ABSTRACT
WEISER, LAURA JEAN. Developing Simulation Models of Peptoid Polymers (Under the direction of Erik E. Santiso).
Peptoids are a type of synthetic, biomimetic foldamers composed of a protein-like, poly-glycine backbone with side chains attached to their nitrogen atoms. Peptoids are promising materials for diverse applications ranging from surfactants, to catalysts, to coatings, because their backbone conformations may be controlled through a careful choice side chains. Additionally, rational combination of side chains can create complex multi-level peptoid folded structures. Understanding how peptoid side chains affect their final folded structure, and therefore their possible applications, is an important research goal.
Experimental studies have made progress decoding peptoid side-chain/conformation relationships and shown that side chain sterics, charges, hydrogen bonds, and n⟶π* bonding, can all be harnessed to stabilize peptoid backbone conformations. Still, only a fraction of the 300+ possible peptoid side chains have been studied experimentally, leaving much room for further characterization.
Computational studies have explored peptoid side-chain/backbone conformation relationships using a variety of methods, and have even identified key peptoid-unique peptoid structures, such as the polyproline type I and type II helices stabilized by bulky, benzene containing compounds. Expanding and improving understanding of side chain/backbone relationships is a goal of this research.
Developing Simulations Models of Peptoid Polymers by
Laura J Weiser
A Dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the degree of
Chemical Engineering
Raleigh, North Carolina 2017
APPROVED BY:
_______________________________ _______________________________
Erik E. Santiso Keith E. Gubbins
Committee Chair
_______________________________ _______________________________
Yaroslava Yingling Balaji Rao
DEDICATION
To my Grandpa Robert “Bud” Weiser - I am proud to follow in your footsteps as an engineer. To my Grandma Lois Gensterblum - You had 5 kids by age 28. I have this dissertation.
BIOGRAPHY
Laura Weiser was born in Lansing, Michigan to Annette and Richard Weiser. She grew up in Okemos Michigan, five miles from the campus Michigan State University, and attended K-12 in the Okemos Public Schools. In 2007, she graduated from Okemos High School where she was a member of the track and Cross Country teams and played viola in the school orchestra. Upon high school graduation, Laura enrolled at the University of Michigan, in Ann Arbor.
Laura got her first lab experience the summer after high school, working for as a lab assistant for the US Forestry Service, and the Michigan State University Department of Entomology. There, she fed beetle larvae, emptied cockroach traps, and was bitten by the research bug – but never bitten by the bugs she researched. As an undergraduate, she worked as a lab assistant at the University of Michigan Medical School in research groups devoted to clinical oncology and molecular immunology. While at Michigan, Laura spent her time running around beautiful Ann Arbor, and was a member of University Students Acting Against Cancer, through which she organized a bone marrow donor registry drive every year she was enrolled.
In 2011 Laura graduated from Michigan, Magna Cum Laude, with a bachelor’s in Chemical Engineering. She enrolled as a graduate student at North Carolina State in 2011, and joined Erik Santiso’s group in 2014.
ACKNOWLEDGMENTS
First, I would like to thank my advisor, Dr. Erik Santiso for his patience, support and positive attitude over the last four years. Thank you for giving me a chance to succeed at NC State, for being supportive of my research, and for building a lab full of colleagues who I enjoy and respect.
I would like to thank my Santiso Group lab members for providing companionship and inspiration. Thank you Deepti, Amit, Charles, Chris, and Amulya, all of your hard work and passion for science inspires me. Thank you Dr. Nathan Duff for technical advice,
research help, coffee, and proofreading; Mariah Ritz for your friendship and your even-keeled attitude towards research and life; and Jen Clark for your friendship and biting sense of humor.
I’d like to thank the members of my committee, Dr. Gubbins, Dr. Rao, Dr.
Pasquinelli, and Dr. Yingling,With special thanks to Dr. Yingling for providinig me extra help, research advice, and encouragement along the way.
I’d also like to thank Dr. Yingling and Dr. Hoshin Kim for their help and guidance on the Graphene Oxide adsorption work described in this dissertation.
Thank you to my parents Annette and Richard Weiser for encouraging me to attend engineering school and then graduate school; for supporting me along the way and for being understanding as I spend so much time so far from home.
Extraordinaire, for providing historical context on computer simulations, and making me appreciate modern technology; and, finally, Katherine Phillips, for your friendship, encouragement, and commiseration along the way.
And lastly, I’d like to thank my boyfriend, Jonathan, for always being, supportive, positive, thoughtful, flexible, and patient. Thank you for proofreading, offering your
TABLE OF CONTENTS
LIST OF TABLES ... viii
LIST OF FIGURES ... ix
CHAPTER 1- Introduction ... 1
1.1. References ... 6
CHAPTER 2- Methods ... 10
2.1. Introduction ... 10
2.2. Description of Methods ... 14
2.2.1. All-atom Molecular Dynamics ... 14
2.2.2. CHARMM General Force Field ... 15
2.2.3. Quantum Methods ... 18
2.2.4. Well-Tempered Metadynamics ... 21
2.3. References ... 24
CHAPTER 3- A Review of Previous Simulation Studies of Peptoids ... 29
Abstract: ... 29
3.1. Introduction ... 31
3.2. Modeling Studies of Peptoids ... 37
3.2.1. Ab initio studies of peptoids ... 38
3.2.2. Peptoid structure prediction with combined methods ... 45
3.2.3. All-atom molecular dynamics studies of peptoids ... 49
3.2.4. Coarse-grained peptoid simulations ... 57
3.2.5. Peptoid simulations in Rosetta ... 60
3.3. Conclusions and future directions ... 62
3.4. References ... 64
CHAPTER 4- CHARMM-Based Peptoid Force Field Development ... 71
4.1. Introduction ... 71
4.2. Background ... 74
4.2.1. The Peptoid Backbone Dihedrals ... 74
4.2.2. The CHARMM Force Field ... 75
4.2.3. CHARMM General Force Field (CGENFF) ... 77
4.3.1. CGENFF ... 78
4.3.2. Quantum Potential Energy Surfaces ... 79
4.3.3. Force Field Potential Energy Surfaces ... 80
4.3.4. Structure Generation for Metadynamics ... 80
4.3.5. Well Tempered Metadynamics ... 81
4.4. Results and Discussion ... 82
4.4.1. MP2/6-31G* Optimized Ramachandran plot of Sarcosine ... 82
4.4.2. CHARMM Parameterization Results ... 83
4.4.3. ψ, ϕ, and ρ Dihedral Fitting ... 93
4.4.4. Extending our Force Field to other Side Chains ... 96
4.4.5. RMSD Energy Comparisons for Minimum Energy Structures ... 98
4.5. Solvated Metadynamics Results ... 99
4.5.1. Sarcosine ... 99
4.5.2. N-Phenyl-Methyl Glycine ... 100
4.5.3. (S)-N-(1-PhenylEthyl)-Glycine ... 101
4.6. Simulations of solvated, unbiased polymers ... 102
4.7. Conclusions ... 104
4.8. References ... 106
CHAPTER 5- Conclusions, Continuing Work and Future Directions ... 112
5.1. Conclusions ... 112
5.2. Continuing Work: Peptoid Adsorption to Graphene Oxide ... 112
5.3. Future Directions ... 114
5.3.1. Adding Additional Side Chains to the Force Field ... 114
5.3.2. Validating the Force Field Against Experiment ... 115
5.3.3. Developing Sampling Methods for Peptide Bond Isomerization ... 116
5.3.4. Study Adsorption Effects of Modified Side Chains ... 117
LIST OF TABLES
