neurotrophin GDNF. J Physiol2004, 560: 391-401.
27. Marceau F, Hess JF, Bachvarov DR:The B1 receptors for kinins. Pharmacol Rev1998, 50:357-386.
28. Liang YF, Haake B, Reeh PW: Sustained sensitization and recruitment of rat cutaneous nociceptors by bradykinin and a novel theory of its excitatory action. J Physiol2001, 532: 229-239.
29. Fox A, Kaur S, Li B, Panasar M, Saha U, Davis C, Dragoni I, Colley S, Ritchie T, Bevan S, et al.: Antihyperalgesic activity of a novel nonpeptide bradykinin B1 receptor antagonist in transgenic mice expressing the human B1 receptor. Br J Pharmacol2005, 144:889-899.
30. Stewart JM: Bradykinin antagonists: discovery and develop-ment. Peptides2004, 25:527-732.
31. Burgess GM, Perkins MN, Rang HP, Campbell EA, Browen MC, McIntyre P, Urban L, Dziadulewicz EK, Ritchie TJ, Hallet A, et al.: Bradyzide, a potent nonpeptide B2 bradykinin receptors antagonist with long lasting oral activity in animal models of inflammatory hyperalgesia. Br J Pharmacol2000, 129:77-86. 32. Gougat J, Ferrari B, Lionel S, Planchenault C, Poncelet M,
Maruani J, Alonso R, Cudennec A, Croci T, Guagnini F, et al.: SSR240612 [(2R)-2-[((3R0-3-(1,3-benzodiol-5-yl)-3-{[(6-methoxy- 2-napthyl)sulfonyl]amino}propanoyl)amino]-3-(4-{[2R,6S)-2,6-dimethylpiperidyl]methyl}phenyl}-N-isoproyl-Nmethylpropana mide hydrochloride], a new nonpeptide antagonist of the bradykinin B1 receptor: biochemical and pharmacological characterization. J Pharmacol Exp Ther2005, 309:661-669. 33. Gabra BH, Sirois P: Beneficial effects of chronic treatment
with the selective bradykinin B1 receptor antagonists, R-715 and R-953, in attenuating streptozotocin-diabetic thermal hyperalgesia. Peptides2003, 24:1131-1139.
34. Flechtenmacher J, Talke M, Veith D, Heil K, Gebauer A, Schoen-harting M: Icatibant induces pain relief in patients with osteoarthritis of the knee. Proceedings of the 9th World Con-gress of the OsteoArthritis Research SocietyInternation (OARSI). December 2-5 2004, Chicago. 12:P332.
35. Fox A, Bevan S: Therapeutic potential of cannabinoid receptor agonists as analgesic agents. Expert Opin Investig Drugs2005, 14:695-703.
36. Rice ASC, Farquhar-Smith WP, Nagy I:Endocannabinoids and pain: spinal and peripheral analgesia in inflammation and neuropathy. Prostaglandins Leukot Essent Fatty Acids2002, 66: 243-256.
37. Mbvundula EC, Bunning RA, Rainsford KD: Arthritis and cannabinoids: HU-210 and Win-55,212-2 prevent IL-1alpha-induced matrix degradation in bovine articular chondrocytes in vitro.J Pharm Pharmacol2006, 58:351-358.
38. Kreitzer Ac, Regeher WG: Retrograde signaling by endo-cannabinoids. Curr Opin Neurobiol2002, 12:324-330. 39. de Novellis V, Mariani L, Palazzo E, Vita D, Marabese I, Scafuro M,
Rossi F, Maione S: Periaqueductal grey CB1 cannabinoid and metabotropic glutamate subtype 5 receptors modulate changes in rostral ventromedial medulla neuronal activities induced by subcutaneous formalin in the rat. Neuroscience 2005, 134:269-281.
40. Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC: Pain modulation by release of the endogenous cannabi-noid anandamide. Proc Natl Acad Sci USA 1999, 96: 12198-12203.
41. Campbell FA, Tramer MR, Carroll D, Reynolds DJ, Moore RA, McQuay HJ: Are cannabinoids an effective and safe treatment option in the management of pain? A quantitative systematic review. BMJ2001, 323:13-16.
