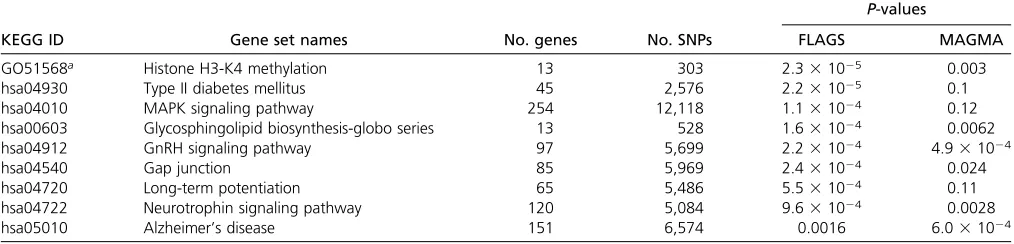| INVESTIGATION
FLAGS: A Flexible and Adaptive Association Test for
Gene Sets Using Summary Statistics
Jianfei Huang,* Kai Wang,†Peng Wei,‡Xiangtao Liu,* Xiaoming Liu,§Kai Tan,**,††Eric Boerwinkle,§,‡‡
James B. Potash,* and Shizhong Han*,††,1
*Department of Psychiatry,†Department of Biostatistics, and **Department of Internal Medicine and††Interdisciplinary Graduate Program in Genetics, University of Iowa, Iowa City, Iowa 52242,‡Department of Biostatistics, University of Texas School of Public Health, Houston, Texas 77225,§Human Genetics Center, University of Texas Health Science Center, Houston, Texas 77030, and ‡‡Human Genome Sequencing Center, Baylor College of Medicine, Houston, Texas 77030
ABSTRACT Genome-wide association studies (GWAS) have been widely used for identifying common variants associated with complex diseases. Despite remarkable success in uncovering many risk variants and providing novel insights into disease biology, genetic variants identified to date fail to explain the vast majority of the heritability for most complex diseases. One explanation is that there are still a large number of common variants that remain to be discovered, but their effect sizes are generally too small to be detected individually. Accordingly, gene set analysis of GWAS, which examines a group of functionally related genes, has been proposed as a complementary approach to single-marker analysis. Here, we propose aflexible andadaptive test forgenesets (FLAGS), using summary statistics. Extensive simulations showed that this method has an appropriate type I error rate and outperforms existing methods with increased power. As a proof of principle, through real data analyses of Crohn’s disease GWAS data and bipolar disorder GWAS meta-analysis results, we demonstrated the superior performance of FLAGS over several state-of-the-art association tests for gene sets. Our method allows for the more powerful application of gene set analysis to complex diseases, which will have broad use given that GWAS summary results are increasingly publicly available.
KEYWORDScomplex disease; gene set; association; GWAS; summary statistics
G
ENOME-WIDE association studies (GWAS), which examine millions of single-nucleotide polymorphisms (SNPs) across the genome, have been widely used for iden-tifying common variants associated with complex diseases (McCarthy et al. 2008). These studies have uncovered nu-merous risk variants and provided novel insights into disease biology. Despite these successes, genetic variants identified to date explain only a small fraction of the heritability for most complex diseases, which raises the question of“missing her-itability” (Manolio et al.2009). One explanation is that a large number of common variants remain to be discovered, but their effect sizes are generally too small to be detected individually. In the search for additional common variants incomplex diseases, more sophisticated analyses of GWAS, rather than SNP-level analysis, can enhance the identification of true genetic signals and advance our understanding of disease biology.
As a complementary approach to SNP-level analysis, gene set analysis, also referred to as pathway analysis, of GWAS has been proposed (Wang et al.2007, 2010). Gene set analysis examines groups of functionally related genes, each of which might contribute a small and individually undetectable effect to the phenotype. The hypothesis is that when examined jointly, the combined effect of all the genes would rise to the detectable level. Gene sets are usually predefined canon-ical pathways, gene ontology terms, or derived from protein– protein interaction networks or other gene networks. Gene set analysis is based on the premise that although many genes may be involved in complex disease, these genes are unlikely to be randomly distributed; rather they should converge on biological pathways and molecular networks underlying the disease. Compared to SNP-level analysis, gene set analysis has several advantages. For example, it can increase power
Copyright © 2016 by the Genetics Society of America doi: 10.1534/genetics.115.185009
Manuscript received November 18, 2015; accepted for publication January 13, 2016; published Early Online January 14, 2016.
Supporting information is available online at www.genetics.org/lookup/suppl/ doi:10.1534/genetics.115.185009/-/DC1.
1Corresponding author: Department of Psychiatry, University of Iowa Carver College of
by reducing multiple testing and aggregating weak signals distributed across a gene set; it is arguably more suited for complex diseases characterized by genetic heterogeneity; and it can provide more insights into disease biology. Indeed, gene set analysis has shown great success in identifying biological pathways underlying many diseases, including au-toimmune diseases (Wang et al. 2009), cancer (Chen and Gyllensten 2014), psychiatric disorders (Network and Path-way Analysis Subgroup of the Psychiatric Genomics Consor-tium 2015), and others.