Table 4.1 The sarcosine backbone charges. The ‘DMA Charge’ column shows the charge obtained from Dimethylacetamide using CGENFF’s fitting procedure. SAR Charge describes the charges of the peptoid subunit derived from using CHARMM’s charge combing rules. ... 87 Table 4.2 The angles describing the minimized sarcosine nitrogen ... 90 Table 4.3 The Fitted CGENFF peptoid backbone parameters. ... 93 Table 4.4 RMSD comparison between force field dihedral energies and MP2 dihedral
LIST OF FIGURES
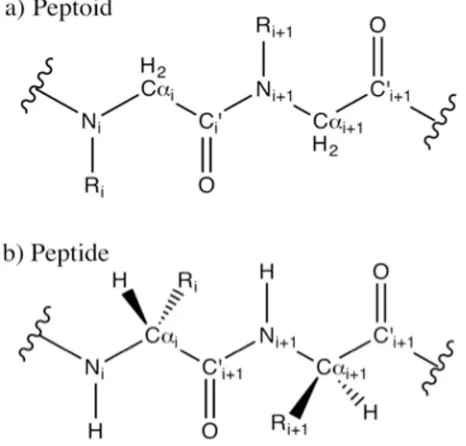
Figure 1.1 The Peptoid Backbone. Peptoids have a protein-like backbone with side chains attached to the Nitrogen atoms, rather than the α-carbons. ... 2 Figure 2.1 The bonded interactions parameterized in the CHARMM force fields ... 16 Figure 3.1 A Peptoid (a) and a peptide (b) Peptoids and peptides share the same backbone,
but peptoids have side chains attached to their nitrogen atoms. This removes backbone chirality and backbone hydrogen bonding capability. Peptoid backbones resemble the polyglycine peptides, with a side chain replacing the nitrogen-bound hydrogen atom. For this reason, peptoids are also called poly-N-substituted glycines. 31
Figure 3.2 Known Peptoid Secondary Structures. (A) The PPI helix (shown here as (S)-N-(1-napthylethyl)glycine) [11], [39], (B) the N-(phenyl)glycine PPII helix [12], (C), the peptoid ribbon [14] (D) the threaded loop [13], and (E) the Σ-strand [15]. Note that the figures are reproduced from different sources, and we have kept the original representations for consistency with them. A,C, and D are shown without their backbone hydrogen atoms, and E is shown without its side chains. The secondary structures are characterized by their patterns of φ and ω dihedrals defined in Figure 3. The PPI and PPII helices have repeating (φ=(-)75°, ω~0°) and (φ=(-)75°,ω=180°) dihedrals, respectively. The threaded loop (D) is created by 9-mer NSPE peptoids and is stabilized by a hydrogen bonding interaction between side chains and their NH2
groups. The ribbon (C) has alternating ω=0° and ω=180° dihedrals and the Σ-strand (E) has ω=180° and alternating φ=-90°,ψ=120° and φ=-120°,ψ=90°- dihedrals. Structure A Reprinted with permission from Stringer JR, Crapster JA, Guzei IA, et al. (2011) J Am Chem Soc 133: 15559–15567. Copyright (2011) American Chemical Society [39]. Structure B reprinted with permission from Shah NH, Butterfoss GL, Nguyen K (2008) J Am Chem Soc 130: 16622–16632. Copyright (2008) American Chemical Society [12]. Structure C reprinted with permission from [14]. Structure D Reprinted (adapted) with permission from Huang K, Wu CW, Sanborn TJ, et al (2006) J Am Chem Soc 128: 1733–1738. Copyright (2006) American Chemical Society [13] . Structure E
reproduced with permission from [15] 37
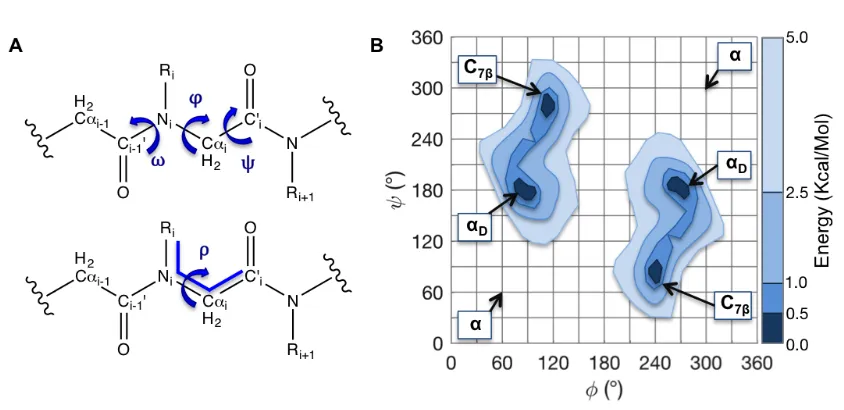
Figure 3.3 The peptoid backbone dihedrals and a peptoid Ramachandran plot. (A) Peptoid backbone conformations can be described by the dihedrals φ, ψ, and ω. The dihedral ω describes the isomerization of the planar peptoid bond. In the cis (ω=0°) configuration, the side chain and the carbonyl oxygen are on the same side of the bond. In the trans (ω=180°) configuration (shown) the side chain and the oxygen lie on opposite sides of the amide bond. Ramachandran plots (B) are used to show backbone (φ-ψ)
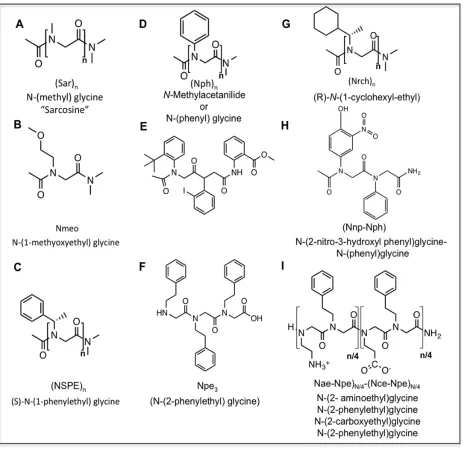
conformational preferences. Ramachandran plots are 2-D histograms that graph either the probability or the conformational energy of each φ/ψ pair of dihedrals. The figure shown corresponds to the trans isomer. This figure is based on data from Butterfoss [41] ... 39 Figure 3.4 A partial summary of the peptoids described in this work. (A) Sarcosine (B)
N-glycine)3, (G) (R)-N-(1-cycloheyl-ethyl), (H) N-(2-nitro-3-hydroxyl
phenyl)glycine-N-(phenyl)glycine, (I) (N-(2-aminoethyl)glycine-N-(2phenylethyl)glycine)N/4
-(N-(2-carboxyethyl)glycine-N-(2-phenylethyl)glycine). ... 41 Figure 3.5 Ab initio Ramachandran plot for cis (A) and trans (B). Both plots represent data at
the HF-6-31G* level of theory. The ‘o’ and ‘+’ markers represent cyclic and linear experimental data. Adapted with permission from Butterfoss GL, Renfrew PD, Kuhlman B, et al. (2009) J Am Chem Soc 131: 16798–16807 Copyright (2009)
American Chemical Society.[41]. ... 42 Figure 3.6 ψ-ω and φ-ω ab initio Ramachandran plots can simultaneously describe both
isomers of the ω dihedral. Butterfoss generated these (A) ψ-ω and (B) φ-ω plots as an alternative (ab initio HF-6-31G* data). ω dihedrals which deviate from 0° or 180° describe pyramidal nitrogen atoms. The ‘o’ and ‘+’ markers represent cyclic and linear experimental data. Adapted with permission from Butterfoss GL, Renfrew PD,
Kuhlman B, et al. (2009) J Am Chem Soc 131: 16798–16807 Copyright (2009)
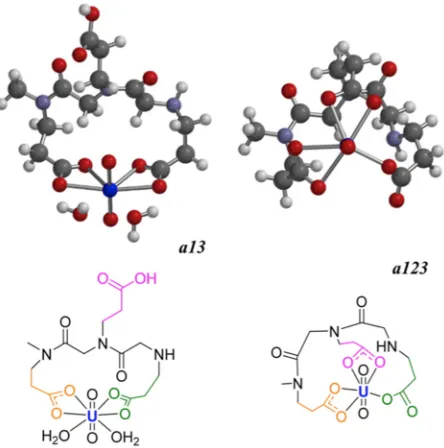
American Chemical Society [41]. ... 43 Figure 3.7 Two possible binding configurations for a uranyl chelating peptoid. The peptoid
can bind the uranyl ion with one, two, or three carboxyl groups. The binding enthalpy released on additional carboxyl binding is counteracted by the entropy loss in the restricted backbone chain. Adapted with permission from Parker BF, Knight AS,
Vukovic S, et al. (2016) Ind Eng Chem Res 55: 4187–4194. Copyright (2009) American Chemical Society [45] ... 45 Figure 3.8 The Σ-strand alternating motif. Mannige et al. [15] described a secondary
struct ure of alternating residues with alternating rotational states. Single-point secondary structures, like α-helices, contain a single repeating dihedral conformation. Two-point or Σ-strand motifs have alternating dihedral pairs, in this example they alternate
between the C7β and C7β’ conformations. This figure is based on a scheme by Mannige
[15] 54
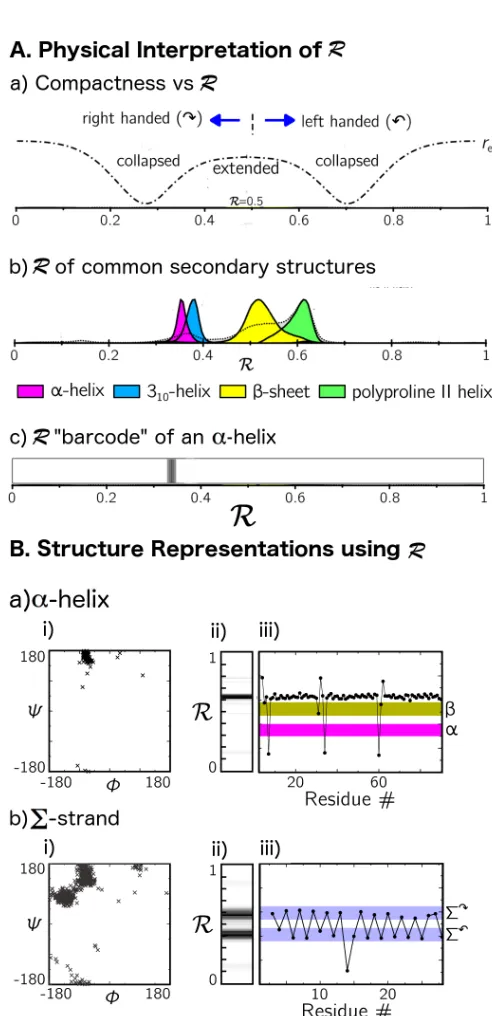
Figure 3.9. The Ramachandran Number. The Ramachandran Number R is a parameter derived by Mannige et al. which combines are residue’s φ and ψ diherals and chain compactness into a single number. A.(a) shows the physical information encoded in R. R>0.5 describes a left handed twist, R < 0.5 describes a right handed twist. R=0,R=0.5, and R=1.0 describe fully extended backbones, while intermediate values are more compact. (b) The distribution of R values for common peptide secondary structures. (c) A Ramachandran number histogram or “barcode” representation of the α-helix, (the distribution in (b) shown as a heat map). B. shows different structure visualizations for an α-helix (a) and for the alternating Σ-strand (b). (i) A Ramachandran plot, (ii) a barcode of the Ramachandran number, (iii) An R versus residue # plot, which, unlike the histograms, can show the alternating pattern of dihedrals in the Σ-strand. This figure is reproduced from figures in [68] ... 56 Figure 3.10 MF-CG-TOID grained peptoid model. (A) The MF-CG-TOID
represented in the diagram by spheres- and a director x – represented by the arrows. All bonded an nonbonded interaction parameters in MF-CG-TOID are functions of both parameters (X(ri, xi)). (B) The all-atom structure upon which this model was based. Figure reproduced from Haxton et. al. [73] ... 59 Figure 3.11 The rotamer dihedrals. Rosetta rotamer libraries describe the side chain
conformation in terms of the dihedrals χ1 and χ2. Renfrew developed rotamer libraries
for 54 peptoid side chains. ... 61 Figure 4.1 The peptoid backbone dihedrals. ... 75 Figure 4.2 MP2/(6-31G*) energy surfaces for the Sarcosine peptoid. The labeled minima
represent the location of the (-φ) conformations. These (+φ) minima are at the same locations, rotated rotated 180° around the origin. The experimental data from Butterfoss et al.39 is overlayed on the potential energy surfaces: ‘o’ markers indicate sarcosine data, and ‘x’ markers indicate data from other peptoids. ... 82 Figure 4.3 The Sarcosine vacuum phase energy conformations. The lowest vacuum phase
