2017 2nd International Conference on Computer Science and Technology (CST 2017) ISBN: 978-1-60595-461-5
Protein Complex Detection by Seed-expansion
Method Based on Random Walk with Restart
Jia-wen JIANG
1, Cheng LUO
2, Jia-hai LIANG
3and Qing-feng CHEN
1,a*1School of Computer, Electronic and Information, Guangxi University,
No.100 Daxue Road, 530004 Nanning, China
2Guangxi University XingJian College of Science and Liberal Arts,
No.75 Xiuling Road, 530005 Nanning, China
3School of Electronics and Information Engineering, Qinzhou University,
Qinzhou, Guangxi, 535000, China
*Corresponding author
Keywords: Protein complex, Protein-protein interaction, Random walk with restart.
Abstract. Proteins form complexes to perform their functions in the living system. Many graph-based clustering methods have been used to detect complexes from protein-protein interaction networks (PPIs). However, it is difficult to find hundreds of complexes of various structures from large and disordered PPIs. This paper proposes a novel approach for complexes detection based on Random Walk with Restart (CDRWR) since RWR can find the global relevance similarity between nodes. CDRWR firstly uses the similarity to weight the edges of PPIs and expands the significant complex seeds from every node to ensure the complexes integrity. The external protein nodes are included by comparing the density, and a merging method is applied to confirm the final protein complexes. Experiments are conducted to compare CDRWR with existing methods on a few yeast PPIs datasets according to two evaluation criteria, F1-score and NMI (Normalized Mutual Information), and the results show that it has the best performance in accuracy.
Introduction
In the biological domain, proteins form complexes to perform their functions. It is important to identify the complexes from a large number of protein-protein interaction networks (PPIs) since it assists in understanding cellular organization and functions [1]. PPI data sets are usually represented as graphs for the purpose of data analysis. And clustering has been the most popular method to detect complexes within the graphs [2]. However, large and noisy PPI networks and complicated complexes structures prevent us from discovering the complexes.
There have been a variety of algorithms proposed in the past few years in terms of the characteristics of PPIs and graph mining techniques. Nevertheless, most of them have limitations in getting the desired results. The results may not only lose complexes but also miss proteins or include redundant proteins in some derived complexes.
the maximal subgraphs as seeds [4]. An early method MCODE also chooses locally dense protein seeds to isolate the dense regions [5].
PPIs weighting is a general measure to enhance the clustering effect. CD-distance and FSWeight [6] are two methods according to the number of common neighbors of two proteins. They are both based on the graph theory. Some other methods use the biology information, like gene sequence, 3D structures, and GO terms, to weight the interactions [7].
Random walk is an effective method to find the property of the whole graph. The RWR, a generalized form of PageRank, offers a good relevance similarity between two nodes in a graph. It has been successfully applied in discovering the correlation between multi-objects, such as words in a text, pixels in an image and frames in a video [8]. This similarity reflects how the topology of two linked nodes is in an entire graph. Thus, weighting the edge by this similarity is helpful to express the interaction clearly. For the nodes in a complex, they are linked strongly but have weak similarities with the nodes in other complexes. It is benefit in generating some seeds which the nodes in them are linked very closely.
In this paper, a new method CDRWR is proposed to detect complexes in PPIs by combing the idea of the Seed-expansion methods and the advantage of the RWR. A graph is constructed in terms of the protein-protein interaction data. RWR is extended to generate the global weighted graph. Based on the RWR result, every node yields a small clique seed by a special threshold. And some small cliques which are a part of the big ones are removed. Then the external nodes are assigned into these suitable seeds based on the density function. At last, a merging operation is used to determine the final correct complexes from the mixed expanded complexes.
Methods
A PPI network is represented as an undirected graph G={ , , }V E W , where G is the
graph, V is the set of the nodes, E is the edge set, and W is the edge weights matrix. In
this paper, according to the PPI graph’s adjacency matrix, the edge weights are all set 1 as their initial values.
Generating the Seeds and Weighting PPIs
Complexes detection usually uses the seed-expansion methods. From a single node or a seed clique, expansion methods are used to expand them to final complexes. The seeds are important in this process. There are many methods to choose the suitable seeds. But they are all not good enough since many of them only consider the dense region. According to the features of RWR which can get the global relevance similarities between all nodes [8], an effective way is to use a similarity threshold to include nodes to form the seeds.
RWR is defined as Eq. 1: suppose there is a walker who starts from the node i. The
walker iteratively moves to its neighbor nodes with the probability which is the normalized edge weight. And at each step, it has the probability c to return to the node i.