42. Dyson A, Peacock M, Chen A, Courade J-P, Yaqoob, Groark A, Brain C, Loong Y, Fox A: Antihyperalgesic properties of the cannabinoid CT-3 in chronic neuropathic and inflammatory pain states in the rat. Pain2005, 116:129-137.
43. Richardson JD, Kilo S, Hargreaves KM: Cannabinoids reduce hyperalgesia and inflammation via interactions with periph-eral CB1 receptors. Pain1998, 75:111-119.
45. Malan TP, Ibrahim MM, Lai J, Vanderah TW, Makriyannis A. Porreca F: CB2 cannabinoid receptor agonists: pain relief without psychoactive effects?Curr Opin Pharmacol 2003, 3: 62-67.
46. Labuda CJ, Koblish M, Little PJ: Cannabinoid CB2 receptor agonist activity in the hindpaw incision model of postopera-tive pain. Eur J Pharmacol2005, 527:172-175.
47. Valenzano KJ, Tafesse L, Lee G, Harrison JE, Bulet JM, Gottshall SL, Mark L, Pearson MS, Miller W, Shan S, et al.: Pharmacologi-cal and pharmacokinetic characterization of the cannabinoid receptor 2 agonist GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neu-ropharmacology2005, 48:658-672.
48. Elmes SJR, Winyard LA, Medhurst A, Clayton SJ, Wilson NM, Kendall AW, Chapman V: Activation of CB1 and CB2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain2005, 118:327-335.
49. Sokal DM, Elmes SJR, Kendall DA, Chapman V: Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurons via activation of Cb2 receptors in anaesthetized rats. Neuropharmacology 2003, 45:404-411. 50. Cravatt BF, Lichtman AH: The endogenous cannabinoid
system and its role in nociceptive behavior. J Neurobiol2004, 61:149-160.
51. Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH: Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolyse. Proc Natl Acad Sci USA2001, 98: 9371-9376.
52. Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BJ: Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprece-dented combination of potency and selectivity. J Pharmacol Exp Ther2004, 311:441-448.
53. Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breit-enbucher JG, Chaplan SR, Webb M: Inhibition of fatty acid amide hydrolase produces analgesia by multiple mecha-nisms. Br J Pharmacol2006, 148:102-113.
54. Cichewicz DL, McCarthy EA: Antinociceptive synergy between D9 tetrahydrocannabinol and opioids after oral administration. J Pharmacol Exp Ther2003, 304:1010- 1015.
55. England S, Bevan S, Dougherty RJ: PGE2 modulate the tetrodotoxin-resistant sodium current in neonatal dorsal root ganglion neurons via the cyclic AMP-protein kinase A cascade. J Physiol1996, 495:429-440.
56. Gold MS, Levine JD, Correa M: Modulation of TTX-R I Na by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci 1998, 18: 10345-10355.
57. Bar K-J, Natura G, Telleria-Diaz A, Tascher P, Vogel R, Vasques E, Schaible H-G, Ebersberger A: Changes in the effect of spinal prostaglandin E2 during inflammation: prostaglandin E9EP1-EP4) receptors in spinal nociceptive processing of input from the normal and inflamed knee joint. J Neurosci2004, 24: 642-651.
58. Yaksh TL, Dirig DM, Conway CM, Svensson C, Luo ZD, Isakson PC: The acute hyperalgesic action of non-steroidal, anti-inflam-matory drugs and release of spinal prostaglandin E2 is medi-ated by the inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J Neurosci2001, 21:5847-5853. 59. Harvey RJ, Depner UB, Wassle H, Ahmadi S, Heidl C, Reinold H,
Smart TG, Harvey K, Schultz B, Abo-Salem OM, et al.: Gly αα3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science2004; 304:884-887.
60. Chandrasekharan NV, Dai H, Roos KLT, Evanson NK, Tomsik J, Elton TS, Simmons DL: COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc Natl Acad Sci USA2002, 99:13926-13931.
61. Mitchell JA, Warner TD: COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov2006, 5:75-85. 62. Jakobsson PJ, Thoen S, Morgenstern R, Samuelsson B:
Identifi-cation of human prostaglandin E synthase: a microsomal , glutathione-dependent, inducible enzyme , constituting a potential novel drug target. Proc Natl Acad Sci USA1999, 96: 7220-7225.