energy structures are cis-α, and trans C7βand are both stabilized by vacuum phase
oxygen-methyl hydrogen bonds. Both of these conformations are destabilized in water in favor of the αD conformation. These conformations were first identified by Möhle and
Hoffman5. ... 83 Figure 4.4 (Figure 4.5) Bonded parameters assigned by the CGENFF utility. Each assigned
parameter involves the tertiary peptoid nitrogen. Parameters assigned force constants of 0, and those with hydrogen dihedrals are not shown. The bonded terms are color coded as follows: Yellow parameters are bonds and angles. Blue parameters are assigned by analogy to the dimethyl formamide (DMF) molecule. The orange parameter is assigned by analogy to the Acetylated-Proline-Glycine dimer. Green parameters are assigned from an alanine dipeptide. ... 86 Figure 4.5 The four ω dihedrals. When no subscript is indicated, ω is equivalent to ω1. In
CGENFF all four ω dihedrals are assigned force constants to enforce the high-energy barrier of peptide bond isomerization. These high force constants lead to a very stiff planar nitrogen. ... 89 Figure 4.6 Adding a dihedral potential with 4-fold periodicity (shown in red) to the existing
ω potenials slightly widens the minimum energy basin, allowing the Nitrogen atom to move further out of plane. ... 92 Figure 4.7 Dihedral fits for the Sarcosine backbone. Frames A-C show the dihedral fits or
peptoids in the cis configurations and frames D-F show the dihedral fits for the trans configurations. A key result of this work was the recognition that the CHARMM cis-α configuration is overstabilized in vacuum, but destabilized in solvent. Accordingly, dihedrals were fit to the αD and C7βconfiguration, and the parameterization did not seek
reproduce α. ... 95 Figure 4.8 Fitting improves the representation of the αD conformation. ... 96
Figure 4.11 The dihedral distribution for the solvated peptoid 20-mer also favor the αD
CHAPTER 1- Introduction
In the last two decades, increased development of complex nanomaterials has driven interest in self-assembling protein-like polymers called foldamers, whose structures are defined by flexible backbones decorated with diverse chemical side-chains1. Just as amino acid sequences drive protein folding behavior and dictate protein function, foldamer side chain sequences induce their backbones to fold into complex structures that may have useful self-assembly2,3 or biomimetic properties4–6. A key goal of foldamer research is to
characterize how side chain sequences influence folding behavior, and to apply this understanding to deliberately design molecules with useful applications such as pharmaceuticals7,8 or solvent responsive nanomaterials9 .
like and non-natural peptoid side chains to create biocompatible or bioactive compounds with unique folding properties and functionality.
Figure 1.1 The Peptoid Backbone. Peptoids have a protein-like backbone with side chains attached to the Nitrogen atoms, rather than the α-carbons.
To understand the folding behavior of peptoids, it is important to understand how changes in the backbone lead peptoids to fold differently from proteins. The first key
difference is that peptoids have increased flexibility of the peptide bond, which connects the carboxyl carbon and the planar nitrogen atom. Proteins almost exclusively have trans peptide bonds, where the nitrogen hydrogen and carboxyl oxygen rest on opposite sides of the bond. In contrast, peptoids can have both cis and trans conformations, depending on their side chains. Peptide bond isomerization is a defining degree of freedom for many peptoid
that stabilize common peptide secondary structures, including α-helices and β-sheets. They instead exhibit peptoid-unique secondary structures that are stabilized by side chain
properties including steric bulk22,23, chirality24, hydrogen-bonding20, and n→π* bonding25,26. Developing peptoids as foldamer-type nanomaterials will require a deeper understanding of how chains affect peptoid conformations. This will enable identification of new side-chains that can induce interesting folding behavior.
Computational studies have played an important role in defining how certain side-chains can induce peptoid backbone folding. In 1998, Armand used conformational search algorithms and all-atom molecular mechanics to show that peptoids with repeating chiral side chains, such as N-(S)-1-phenylethylglycine (NSPE), form stable poly-proline type I (PPI) helices24. Ten years later, Shah used quantum semi-empirical methods to show that
N-aromatic side chains stabilize poly-proline type II helices22. Quantum mechanics calculations have revealed that side chain influences on the peptoid backbone can be seen at the monomer level22,27. For example, a single monomer containing a helix-inducing NSPE side-chain shows an energetic preference for a left handed PPI turn27.
studies. All-atom simulations can also build upon previous studies by using quantum mechanics calculations to develop simulation parameters.
The accuracy of all-atom MD depends on the force field parameters used to calculate the system energies. Biomolecule force field parameters are often derived from quantum mechanics calculations and empirically tuned to databases of experimental structures32. An empirical force field has not yet been developed for peptoids, as little experimental data on peptoid structures exists. As a first approach, peptoid simulations have been performed using existing protein force fields or generalized force fields without any tuning to peptoids. For molecules with strong steric side-chain/backbone interactions, these force fields have successfully reproduced NMR and crystal structure data33,34. However, simulations using untuned force fields were unable predict structures stabilized by solvent effects34, and also failed to predict the helical twist of the well-characterized NSPE helix33.
Recently, researchers have parameterized all-atom MD force fields specific to peptoids, allowing a more precise representation of peptoid conformational preferences. Following this approach, Mukherjee tuned the Generalized Amber Force Field (GAFF) to favor NSPE helices 35, and Mirijanian developed a CHARMM22 based peptoid force field36.
Each of these force fields were developed by tuning a single dihedral energy term and a more rigorous parameterization has not been performed. Additionally, published data shows that the CHARMM22-based peptoid force-field correctly reproduces peptoid
In this work we seek to develop force field parameters for improved all-atom simulations of peptoid chains. First, we derive all-atom, CHARMM General Force Field37 parameters for peptoids, which accurately represent the minimum energy configurations for both cis and trans peptoid isomers. We then validate these parameters through MD
simulations of peptoids solvated in TIP3P water. Finally, we show an application of our parameters by simulating the adsorption of peptoid chains to graphene oxide sheets. We expect that these methods can be used as a template for future peptoid force field
development and simulation.
1.1. References
(1) Gellman, S. H. Foldamers: A Manifesto. Acc. Chem. Res. 1998, 31 (4), 173–180. (2) Well-defined secondary structures - Sanford - 2004 - The FEBS Journal - Wiley
Online Library http://onlinelibrary.wiley.com.prox.lib.ncsu.edu/doi/10.1111/ j.14321033.2004.04062.x/abstract;jsessionid=4A0829EAE9B208E277E84380291 42C59.f02t04 (accessed Jul 31, 2017).
(3) Kwon, S.; Jeon, A.; Yoo, S. H.; Chung, I. S.; Lee, H.-S. Unprecedented Molecular Architectures by the Controlled Self-Assembly of a Beta-Peptide Foldamer. Angew. Chem.-Int. Ed. 2010, 49 (44), 8232–8236.