The relevance similarity of node jwrt node i is defined as the steady-state probability
ij
r that the walker will finally stay at node j.
1( ) ( ) (1 )
t t
i
R+ i =cWR i + −c e (1)
1 1
'
2 2
where the W is the normalized adjacency matrix, c is the restart probability, ei is the
restart vector of node i where the entry corresponding to node i is 1 and others 0. W is
normalized by Lapalician method[8], as the Eq. 2, the '
W is the original adjacency
matrix of the graph, D is the diagonal matrix with its element located in the row i and
column i is the sum of the elements in the row i of W'. The Lapalician normalized
method can make result has a symmetrical characteristic, rij =rij.
The steady-state is Rt+1( )i =R it( ) , so for fast computation, the fast RWR [8] is
computed as the Eq. 3.
1
( ) ( ) (1 )
t
i
R i = −I cW − × × −e c (3)
The computation of RWR generates a global relevance similarities vector between this start node i with other nodes. It is easy to find the closest nodes of node i according
to this vector. When giving a fixed threshold, if the similarities between this node i and
other nodes are greater than this threshold, these closet nodes are viewed as a small clique seed.
In contrast, given different start nodes, the RWR result vectors cannot be cut by a same threshold. The RWR result is based on the characteristic of the whole graph, for a start node, its close nodes’ relevance similarities with respect to it may be some small numbers, but for other start nodes, the similarities may be some bigger numbers. In PPIs, the smallest complex has two proteins at least, that is to say, one node must have one linked node to form a complex. So, for looking for the suitable nodes to form seed, the threshold is decided by the closest node of the start node. And the other close nodes’ similarities scores are close to this value actually. Then for a node, its threshold is defined as the Eq. 4.
( )
i i
Th = ×α Max R (4)
where Max R( )i is the maximum value in the relevance similarities for node i, α is the
tuning value, its range is lower than 1.
Every node yields one small clique. And there are many small cliques are part of some bigger ones, they should be deleted. This pruning process is according to common nodes between the smaller cliques, as the Eq. 5.
(5)
where Ci and Cj are two small cliques, computes their common nodes,
min{|Ci|,|Cj|} is the size of the smaller one of these two cliques.
In this paper, CoC=1 means the smaller one is the redundant clique to delete. So, the
small cliques are sorted by their size from the biggest to the smallest. And then compute their CoC values between these cliques according to the sort, if the CoC=1, the smaller
one is deleted from the queue. The last small cliques are the real seeds. Of course, there are also some seeds has some common nodes among them.
Expanding Process and Complexes Merge Disposal
These seeds are needed to be expanded to the final protein complexes. Usually, a general density function is used to include the external nodes in the seeds. The density function is the Eq. 6.
( ) in
in out
W density C
W W
=
+ (6)
where C is the seed clique, Win is the sum of the weight of the edge that its two ends are
both in C, Woutis the sum of the weight of the edge that has only one end in C, and the
edge weight is the RWR result.
For an external node, compare the density of a seed adding it with not adding it, and if it can increase the density, it should be the member of the complex expanded from this seed, otherwise not. According to this rule, all external nodes which link to the seed are checked, the final results are the expanded complexes.
Due to the tuning valueα, it may generate too many seeds, and then the number of expanded complexes is big. They must be merged.
A descending sort by the size is used in the expanded complexes. The first biggest one is chosen as the first complex. The rest expanded complexes are compared with it by a similarity between them. If the similarity is more than the threshold, they are merged. This disposal is for all the rest complexes which are not merged. The similarity is same as the Eq. 5, but the threshold is not 1 here.
The global process of the method CDRWR is as follows:
(1) Use RWR to get global relevance similarities between all nodes.
(2) Use the threshold setting to get the small clique for every node, and delete the small cliques are a part of some bigger ones. The rest are the seeds.
(3) Expand these seeds by the density.
(4) Merge the expanded complexes to the final complexes.
Experiments
Datasets
Two famous yeast PPIs are tested in the experiments, they are Collins [9], Gavin [1]. Collins has 1622 proteins and 9070 interactions and Gavin has 1430 proteins and 6531 interactions. The reference protein complexes data is the CYC2008 [10], an annotated yeast high-throughput complexes dataset composing of 399 complexes. In the experiment, we choose the common proteins from the yeast PPIs and the reference complexes dataset.
Evaluation Methods
(7) where CA and CB are two different complexes.