63. Claveau D, Sirinyan M, Guay J, Gordon R, Chan C-C, Bureau Y, Riendeau D, Mancini JA: Microsomal prostaglandin synnthase-1 is a major terminal synthase that is selectively upregulated during cyclooxygenase-2-dependent prostaglandin E2 pro-duction in the rat adjuvant-arthritis model. J Immunol2003, 170:4738-4744.
64. Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, et al.: Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA 2003, 100:9044-9049.
65. Sommer C, Kress M: Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett 2004, 361: 184-187.
66. Ohtori S, Takahashi K, Moriya H, Myers RR: alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine2004, 29:1082-1088.
67. Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH: TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurons. Neuropharmacology2002, 42:93-106. 68. Schafers M, Marziniak M, Sorkin LS, Yaksh TL, Sommer C:
Cyclooxygenase inhibition in nerve-injury- and TNF-induced hyperalgesia in the rat. Exp Neurol 2004, 185:160-168. 69. Schafers M, Sorkin LS, Geis C. Shubayev VI: Spinal ligation
induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia of rat. Neurosci Lett 2003, 347:179-182.
70. Grunke M, Schulze-Koops H: Successful treatment of inflam-matory knee osteoarthritis with tumour necrosis factor block-ade. Ann Rheum Dis2006, 65:555-556.
71. Watkins LR, Maier S: Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev2002, 82:981-1011.
72. Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ: A proinflammatory chemokine, CCL3, sensi-tizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci USA2005, 102:4536-4541.
73. Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ: Chemokines and glycoprotein 120 produce pain hypersensi-tivity by directly exciting primary nociceptive neurons. J Neu-rosci2001, 21:5027-5035.
74. Weidler C, Holzer C, Harbuz M, Hofbauer R, Angele P, Scholmerich J, Straub RH: Low density of sympathetic nerve fibres and increased density of brain derived neurotrophic factor positive cells in RA synovium. Ann Rheum Dis2005, 64:13-20.
75. Miller LE, Wiedler C, Falk W, Angele P, Schaumberger J, Scholmerich J, Straub R: Increased prevalence of sema-phorin3C, a repellent of sympathetic nerve fibres in synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum 2004, 50:1156-1163.
76. Niissalo S, Hukkanen M, Imai S, Tornwall J, Konttinen YT: Neu-ropeptides in experimental and degenerative arthritis. Ann New York Acad Sci2002, 966:384-399.
77. Zhang JM, LI H, Munir MA: Decreasing sympathetic sprouting in pathological sympathetic ganglia: a new mechanism for treating neuropathic pain using lidocaine. Pain2004, 109: 143-149.
78. Shinder V, Govrin-Lippmann R, Cohen S, Belenky M, Ilin P, Fried K, Wilkinson HA, Devor M: Structural basis of sympathetic-sensory coupling in rat and human dorsal root ganglia follow-ing peripheral nerve injury. J Neurocytol1999, 28:743-761. 79. Sato J, Perl ER: Adrenergic excitation of cutaneous pain
recep-tors induced by peripheral nerve injury. Science1991, 251: 1608-1610.
80. Lee DH, Liu X, Kim HT, Chung K, Chung JM: Receptor subtype mediating the adrenergic sensitivity of pain behavior and ectopic discharges in neuropathic Lewis rats. J Neurophysiol 1999, 81:2226-2233.
81. Ali Z, Raja SN, Wesselmann U, Fuchs PN, Meyer, RA, Campbell JN: Intradermal injection of norepinephrine evokes pain in patients with sympathetically maintained pain. Pain 2000, 88: 161-168.
synovial fluids of patients with active arthropathies. Clin Exp Immunol2004, 137:621-627.
83. Neugebauer V: Glutamate receptor ligands. Hand Exp Pharma-col2007, 177:217-249.
84. Szekely JI, Torok K, Mate G: The role of ionotropic glutamate receptors in nociception with special regard to the AMPA binding sites. Curr Pharmaceut Des2002, 8:887-912.
85. Merle B, Itzstein C, Delmas PD, Chenu C: NMDA glutamate receptors are expressed by osteoclast precursors and involved in the regulation of osteoclastogenesis. J Cell Biochem2003, 90:424-436.