(4) Gibney, K. A.; Sovadinova, I.; Lopez, A. I.; Urban, M.; Ridgway, Z.; Caputo, G. A.; Kuroda, K. Poly(ethylene Imine)s as Antimicrobial Agents with Selective Activity. Macromol. Biosci. 2012, 12 (9), 1279–1289.
(5) Li, Z.-T.; Hou, J.-L.; Li, C. Peptide Mimics by Linear Arylamides: A Structural and Functional Diversity Test. Acc. Chem. Res. 2008, 41 (10), 1343–1353.
(6) Wu, C. W.; Seurynck, S. L.; Lee, K. Y. C.; Barron, A. E. Helical Peptoid Mimics of Lung Surfactant Protein C. Chem. Biol. 2003, 10 (11), 1057–1063.
(7) Mandity, I. M.; Fueloep, F. An Overview of Peptide and Peptoid Foldamers in Medicinal Chemistry. Expert Opin. Drug Discov. 2015, 10 (11), 1163–1177.
(8) Bautista, A. D.; Craig, C. J.; Harker, E. A.; Schepartz, A. Sophistication of Foldamer Form and Function in Vitro and in Vivo. Curr. Opin. Chem. Biol. 2007, 11 (6), 685– 692.
(9) Zhang, Z.; Che, Y.; Smaldone, R. A.; Xu, M.; Bunes, B. R.; Moore, J. S.; Zang, L. Reversible Dispersion and Release of Carbon Nanotubes Using Foldable Oligomers. J. Am. Chem. Soc. 2010, 132 (40), 14113–14117.
(10) Seo, J.; Lee, B.-C.; Zuckermann, R. N. Peptoids: Synthesis, Characterization, and Nanostructures. In Comprehensive Biomaterials; Elsevier, 2011; Vol. 2, pp 53–76. (11) Sun, J.; Zuckermann, R. N. Peptoid Polymers: A Highly Designable Bioinspired
Material. Acs Nano 2013, 7 (6), 4715–4732.
Function, and Mechanism of Helical Antimicrobial Peptides. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (8), 2794–2799.
(13) Li, N.; Zhu, F.; Gao, F.; Wang, Q.; Wang, X.; Li, H.; Ma, C.; Sun, W.; Xu, W.; Wang, C.; Zhang, L. Blockade of CD28 by a Synthetical Peptoid Inhibits T-Cell Proliferation and Attenuates Graft-versus-Host Disease. Cell. Mol. Immunol. 2010, 7 (2), 133–142.
(14) Luo, Y.; Vali, S.; Sun, S.; Chen, X.; Liang, X.; Drozhzhina, T.; Popugaeva, E.;
Bezprozvanny, I. A Beta 42-Binding Peptoids as Amyloid Aggregation Inhibitors and Detection Ligands. Acs Chem. Neurosci. 2013, 4 (6), 952–962.
(15) Dohm, M. T.; Seurynck-Servoss, S. L.; Seo, J.; Zuckermann, R. N.; Barron, A. E. Close Mimicry of Lung Surfactant Protein B by “clicked” Dimers of Helical, Cationic Peptoids. Pept. Sci. 2009, 92 (6), 538–553.
(16) Seurynck, S. L.; Patch, J. A.; Barron, A. E. Simple, Helical Peptoid Analogs of Lung Surfactant Protein B. Chem. Biol. 2005, 12 (1), 77–88.
(17) Zuckermann, R.; Kerr, J.; Kent, S.; Moos, W. Efficient Method for the Preparation of Peptoids [Oligo(n-Substituted Glycines)] by Submonomer Solid-Phase Synthesis. J. Am. Chem. Soc. 1992, 114 (26), 10646–10647.
(18) Olivier, G. K.; Cho, A.; Sanii, B.; Connolly, M. D.; Tran, H.; Zuckermann, R. N. Antibody-Mimetic Peptoid Nanosheets for Molecular Recognition. ACS Nano 2013, 7 (10), 9276–9286.
(19) Krieger, V.; Ciglia, E.; Thoma, R.; Vasylyeva, V.; Frieg, B.; de Sousa Amadeu, N.; Kurz, T.; Janiak, C.; Gohlke, H.; Hansen, F. K. α-Aminoxy Peptoids: A Unique Peptoid Backbone with a Preference for Cis-Amide Bonds. Chem. – Eur. J. 2017, 23 (15), 3699–3707.
(20) Stringer, J. R.; Crapster, J. A.; Guzei, I. A.; Blackwell, H. E. Construction of Peptoids with All Trans-Amide Backbones and Peptoid Reverse Turns via the Tactical
Incorporation of N-Aryl Side Chains Capable of Hydrogen Bonding. J. Org. Chem. 2010, 75 (18), 6068–6078.
(21) Crapster, J. A.; Guzei, I. A.; Blackwell, H. E. A Peptoid Ribbon Secondary Structure. Angew. Chem.-Int. Ed. 2013, 52 (19), 5079–5084.
(23) Stringer, J. R.; Crapster, J. A.; Guzei, I. A.; Blackwell, H. E. Extraordinarily Robust Polyproline Type I Peptoid Helices Generated via the Incorporation of Alpha-Chiral Aromatic N-1-Naphthylethyl Side Chains. J. Am. Chem. Soc. 2011, 133 (39), 15559– 15567.
(24) Armand, P.; Kirshenbaum, K.; Falicov, A.; Dunbrack, R. L.; Dill, K. A.;
Zuckermann, R. N.; Cohen, F. E. Chiral N-Substituted Glycines Can Form Stable Helical Conformations. Fold. Des. 1997, 2 (6), 369–375.
(25) Gorske, B. C.; Stringer, J. R.; Bastian, B. L.; Fowler, S. A.; Blackwell, H. E. New Strategies for the Design of Folded Peptoids Revealed by a Survey of Noncovalent Interactions in Model Systems. J. Am. Chem. Soc. 2009, 131 (45), 16555–16567. (26) Benjamin C. Gorske; Nelson, R. C.; Bowden, Z. S.; Kufe, T. A.; Childs, A. M.
“Bridged” N→π* Interactions Can Stabilize Peptoid Helices. J. Org. Chem. 2013, 78 (22), 11172–11183.
(27) Butterfoss, G. L.; Renfrew, P. D.; Kuhlman, B.; Kirshenbaum, K.; Bonneau, R. A Preliminary Survey of the Peptoid Folding Landscape. J. Am. Chem. Soc. 2009, 131 (46), 16798–16807.
(28) Scheraga, H. A.; Khalili, M.; Liwo, A. Protein-Folding Dynamics: Overview of Molecular Simulation Techniques. In Annual Review of Physical Chemistry; Annual Reviews: Palo Alto, 2007; Vol. 58, pp 57–83.
(29) Klauda, J. B.; Venable, R. M.; Freites, J. A.; O’Connor, J. W.; Tobias, D. J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A. D.; Pastor, R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114 (23), 7830–7843.
(30) Guvench, O.; Mallajosyula, S. S.; Raman, E. P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T. J.; Jamison, F. W.; MacKerell, A. D. CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and
Carbohydrate–Protein Modeling. J. Chem. Theory Comput. 2011, 7 (10), 3162–3180. (31) MacKerell, A. D.; Banavali, N. K. All-Atom Empirical Force Field for Nucleic
Acids: II. Application to Molecular Dynamics Simulations of DNA and RNA in Solution. J. Comput. Chem. 2000, 21 (2), 105–120.
(33) Voelz, V. A.; Dill, K. A.; Chorny, I. Peptoid Conformational Free Energy Landscapes from Implicit-Solvent Molecular Simulations in AMBER. Pept. Sci. 2011, 96 (5), 639–650.
(34) Butterfoss, G. L.; Yoo, B.; Jaworski, J. N.; Chorny, I.; Dill, K. A.; Zuckermann, R. N.; Bonneau, R.; Kirshenbaum, K.; Voelz, V. A. De Novo Structure Prediction and Experimental Characterization of Folded Peptoid Oligomers. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (36), 14320–14325.
(35) Mukherjee, S.; Zhou, G.; Michel, C.; Voelz, V. A. Insights into Peptoid Helix
Folding Cooperativity from an Improved Backbone Potential. J. Phys. Chem. B 2015, 119 (50), 15407–15417.
(36) Mirijanian, D. T.; Mannige, R. V.; Zuckermann, R. N.; Whitelam, S. Development and Use of an Atomistic CHARMM-Based Forcefield for Peptoid Simulation. J. Comput. Chem. 2014, 35 (5), 360–370.
CHAPTER 2- Methods 2.1. Introduction
Peptoids are promising materials for diverse applications ranging from surfactants, to catalysts, to coatings, because their backbone conformations may be controlled through a careful choice side chains. This precise control of folding has led to peptoid applications, including lung surfactant mimics built from combinations of amino acid-like and helix inducing side chains1, and antibacterial peptoids whose target specificity depends on their
backbone helicity2. Other peptoid applications such as scaffolds3 and collagen mimics4 are stabilized by ordered and predictable secondary structures. Pharmaceutical applications incorporating functional or drug-like groups onto these scaffolds will require knowledge of how side chain substitutions affect secondary structure stability. Therefore, a goal of peptoid research is to characterize how specific side chains may be combined to build complex and useful materials. A key aspect of this goal is characterizing side chain sequence effects on peptoid structure.