Then the F1-score is 2 (× precision+recall) / (precision recall+ ) . The proportion is usually varied from 0.2 to 1.0. The NMI method is derived from Lancichinetti’s paper [11].
Results and Analysis
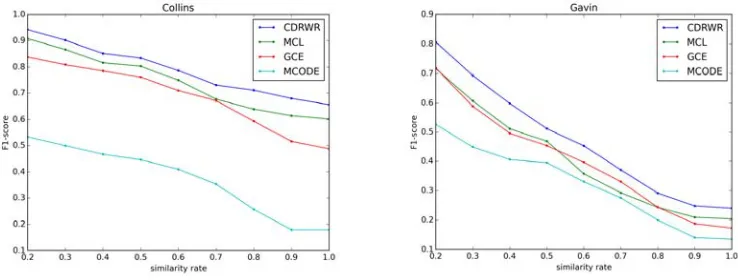
Our method CDRWR is compared with other 3 famous methods MCL [12], MCODE and GCE. Fig 1. are the F1-score results on the Collins dataset and Gavin dataset.
Figure 1. F1-score results on Collins and Gavin PPIs.
On two datasets, our method CDRWR performs the best on the similarity rate from 0.2 to 1.0. On the Collins data, its F1-score is 0.656, MCL is 0.602 at 1.0. On the Gavin data, its F1-score is 0.2402, MCL is 0.2044. So, our method CDRWR is the best one among these four algorithms on F1-score evaluation method.
For NMI, the result is described in Table 1. CDRWR’S NMI value is both the highest on two datasets which refer to CYC2008 complexes. So, our method also performs well on the NMI test.
[image:5.612.121.490.242.382.2]The tuning parameter αin this experiment is 0.8 to choose the very close nodes to form the seeds. If this value is too small, some redundant nodes are included, and if this value is close to 1, the seeds are too many and the algorithm might consume a lot of computation time.
Table 1. The NMI results on two PPIs.
Data sets DCDRW MCL GCE MCODE
Collins 0.805576 0.769323 0.612986 0.513926
Gavin 0.479573 0.460939 0.469196 0.417044
Conclusion and Discussion
in which the nodes are very close, and are taken as the weights of the edges to enhance the expanding process to get perfect final complexes. The experimental result shows that our method is the best compared with some algorithms according to two evaluation methods, F1-score and NMI. But our algorithm will generate too many seeds and consume too much computation time in the expanding process. In the future work, we will find some heuristics to reduce redundant seeds.
Acknowledgement
The work reported in this paper was partially supported by a National Natural Science Foundation of China project 61363025, and two key projects of Natural Science Foundation of Guangxi 2012GXNSFCB053006 and 2013GXNSFDA019029.
References
[1] Gavin, A. C., et al. Proteome survey reveals modularity of the yeast cell machinery, Nature 440.7084 (2006): 631-636.
[2] Pizzuti, C. and Simona E. R. Algorithms and tools for protein–protein interaction networks clustering, with a special focus on population-based stochastic methods, Bioinformatics 30.10 (2014): 1343-1352.
[3] Li, X. L., et al. Interaction graph mining for protein complexes using local clique merging, Genome Informatics 16.2 (2005): 260-269.
[4] Lee, C., et al. Detecting highly overlapping community structure by greedy clique expansion, arXiv preprint arXiv:1002.1827 (2010).
[5] Bader, G. D. and Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks, BMC bioinformatics 4.1 (2003): 2. [6] Chua, H. N., Sung, W. K. and Wong, L. Exploiting indirect neighbours and topological weight to predict protein function from protein–protein interactions, Bioinformatics 22.13 (2006): 1623-1630.
[7] Ho, Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry." Nature 415.6868 (2002): 180-183.
[8] Tong, H., Faloutsos C. and Pan J. Y. Fast Random Walk with Restart and Its Applications, International Conference on Data Mining IEEE Computer Society, 2006:613-622.
[9] Collins, S. R., et al. Toward a Comprehensive Atlas of the Physical Interactome of Saccharomyces cerevisiae, Molecular & Cellular Proteomics Mcp 6.3 (2007):439. [10] Pu, S., et al. Up-to-date catalogues of yeast protein complexes, Nucleic Acids Research 37.3 (2009):825-31.
[11] Lancichinetti, A., Fortunato S. and Kertész J. Detecting the overlapping and hierarchical community structure of complex networks, New Journal of Physics 11.3(2009):19-44.