86. Salter DM, Wright MO, Millward-Sadler SJ: NMDA receptor expression and roles in human articular chondrocyte mechan-otransduction.Biorheology2004, 41:273-281.
87. Danysz W, Parsons CG: GlycineB recognition site of NMDA receptors and its antagonists. Amino Acids1998, 14:205-206. 88. Quartaroli M, Fasdelli N, Bettelini L, Maraia G, Corsi M:
GV196771A, an NMDA receptor/glycine site antagonist, atten-uates mechanical allodynia in neuropathic rats and reduces tolerance induced by morphine in mice. Eur J Pharmacol2001, 430:219-227.
89. Wallace MS, Rowbotham MC, Katz NP, Dworkin RH, Dotson RM, Galer BS, Rauck RL, Backonja MM, Quessy SN, Meisner PD: A randomized, double blind, placebo-controlled trial of a glycine antagonist in neuropathic pain. Neurology2002, 59:1694-1700. 90. Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti F, Nagahisa A: Antinociceptive actions of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br J Pharmacol 1997, 12:809-812.
91. Hudson LJ, Bevan S, McNair K, Gentry C, Fox A, Kuhn R, Winter J: Metabotropic glutamate receptor 5 up-regulation in A-fibers after spinal nerve injury: 2-Methyl-6-(phenylethynyl)-pyridine (MPEP) reverses the induced thermal hyperalgesia. J Neu-rosci2002, 22:2660-2668.
92. Dogrul A, Ossipov MH, Lai J, Malan TP, Porecca F: Peripheral and spinal antihyperalgesic activity of SIB-1757, a metabo-tropic glutamate receptor (mGluR5) antagonist , in experi-mental neuropathic pain in rats. Neurosci Letts 2000, 292: 115-118.
93. Zhu CZ, Wilson SG, Mikusa JP, Wismer CT, Gauvin DM, Lynch JJ 3rd, Wade CL, Decker MW, Honore P: Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur J Pharmacol2004, 506:107-118.
94. Boxall SJ, Berthele A, Laurie DJ, Sommer B, Zieglgansberger W, Urban L, Tolle TR: Enhanced expression of metabotropic gluta-mate receptor 3 messenger RNA in the rat spinal cord during ultraviolet irradiation induced peripheral inflammation. Neuro-science1998, 82:591-602.
95. Chiechio S, Caricasole A, Barletta E, Storto M, Catania MV, Copani A, Vertechy M, Nicolai R, Calvani M, Melchiorri D, Nicoletti F: L-Acetylcarnitine induces analgesia by selectively upregu-lating mGlu2 metabotropic glutamate receptors. Mol Pharm 2002, 61:989-996.
96. Vellani V, Mappleback S, Moriondo A, Davis JB, McNaughton PA: Protein kinase C activation gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol 2001, 534:813-825.
97. Szallasi A, Blumberg PM: Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol Rev1999, 51:159-212.
98. Cantillon M, Vause E, Sykes D, Moon A, Hughes S: Safety, toler-ability and efficacy of ALGRX 4975 in osteoarthritis (OA) of the knee. Journal of Pain 2005, 6(3): S39.
99. Garcia-Martiez C, Humet M, Planells-Casas R, Gomis A, Capri M, Viaa F, de la Pena, E, Sachez-Baez F, Carbonell T, de Felipe C, et al.: Attenuation of thermal nociception and hyperalgesia by VR1 blockers. Proc Natl Acad Sci USA2002, 99:2374-2379. 100. Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang
J, Zhu D, Vanderah TW, Porreca F, Doherty EM, et al.: AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther2005, 313:473-484.
101. Chizh B, Napolitano A, O’Donnell M, Wang J, Brooke A, Lai R, Aylott M, Bullman J, Gray E, Williams P, Appleby J: The TRPV1 antagonist SB705498 attenuates TRPV1 receptor mediated activity and inhibits inflammatory hyperalgesia in humans: Results from a Phase 1 study. Journal of Pain2006, 7 (4): S42.
102. Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Pataapouian A: Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004, 41:849-853.
103. Band-Ward PA, Humphrey PP: P2x receptor mediated ATP-induced primary nociceptive neurone activation. J Autonom Nerv Syst2000, 81:146-151.