Experimental studies have made progress decoding peptoid side-chain/conformation relationships and, in particular, have shown how side chains can influence cis/trans
isomerization of the peptoid bond. Nuclear Overhauser effect spectroscopy5 (NOESY) can
measure the isomerization state of the peptoid bond6 and has been used to show that side chain sterics7, charges, hydrogen bonds8, and n⟶π* bonding9 can all be harnessed to
stabilize cis or trans states. Circular Dichroism (CD)10 and X-ray spectra11 have shown that
built by alternating cis-inducing and trans-inducing side-chains 14. Still, experimental work has done little to identify how side chains can control the more flexible peptoid degrees of freedom including other backbone dihedrals. Additionally, just a fraction of the 300+ possible peptoid side chains have been studied experimentally, leaving much room for further characterization.
Previous computational studies have explored peptoid side-chain/backbone
conformation relationships using a variety of methods including quantum mechanics15,16, all-atom MD17–21, coarse-grained Monte Carlo22–24, conformational search25, and knowledge-based methods26,27. Results have been promising, for example: Butterfoss et al. showed that combined all-atom MD and quantum mechanics methods could successfully predict
structures of peptoids stabilized by strong steric effects16. Butterfoss also used quantum mechanics simulations to show that monomer level side chain effects correspond to macromolecular structures15. This promising result suggests that methods that
parameterization of all-atom force fields using quantum data might be effective for structure prediction of longer chains.
Our work uses all-atom MD simulations to study peptoid systems in explicit solvent. Molecular dynamics simulations reproduce system behavior by numerically solving
force on each of the components, and uses them to propagate the positions and velocities in finite time steps using numerical integration methods such as the Verlet or velocity Verlet algorithms29. In all-atom MD, the interacting entities are the atoms that make up the system. Force field interaction parameters may also describe molecular interactions in terms of clusters of atoms. This approach is called coarse-graining and sacrifices simulation resolution for the ability to sample larger groups of molecules at longer timescales. Peptoid secondary structures are stabilized by atomic-scale interactions including hydrogen bonds and n⟶π* bonding, and thus we have chosen to model them using all-atom simulations.
The accuracy of an all-atom MD simulation depends on the quality of the force field used. Several classes of force fields exist. The simplest, Class I force fields include
intramolecular interactions such as bond stretching, angle bending, and dihedral potentials, as well as intermolecular interactions such as van der Waals forces and electrostatics30.
Commonly used Class I force fields include AMBER and CHARMM. Other force fields, such as Class II31 force fields use higher order terms such as cross coupling. But these are not
discussed here.
We have chosen to use the CHARMM General force field, because it has a clearly defined parameterization protocol. We carefully followed these procedures to obtain force field parameters for the peptoid backbone and three common peptoid side chains from quantum calculations. and so we have developed highly-tuned peptoid force field parameters based on the CHARMM general force field (CGENFF)32. GGENFF is a force field
parameters to new structures via analogy to the compounds in the database32, and clearly defined procedures for tuning these assigned parameters using quantum mechanical calculations, which are described in section 2.2.3.
Our work is complementary to previous work developing parameters for the CHARMM22 force field20. However, that work required experimental tuning of Lennard-Jones parameters, a procedure not required for fitting CGENFF parameters. Our work
presents a completely computational approach to peptoid force field development, and shows that peptoid parameters fit to quantum mechanics calculations are able to reproduce
experimental data. Additionally, it offers a template showing how CGENFF might be used to characterize other side-chains in the future.
To understand how solvation affects peptoids simulated with our force field, we use the metadynamics method33 to directly calculate the conformation-dependent free energies for solvated peptoid monomers. Metadynamics uses collective variables, which are
combinations of coordinates that describe important motions of a system. The collective variables used in this work are the backbone dihedrals φ and ψ, which have historically been used to describe peptide conformations in the form of Ramachandran plots. Metadynamics can be used to calculate conformation-dependent free energies. Calculating a conformation’s free energy allows direct calculation of its probability using the equation F=-kBTln(P).
and allows us to restrict the region of the free energy surface explored by changing how fast the hills decay34,35. Converged WT-MetaD should yield a fully sampled system, and the underlying biasing force profile. From this profile, the underlying free energy of the system can be calculated. More details on this method are given in section 2.2.4
2.2. Description of Methods
2.2.1.All-atom Molecular Dynamics
Molecular dynamics (MD) is a simulation method used to study the dynamic and equilibrium properties of chemical systems through integration of Newton’s laws of motion. As described in section 2.1, all-atom MD represents the chemical system at atomic
resolution. MD calculates motions and energies of a system of molecules over a series of timesteps. The time and length scales accessible by MD depend on the computer hardware used, and the time for each simulation step generally scales in a quasilinear manner with system size. Timesteps must be less than the system’s fastest degree of freedom, and for all-atom MD, are typically on the order of 1 fs ≡ 10-15 s. Massively parallel simulation software including NAMD36, GROMACS37, CHARMM38, AMBER39, and LAMMPS40 increase the time and length scales accessible to all-atom MD simulations. MD simulations in this work were performed using NAMD.
system becomes caught in a metastable state and is unable to sample over high energy barriers To escape these energy barriers, accelerated sampling methods such as replica exchange MD (REMD)41, umbrella sampling42, or metadynamics may be used33.
2.2.2.CHARMM General Force Field
2.2.2.1.The CHARMM Potential Function
The CHARMM family of force fields are built on Class I additive potential energy function, where contributions from each bonded and non-bonded interaction are summed to yield he total potential energy of the system:
𝑈*+,-.. = 𝑈010210343+ 𝑈210343 (2.1) CHARMM describes the nonbonded van der Waals, overlap and electrostatic interactions, using Lennard-Jones type potential energy functions and a Coulomb potential term that uses effective “partial charges” for all atoms in the system:
𝑈010210343= 𝜀78 𝑅:70;< 𝑟78
>?
− 𝑅:70;< 𝑟78
A
7B8
+ 𝑞7𝑞8
4𝜋𝐷𝑟78 (2.2)
Where the force field parameters are:
𝑅:70;<-the Lennard-Jones radius εij - the interaction energy
qi and qj- the partial charges
rij - the distance between atoms
𝑈210343= 𝑘2 𝑏 − 𝑏1 ? 2103I
+ 𝑘J 𝜃 − 𝜃1 ? L0MN4I
+
𝑘O 37P43QLNI
1 + cos 𝑛𝜙 − 𝛿 + 𝑘Y 𝜔 − 𝜔1 ?
7:[Q1[4QI (2.3) + 𝑘\ 𝑢 − 𝑢1 ?
^Q4_`aQL3N4_
Where kb, kθ, kω, ku are force constants, and bo, θo, ωo, and uo are equilibrium values. Figure
2.1 shows the interactions described by the bonded force field terms. Unique force constants and equilibrium terms are defined for each combination of atom types. These parameters together comprise the force field.
Figure 2.1 The bonded interactions parameterized in the CHARMM force fields
Two of the interaction terms shown in equation (2.3) are not discussed in this work: Improper dihedrals are used to hold chemical groups in a plane, and in CGENFF they are only used to describe the planar carboxyl group. Urey-Bradley terms place a harmonic restraint interaction distance between atoms separated by a single angle, and are typically redundant with the angle potential.
𝑘O b7P43QLNI
1 + cos 𝑛𝜙 − 𝛿 (2.4) where:
kd –is the dihedral force constant in kcal/Mol n – is an integer periodicity of the angle and:
δ – is a shift factor equaling either 0° or 180°
The periodicity, n, of the dihedral terms should relate to the rotational symmetry of the rotated dihedral term. Multiple dihedral potential terms may be tuned for each dihedral fit. An example of this is seen in section 4.5.
2.2.2.2.The CHARMM General Force Field
The CHARMM General Force Field (CGENFF)32 was developed to enable quick parameterization of complex drug molecules within the CHARMM framework. CGENFF contains a set of atom types, a set of previously parameterized bonded and nonbonded parameters, and an automated fitting utility that assigns previously undefined parameters based on analogy to a database of parameterized compounds.
Force-field fitting via CGENFF consists of two steps: 1. Submitting a chemical structure to CGENFF’s automated fitting utility CGENFF43–45, which automatically assigns nonboded parameters based on similarity to previously parameterized molecules in a
verified. For our system, CGENFF recommended that we fit a single dihedral parameter but properly reproducing backbone energetics actually required that we refit partial charges and three dihedral potentials.
An appealing aspect of CGENFF parameterization is that all CGENFF parameters are fitted to quantum mechanics calculations rather than to experimental data. The van der Waals parameters, originally fit to experimental liquid densities, are assigned by CGENFF and left unchanged during force field tuning. Angle, bond, and improper dihedral parameters are refined using quantum vibrational spectra. Dihedral parameters are fit to energy scans of dihedrals at the MP2/(G-31G*) level of theory. In these scans, the target dihedral is adjusted in 5-15° intervals and the system is minimized, and energy is calculated with the dihedral constrained. Dihedral force constants are then fit to minimize the root mean square distance between force field and the quantum energy values.
All of the CGENFF parameter tuning procedures are implemented in the Visual Molecular Dynamics (VMD) 1.9.146 Force Field Toolkit (FFTK)47, and all quantum calculations are performed using the Gaussian09 software package48.
2.2.3.Quantum Methods
2.2.3.1.Hartree-Fock and MP2 Methods
Quantum mechanics simulation methods estimate the electronic energy by finding approximate solutions to the many-body Shcrödinger equation:
𝐻𝜓 = Ε𝜓 (2.5) Where ψ is the wave function, E is the system energy, and H is the Hamiltonian:
𝐻 = − ∇7? i 7j> − 1 2𝑀, . ,j> ∇,?− 𝑍, 𝑟7, . ,j> i 7j> + 1 𝑟78 i 8n> i 7j>
+ 𝑍,𝑍a 𝑟,a . an, . ,j> (2.6)
The ∇s are position gradients:
∇ = 𝜕? 𝜕𝑥?+ 𝜕? 𝑑𝑦?+ 𝜕? 𝑑𝑧? (2.7) MA,ZA, ZB are the mass and charges of nuclei, and rAB, rij, and riA are distances between two
nuclei, between two electrons, and between a nuclei and an electron respectively.
Solving the Schrödinger equation is an eigenvalue problem whose solution requires determining both the wave function ψ and the energy E. The Schrödinger equation can only be solved exactly for single-particle systems, however, useful approximations have been derived by applying appropriate assumptions, approximations, and constraints. One
important assumption is the Born-Oppenheimer approximation, which assumes the nuclei are stationary and that the energy of the system depends on the motions of the electrons. This assumption means that QM simulations estimate energies at 0 K.
𝑓7𝜒7 = 𝜀7𝜒7 (2.8)
In these equations fi is the Fock operator which calculates the Hamiltonian for an electron in
the field of the system’s other electrons. The Fock operator incorporates interactions with the electron core and other electrons of the system subject to constraints including the Pauli Exclusion principal. 𝜒7 is the spin orbital containing electron i, and εi is the energy of that
orbital.
Hartree-Fock equations are solved iteratively using the Self Consistent Field approach where the interdependent fi and χi are solved iteratively, until both equations yield consistent
results. Approximate solutions are usually achieved using the representation of an orbital with a linear combination of atomic orbitals (LCAO):
𝜓7 = 𝑐w7𝜙w x
wj>
(2.9)
Where 𝜙y called the “basis” functions, and c are constants of a matrix called the slater determinant. The choice of basis sets affects the quality of the calculation. Many basis sets have been developed, such as the Gaussian basis sets and the Dunning correlation-consistent basis sets.
HF by performing a second order Taylor Series expansion using the Hartree Fock solution as a reference.
2.2.4.Well-Tempered Metadynamics
Metadynamics is an enhanced sampling method designed to increase sampling of rare events, therefore improving system sampling, and encouraging the system to explore new configurations based on a biasing force that is dynamically evolved throughout the
simulation. In metadynamics, biasing force is added in the form of Gaussian hills, according to the equations:
𝑉 = 𝜔 𝜏M𝑒
`I } `I } ~• € ?ð ‚€
(2.10)
Where:
𝑉 is the change in biasing force with respect to time
ω is the frequency of hill deposition
τg is a characteristic time describing the rate of bias evolution
s(x) is a collective variable as a function of geometric coordinates x and
δ is the width of the deposited hill
The system coordinates move according to the sum of the system free energy profile and the biasing potential. Giving an effective potential:
A metadynamics simulation where F=V will have a VEffectuve of zero. A system under this
condition will diffuse randomly with respect to the biased variables. Random diffusion with respect tot the biased variable is one convergence test for metadynamics simulations. At this point, the added biased can be calculated to determine determine the underlying free energy surface of the system.
In this work, we use metadynamics of the well-tempered variety, where the size of the hills declines based on the amount of previous bias added to the coordinate measure. The Well-Tempered Metadynamics (WT-MetaD) variation deposits hills of the form:
𝑉 =𝜔𝐾a∆𝑇 𝜏M 𝑒
`(Š I,~ )‹
Œ∆• 𝑒
`I } `I } ~• € ?ð ‚€
(2.12)
Where the variables have the same definitions as in equation2.10 And 𝐾a is the Boltzman constant
∆𝑇 is a constant in temperature units that describes the rate of hill decay
WT-MetaD is an improvement on plain metadynamics in that it can be rigorously proven to converge34. Additionally, WT-MetaD can overcome metadynamics’ tendency to drive exploration of unfavorable, high energy states. The bias temperature controls the amount that the simulation is biased, effectively giving control of the temperature “experienced” by the collective variables.
In addition to increasing sampling of transition states and slow degrees of freedo the metadynamics bias potential essentially creates a mold of the underlying free energy surface. This can be used to calculate solvated free energy plots and develop free energies of
Careful choice of simulation parameters is key to a successful metadynamics calculation35. The tunable parameters have the following effects: The Gaussian hill width determines the resolution of the free energy surface. A small hill width will create finer resolution surface but will slow the convergence of the simulation. The Gaussian hill height and thee rate of hill deposition together determine the characteristic time (τ) of bias evolution and control convergence rate. Too quick of a characteristic time, a small τ, and the simulation will deposit hills on top of each other, creating errors in the bias. A very large τ will cause the bias to converge very slowly.
Unbiased, slow degrees of freedom can lead to hysteresis in metadynamics and severely disrupt convergence. In our metadynamics scans of peptoids with large side chains, occasional reorientation of the side chainslead to slow convergence of the simulations. In situations where our metadynamics simulations involved a slow degree of freedom we used harmonic restraints to restrict the additional collective variable to a reasonable value.
2.3. References
(1) Wu, C. W.; Seurynck, S. L.; Lee, K. Y. C.; Barron, A. E. Helical Peptoid Mimics of Lung Surfactant Protein C. Chem. Biol. 2003, 10 (11), 1057–1063.
(2) Chongsiriwatana, N. P.; Patch, J. A.; Czyzewski, A. M.; Dohm, M. T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R. N.; Barron, A. E. Peptoids That Mimic the Structure, Function, and Mechanism of Helical Antimicrobial Peptides. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (8), 2794–2799.
(3) Olivier, G. K.; Cho, A.; Sanii, B.; Connolly, M. D.; Tran, H.; Zuckermann, R. N. Antibody-Mimetic Peptoid Nanosheets for Molecular Recognition. ACS Nano 2013, 7 (10), 9276–9286.
(4) Johnson, G.; Jenkins, M.; McLean, K. M.; Griesser, H. J.; Kwak, J.; Goodman, M.; Steele, J. G. Peptoid-Containing Collagen Mimetics with Cell Binding Activity. J. Biomed. Mater. Res. 2000, 51 (4), 612–624.
(5) Schnell, I. Basic One- and Two-Dimensional NMR Spectroscopy. Fourth,
Completely Revised and Updated Edition. By Horst Friebolin. ChemPhysChem 2005, 6 (6), 1187–1188.
(6) Huang, K.; Wu, C. W.; Sanborn, T. J.; Patch, J. A.; Kirshenbaum, K.; Zuckermann, R. N.; Barron, A. E.; Radhakrishnan, I. A Threaded Loop Conformation Adopted by a Family of Peptoid Nonamers. J. Am. Chem. Soc. 2006, 128 (5), 1733–1738.
(7) Shah, N. H.; Butterfoss, G. L.; Nguyen, K.; Yoo, B.; Bonneau, R.; Rabenstein, D. L.; Kirshenbaum, K. Oligo(N-Aryl Glycines): A New Twist on Structured Peptoids. J. Am. Chem. Soc. 2008, 130 (49), 16622–16632.
(8) Stringer, J. R.; Crapster, J. A.; Guzei, I. A.; Blackwell, H. E. Construction of Peptoids with All Trans-Amide Backbones and Peptoid Reverse Turns via the Tactical
Incorporation of N-Aryl Side Chains Capable of Hydrogen Bonding. J. Org. Chem. 2010, 75 (18), 6068–6078.
(9) Benjamin C. Gorske; Nelson, R. C.; Bowden, Z. S.; Kufe, T. A.; Childs, A. M. “Bridged” N→π* Interactions Can Stabilize Peptoid Helices. J. Org. Chem. 2013, 78 (22), 11172–11183.
(10) Berova, N.; Nakanishi, K.; Woody, R. Circular Dichroism : Principles and Applications, 2nd ed.; Wiley-VCH,: New York, c2000.
(12) Wu, C. W.; Sanborn, T. J.; Huang, K.; Zuckermann, R. N.; Barron, A. E. Peptoid Oligomers with Alpha-Chiral, Aromatic Side Chains: Sequence Requirements for the Formation of Stable Peptoid Helices. J. Am. Chem. Soc. 2001, 123 (28), 6778–6784. (13) Shah, N. H.; Butterfoss, G. L.; Nguyen, K.; Yoo, B.; Bonneau, R.; Rabenstein, D. L.;
Kirshenbaum, K. Oligo(N-Aryl Glycines): A New Twist on Structured Peptoids. J. Am. Chem. Soc. 2008, 130 (49), 16622–16632.