104. Honore P, Kage K, Mikusa J, Watt AT, Johnston JF, Wyatt JR, Fal-tynek CR, Jarvis MF, Lynch K: Analgesic profile of intrathecal P2X(3) antisense oligonucleotide treatment in chronic inflam-matory and neuropathic pain states in rats. Pain2002, 99: 11-19.
105. Jarvis MF, Burgard EC, McGaraughty S, Honore P, Lynch K, Brennan TJ, Subieta A, van Biesen T, Cartmell J, Bianchi B, et al.: A-317491, a novel potent and selective non nucleotide antag-onist of P2X(3) and P2X(2/3) receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA2002, 99:17179-17184.
106. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K: P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature2003, 424:778-783.
107. Deuchars SA, Atkinson L, Brooke RE, Musa H, Milligan CJ, Batten TFC, Buckley NJ, Parson SH, Deuchars J: Neuronal P2X7 recep-tors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci2001, 21:7143-7152. 108. Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green
P, Egerton J, Murfin M, Richardson J, Peck WL, et al.: Disruption of the P2X7 purinoceptor gene abolishes chronic inflamma-tory and neuropathic pain. Pain 2005, 114:386-396.
109. Chaplan SR, Guo H-Q, Lee DH, Luo L, Liu C, Kuei C, Velumian AAL, Butler MP, Brown SM, Dubin AE: Neuronal hyperpolariza-tion-activated pacemaker channels drive neuropathic pain. J Neurosci2003, 23:1169-1178.
110. Yao H, Donnelly DF, Ma C, LaMotte RH: Upregulation of the hyperpolarization activation cation current after chronic com-pression of the dorsal root ganglion. J Neurosci 2003, 23: 2069-2074.
111. Giatromanolaki A, Sivridis E, Maltezos E, Athanassou N, Papa-zoglou D, Gatter KC, Harris AL, Koukourakis MI: Upregulated hypoxia inducible factor-1alpha and -2alpha pathway in rheumatoid arthritis and osteoarthritis.Arthritis Res Ther2003, 5:R193-201.
112. Leffler A, Monter B, Koltzenburg M: The role of the capsaicin receptor TRPV1 and acid sensing ion channels (ASICs) in proton sensitivity of subpopulation of primary nociceptive neurons in rats and mice. Neuroscience2006, 139:699-709. 113. Dube GR, Lehto SG, Breese NM, Baker SJ, Wang X, Matulenko
MA, Honore P, Stewart AO, Moreland RB, Brioni JD: Electro-physiological and in vivocharacterization of A-317567, a novel blocker of acid sensing ion channels.Pain2005, 117:88-96. 114. Matzner O, Devor M: Hyperexcitability at sites of nerve injury
depends on voltage sensitive sodium channels. J Neurophys-iol1994, 72:349-359.
115. Eglen RM, Hunter JC, Dray A: Ions in the fire: recent ion-channel research and approaches to pain therapy. Trends Pharmacol Sci1999, 8:337-342.
116. Devor M: Sodium channels and mechanisms of neuropathic pain.Pain2005, 7(Suppl 1):S3-S12.
117. Waxman SG, Dib-Hajj S: Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol Med2005, 11:555-562. 118. Gould HJ 3rd, England JD, Soignier RD, Nolan P, Minor LD, Liu
ZP, Levinson SR, Paul D: Ibuprofen blocks changes in NaV1.7 and 1.8 sodium currents associated with complete Freunds adjuvant induced inflammation in rat. Pain 2004, 5:270-280. 119. Araujo MC, Sinnott CJ, Strichartz GR: Multiple phases of relief
from experimental mechanical allodynia by systemic lido-caine: responses to early and late infusions.Pain2003, 103: 21-29.
120. Kastrup J, Petersen P, Dejgard A, Angelo FR: Intravenous lido-caine infusion: a new treatment of chronic painful diabetic neuropathy. Pain1987, 28:69-75.
121. Okuse K, Malik-Hall M, Baker MD, Poon W-YL, Chao MV, Wood JN: Annexin II light chain regulates sensory neurone-specific sodium channel expression. Nature2002, 417:653-656. 122. Rush AM, Craner MJ, Kageyama T, Dib-Haj SD, Waxman SG:
![{4 Bromo 2 [2 (isopropylamino)ethyliminomethyl]phenolato}thiocyanatocopper(II)](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)