(14) Crapster, J. A.; Guzei, I. A.; Blackwell, H. E. A Peptoid Ribbon Secondary Structure. Angew. Chem.-Int. Ed. 2013, 52 (19), 5079–5084.
(15) Butterfoss, G. L.; Renfrew, P. D.; Kuhlman, B.; Kirshenbaum, K.; Bonneau, R. A Preliminary Survey of the Peptoid Folding Landscape. J. Am. Chem. Soc. 2009, 131 (46), 16798–16807.
(16) Butterfoss, G. L.; Yoo, B.; Jaworski, J. N.; Chorny, I.; Dill, K. A.; Zuckermann, R. N.; Bonneau, R.; Kirshenbaum, K.; Voelz, V. A. De Novo Structure Prediction and Experimental Characterization of Folded Peptoid Oligomers. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (36), 14320–14325.
(17) Voelz, V. A.; Dill, K. A.; Chorny, I. Peptoid Conformational Free Energy Landscapes from Implicit-Solvent Molecular Simulations in AMBER. Pept. Sci. 2011, 96 (5), 639–650.
(18) Mukherjee, S.; Zhou, G.; Michel, C.; Voelz, V. A. Insights into Peptoid Helix
Folding Cooperativity from an Improved Backbone Potential. J. Phys. Chem. B 2015, 119 (50), 15407–15417.
(19) Park, S. H.; Szleifer, I. Structural and Dynamical Characteristics of Peptoid Oligomers with Achiral Aliphatic Side Chains Studied by Molecular Dynamics Simulation. J. Phys. Chem. B 2011, 115 (37), 10967–10975.
(20) Mirijanian, D. T.; Mannige, R. V.; Zuckermann, R. N.; Whitelam, S. Development and Use of an Atomistic CHARMM-Based Forcefield for Peptoid Simulation. J. Comput. Chem. 2014, 35 (5), 360–370.
(21) Mannige, R. V.; Haxton, T. K.; Proulx, C.; Robertson, E. J.; Battigelli, A.; Butterfoss, G. L.; Zuckermann, R. N.; Whitelam, S. Peptoid Nanosheets Exhibit a New
Secondary-Structure Motif. Nature 2015, 526 (7573), 415–420.
(23) Haxton, T. K.; Mannige, R. V.; Zuckermann, R. N.; Whitelam, S. Modeling Sequence-Specific Polymers Using Anisotropic Coarse-Grained Sites Allows Quantitative Comparison with Experiment. J. Chem. Theory Comput. 2015, 11 (1), 303–315.
(24) Sanii, B.; Haxton, T. K.; Olivier, G. K.; Cho, A.; Barton, B.; Proulx, C.; Whitelam, S.; Zuckermann, R. N. Structure-Determining Step in the Hierarchical Assembly of Peptoid Nanosheets. ACS Nano 2014, 8 (11), 11674–11684.
(25) Armand, P.; Kirshenbaum, K.; Falicov, A.; Dunbrack, R. L.; Dill, K. A.;
Zuckermann, R. N.; Cohen, F. E. Chiral N-Substituted Glycines Can Form Stable Helical Conformations. Fold. Des. 1997, 2 (6), 369–375.
(26) Renfrew, P. D.; Craven, T. W.; Butterfoss, G. L.; Kirshenbaum, K.; Bonneau, R. A Rotamer Library to Enable Modeling and Design of Peptoid Foldamers. J. Am. Chem. Soc. 2014, 136 (24), 8772–8782.
(27) Drew, K.; Renfrew, P. D.; Craven, T. W.; Butterfoss, G. L.; Chou, F.-C.; Lyskov, S.; Bullock, B. N.; Watkins, A.; Labonte, J. W.; Pacella, M.; Kilambi, K. P.; Leaver-Fay, A.; Kuhlman, B.; Gray, J. J.; Bradley, P.; Kirshenbaum, K.; Arora, P. S.; Das, R.; Bonneau, R. Adding Diverse Noncanonical Backbones to Rosetta: Enabling Peptidomimetic Design. Plos One 2013, 8 (7), e67051.
(28) Leach, A. R. Molecular Modelling : Principles and Applications, 2nd ed.; Pearson/Prentice Hall,: Harlow, England, 2001.
(29) Frenkel, D.; Smit, B. Understanding Molecular Simulation : From Algorithms to Applications, 2nd ed.; Computational science ; v. 1; Academic,: San Diego, Calif., c2002.
(30) Guvench, O.; MacKerell, A. D. Comparison of Protein Force Fields for Molecular Dynamics Simulations. In Molecular Modeling of Proteins; Methods Molecular BiologyTM; Humana Press, 2008; pp 63–88.
(31) Maple, J.; Hwang, M.; Stockfisch, T.; Dinur, U.; Waldman, M.; Ewig, C.; Hagler, A. Derivation of Class-Ii Force-Fields .1. Methodology and Quantum Force-Field for the Alkyl Functional-Group and Alkane Molecules. J. Comput. Chem. 1994, 15 (2), 162– 182.
CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31 (4), 671–690.
(33) Laio, A.; Gervasio, F. L. Metadynamics: A Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemistry and Material Science. Rep. Prog. Phys. 2008, 71 (12), 126601.
(34) Dama, J. F.; Parrinello, M.; Voth, G. A. Well-Tempered Metadynamics Converges Asymptotically. Phys. Rev. Lett. 2014, 112 (24), 240602.
(35) Bussi, G.; Branduardi, D. Free-Energy Calculations with Metadynamics: Theory and Practice. In Reviews in Computational Chemistry, Vol 28; Parrill, A. L., Lipkowitz, K. B., Eds.; John Wiley & Sons Inc: Hoboken, 2015; Vol. 28, pp 1–49.
(36) Phillips, J. C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R. D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802.
(37) Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4 (3), 435–447.
(38) Brooks, B. R.; Brooks, C. L.; Mackerell, A. D.; Nilsson, L.; Petrella, R. J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; Caflisch, A.; Caves, L.; Cui, Q.; Dinner, A. R.; Feig, M.; Fischer, S.; Gao, J.; Hodoscek, M.; Im, W.; Kuczera, K.; Lazaridis, T.; Ma, J.; Ovchinnikov, V.; Paci, E.; Pastor, R. W.; Post, C. B.; Pu, J. Z.; Schaefer, M.; Tidor, B.; Venable, R. M.; Woodcock, H. L.; Wu, X.; Yang, W.; York, D. M.; Karplus, M. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30 (10), 1545–1614.
(39) Pearlman, D.; Case, D.; Caldwell, J.; Ross, W.; Cheatham, T.; Debolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. Amber, a Package of Computer-Programs for Applying Molecular Mechanics, Normal-Mode Analysis, Molecular-Dynamics and Free-Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput. Phys. Commun. 1995, 91 (1–3), 1–41.
(40) Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Comput. Phys. 1995, 117 (1), 1–19.
(42) Torrie, G. M.; Valleau, J. P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23 (2), 187– 199.
(43) Vanommeslaeghe, K.; MacKerell, A. D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52 (12), 3144–3154.
(44) Vanommeslaeghe, K.; Raman, E. P.; MacKerell, A. D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52 (12), 3155–3168.
(45) ttps://Cgenff.paramchem.org.
(46) Humphrey, W.; Dalke, A.; Schulten, K. VMD – Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38.
(47) Mayne, C. G.; Saam, J.; Schulten, K.; Tajkhorshid, E.; Gumbart, J. C. Rapid Parameterization of Small Molecules Using the Force Field Toolkit. J. Comput. Chem. 2013, 34 (32), 2757–2770.
(48) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox. Gaussian˜09 Revision A.02; Wallingford CT: Gaussian, Inc, 2016.
CHAPTER 3- A Review of Previous Simulation Studies of Peptoids A version of this chapter has been published in:
L.J. Weiser and E.E. Santiso, “Molecular Modeling Studies of Peptoid Polymers”, AIMS Biomaterials, 2017, 4(5)
Abbreviations
AMBER Assisted Model Building with Energy Refinement B3LYP Becke, 3-parameter, Lee-Yang-Parr hybrid functional CHARMM Chemistry at Harvard Macromolecular Mechanics
BCC bond centered charges
CD Circular Dichroism
GAFF General AMBER Force Field
GBSA Generalized Born Surface Area implicit solvent
HF Hartree Fock
IEFPCM integral equation formalism for the polarizable continuum model MF-CG-TOID Molecular Foundry Coarse-grained Peptoid model
MFTOID Molecular Foundry Peptoid Force Field MP2 Møller-Plesset perturbation theory
NMR Nuclear magnetic resonance spectroscopy
PCM polarized continuum model
PPI Helix polyproline type I helix PPII Helix polyproline type II helix
3.1. Introduction
Peptoids are synthetic, protein-like polymers composed of poly-glycine backbones with side chains attached to their backbone nitrogen atoms [1][2]. (See Figure 1). Peptoids are protease resistant and biocompatible and can be designed to mimic peptides[3], diffuse through membranes [4], and bind biological targets [5]. They have potential applications as drug molecules [6], antimicrobials[7], lung surfactants [8], and catalysts [9]. Peptoids can be synthesized with specific sequences in a step-by-step approach. Their synthesis, applications, and experimental characterizations have been extensively reviewed elsewhere [1][2].
Peptoids are a kind of foldamer [10], i.e. linear polymers that fold into diverse 3D structures depending on the sequence of their attached side chains – just as amino acids combine to yield a diverse library of proteins. Single peptoid chains have been shown to adopt ordered structures including polyproline type I helices (PPI) [11], polyproline type II helices (PPII) [12], a threaded loop [13], flat ‘ribbons’ [14] and ‘Σ-strands’ [15], which are shown in Figure 2. Groups of peptoid chains can further self-assemble into nanostructures including microspheres [16], superhelices [17], and thin, bilayered nanosheets [18]. The chief focus of this review are simulation studies that enhance our understanding of peptoids as foldamers, i.e. those studies that describe the relation between side chains and backbone conformations.
As researchers attempt to develop peptoids into useful materials, a key challenge is identifying side chains, and combinations of side chains, that drive backbone folding in a predictable way. This challenge is closely related to the well-known protein folding problem [19], whereby researchers aspire to predict folded protein structures from sequences of amino acids. Peptoid researchers have mainly approached this problem by identifying side chains expected to exert local structural preferences through sterics [11,12], electrostatics [20], hydrogen bonding [21], or n-π* bonding [22]. These candidates are then synthesized into peptoid polymers and characterized using experimental methods such as X-ray crystallography [23], circular dichroism [24], and NMR [25].
(potentially synthesized from over 300 commercially available primary amines [2]), and thus
the astronomical number of possible combinations of side chains, molecular simulations will continue to play a crucial role in peptoid development, and will benefit from the extensive body of work devoted to the protein folding problem.
Computational approaches to the protein folding problem consist of methods, models, and algorithms that read in amino acid sequences and return 3-D structure predictions. These predictions can be based on physical, force field models with parameters derived from theory or tuned to experimental data [26-27], and/or they can be heuristically scored according to similarity with existing sequences [28-30]. We focus our discussion on the former approach, as it is most transferrable to peptoids: little experimental data exists for most side chains and theoretical data is easier to obtain.
larger simulations involving multiple chains, such as aggregation. More details on the different levels of molecular models can be found in [31] and [32].
Peptoid modeling can be approached using a similar framework: using quantum methods to characterize monomer subunits, and building these into force fields that can describe longer chains, or systems of chains. Protein force fields may sometimes be used for peptoid simulation because the backbones of peptides and peptoids are composed from the same atoms. However, some key differences between peptoids and peptides can limit this transferability. Force fields for peptoids should be carefully verified and parameterized when appropriate.
Three key features of peptoid backbones can grant them local folding behavior significantly different from that of proteins: (1) peptoid backbones do not form hydrogen bonds, (2) their backbones are achiral, and (3) the peptoid sp2 amide bond can isomerize and exist as both a cis or a trans conformer. In proteins, hydrogen bonding between nearby backbone amide and carboxyl groups stabilizes highly ordered, localized secondary structures, including α-helices and β-sheets. Because the peptoid side chain replaces the amide hydrogen, peptoids do not form the same secondary structures. Furthermore, in proteins, each Cαatom (with the exception of glycine) is a chiral center with (L) chirality,
always in a trans configuration, where the carboxyl oxygen and amide hydrogen rest on opposite sides of the peptide bond, as the cis conformation leads to a steric clash between side chains. The peptoid amide bond, in contrast, can exist as a cis or a trans conformer, generally determined by the size of the side chain residue.
The differences mentioned above must be addressed when developing peptoid models. In the case of chirality and hydrogen bonding differences, this can be achieved by careful adaption of protein models. Protein force field parameters can be tuned to favor secondary structures not present in peptoids [33], so protein force fields transferred to peptoids should be verified and tuned against peptoid experimental data. Addressing the amide bond isomerization can be more challenging. The effect of a side chain on a peptoid secondary structure can differ depending on the isomerization state of the peptide bond. Due to this issue, all ab initio characterizations of side chain energetics should be examined in both the cis and the trans states. Ideally, parameters in all-atom and coarse-grained force fields should be tuned to accurately represent both configurations.
between cis and trans structures, this isomerization must be studied through enhanced sampling techniques that force the system across the barrier. These include umbrella sampling [37] and replica exchange molecular dynamics [38].
Figure 3.2 Known Peptoid Secondary Structures. (A) The PPI helix (shown here as (S)-N-(1-napthylethyl)glycine) [11], [39], (B) the N-(phenyl)glycine
PPII helix [12], (C), the peptoid ribbon [14] (D) the threaded loop [13], and (E) the Σ-strand [15]. Note that the figures are reproduced from different sources, and we have kept the original representations for consistency with them. A,C, and D are shown without their backbone hydrogen atoms, and E is shown without its side chains. The secondary structures are characterized by their patterns of φ and ω dihedrals defined in Figure 3. The PPI and PPII helices have repeating (φ=(-)75°, ω~0°) and (φ=(-)75°,ω=180°) dihedrals, respectively. The threaded loop (D) is created by 9-mer NSPE peptoids and is stabilized by a hydrogen bonding interaction between side chains and their NH2 groups. The ribbon (C) has alternating ω=0° and
ω=180° dihedrals and the Σ-strand (E) has ω=180° and alternating φ=-90°,ψ=120° and φ=-120°,ψ=90°- dihedrals. Structure A Reprinted with permission from Stringer JR, Crapster JA, Guzei IA, et al. (2011) J Am Chem Soc 133: 15559–15567.
Copyright (2011) American Chemical Society [39]. Structure B reprinted with permission from Shah NH, Butterfoss GL, Nguyen K (2008) J Am Chem Soc 130: 16622–16632. Copyright (2008) American Chemical Society [12]. Structure C reprinted with permission from [14]. Structure D Reprinted (adapted) with
3.2.1. Ab initio studies of peptoids
Ab initio simulations can be used to calculate highly accurate energy minimized structures for small molecules and have been used to characterize monomer-level structural preferences for peptoids. An advantage to this approach is that ab initio calculations are based wholly on quantum theory and do not rely on system-specific force field parameters or tuning to experimental data. A disadvantage is that ab initio simulations are computationally expensive for all but the smallest molecules. Therefore, ab initio peptoid modeling has mostly been used to characterize short-range energy preferences of peptoid monomers and to refine structure predictions from all-atom simulations (see section 2.2). This section focuses on ab initio characterization of peptoid monomers, but also describes the ab initio characterization of the binding structure of a peptoid trimer.
conformers. A 3rd backbone dihedral, ω, describes the cis/trans isomerization. To compare cis (ω = 0° ) and trans (ω = 180° ) probabilities in the same figure, plots of ω versus φ or ω versus ψ, may be more appropriate (See Figure 4) [41]. By convention, peptoid Ramachandran plots may also be plotted on 0°-360° axes rather than -180°-180° axis so that the αD minimum is centered.
Figure 3.3 The peptoid backbone dihedrals and a peptoid Ramachandran plot. (A) Peptoid backbone conformations can be described by the dihedrals φ, ψ, and ω. The dihedral ω describes the isomerization of the planar peptoid bond. In the cis (ω=0°) configuration, the side chain and the carbonyl oxygen are on the same side of the bond. In the trans (ω=180°) configuration (shown) the side chain and the oxygen lie on opposite sides of the amide bond. Ramachandran plots (B) are used to show backbone (φ-ψ) conformational preferences. Ramachandran plots are 2-D histograms that graph either the probability or the conformational energy of each φ/ψ pair of dihedrals. The figure shown corresponds to the trans isomer. This figure is based on data from Butterfoss [41]
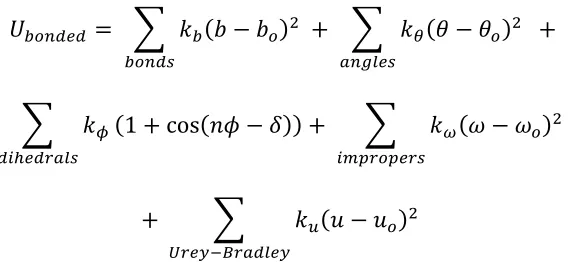
![Figure 3.2 Known Peptoid Secondary Structures. (A) The PPI helix (shown here as (S)-N-(1-napthylethyl)glycine) [11], [39], (B) the N-(phenyl)glycine PPII helix [12], (C), the peptoid ribbon [14] (D) the threaded loop [13], and (E) the Σ-strand [15]](https://thumb-us.123doks.com/thumbv2/123dok_us/1335920.1166559/52.612.210.418.66.320/figure-peptoid-secondary-structures-napthylethyl-glycine-peptoid-threaded.webp)


![Figure 3.8 The Σ-strand alternating motif. Mannige et al. [15] described a secondary structure of alternating residues with alternating rotational states](https://thumb-us.123doks.com/thumbv2/123dok_us/1335920.1166559/69.612.195.444.170.488/alternating-mannige-described-secondary-structure-alternating-alternating-rotational.webp)