A variety of methods exist for gene set analysis of GWAS, many of which were evolved from those for gene expression data. Gene set analysis approaches can be broadly catego-rized into competitive or self-contained methods based on the null hypothesis tested (Wanget al.2010). The null hy-pothesis for competitive methods is that genes in a gene set are not more strongly associated with the disease than genes outside the gene set. Examples of competitive tests include gene set enrichment analysis (Wang et al. 2007), the hypergeometric test, and their extensions, such as ALIGATOR (Holmanset al.2009). For self-contained gene set analysis, the null hypothesis assumes that none of the genes in a gene set are associated with the disease. Therefore, it directly tests association for a gene set and does not depend on genes outside the set. Some self-contained methods in-clude the SNP-ratio test (SRT) (O’Dushlaineet al.2009), the sequence kernel association test (SKAT) (Wuet al.2010), PLINK-set (PLINK set-based test) (Purcell et al. 2007), GRASS (gene set ridge regression in association studies) (Chen et al.2010), aSPUpath (adaptive pathway-based sum of powered score test) (Pan et al. 2015), MAGMA (multi-marker analysis of genomic annotation) (de Leeuw et al.2015), and methods combiningP-values. Comprehen-sive reviews and comparisons of existing methods have been offered by several authors (Wang et al.2010, 2011; Atias et al. 2013; Mooney et al. 2014). No consensus has been reached as to which method is optimal, but it is generally believed that competitive test approaches are more conser-vative than self-contained tests (Goeman and Bühlmann 2007).
Existing self-contained methods for gene set analysis generally suffer from several important statistical and com-putational issues. First, most current methods test each gene set equally and do not consider their unique association patterns. It is well known that not all genes in a gene set are causal and the proportion of risk genes may also vary across gene sets. A test that is optimal for one gene set may be underpowered for another if the two sets have very different proportions of risk genes. For instance, if a gene set contains a large proportion of risk genes, a test that combines all genes may be the most powerful; in contrast, the same test will be less powerful for another set that contains only a few causal genes. As we never know the true association pattern of a gene set, it is statistically important and challenging to adaptively aggregate information over causal genes while minimizing the noise of noncausal genes. Second, many current methods use
a permutation-based approach to deriveP-values. Not only is this computationally demanding, but also there are situations in which permutation is not easy or not possible, such as in family-based GWAS designs and gene set analyses for GWAS meta-analytic results from large consortia. Third, current methods often require raw genotype data as input, but it is not always possible to obtain such data.
To address the limitations of existing methods, we propose a flexible and adaptive test for gene sets (FLAGS), using summary statistics. FLAGS takes into account the unique as-sociation pattern of each gene set by adaptively aggregating gene signals derived from SNP-level summary statistics. Ex-tensive simulations show that FLAGS has an appropriate type I error rate and outperforms existing methods with increased power. We further demonstrate by comparative analysis of two real data sets that FLAGS is competitive with and complementary to existing methods. In summary, FLAGS has several advantages over existing methods, including the following:
1. It does not require raw genotype data; rather, it needs only SNP-levelP-values and genotype data of ancestry-matched reference samples.
2. Permutation is not needed to calculate the gene set P-values. Instead, it uses a computationally efficient simulation approach based on a multivariate normal distribution.
3. It is moreflexible and can be applied to almost any GWAS design that has SNP-levelP-values available.
4. It can adaptively choose the most likely subset of causal genes (if any) and maintain high performance across a wide range of disease models.
Materials and Methods
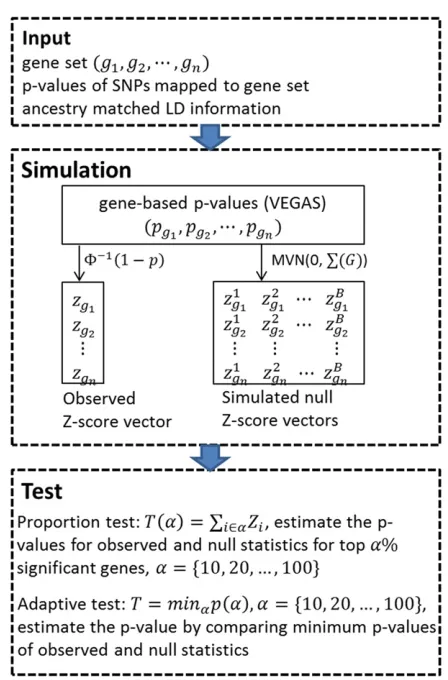
An overview of FLAGS
distribution of joint z-scores of the gene set. To adaptively combine gene signals, FLAGS will construct a class of test statistics that sum over thez-scores for various proportions of the most significant genes (for example, the top 10%, 20%, . . ., 100% of most significant genes). The empirical P-value of each test statistic is then the proportion of simu-lated null statistics that are equal to or larger than the ob-served statistic. To capture the most likely risk genes, FLAGS will scan the series of test statistics aforementioned and take their minimumP-value as an adaptive test statistic. The null distribution of the adaptive test statistic (minimumP-value) can be obtained from the same set of simulated z-score vectors. TheP-value of the adaptive statistic is the propor-tion of null minimumP-values that are equal to or less than the observed minimumP-value.
Estimation of gene correlation matrix
To obtain the null distribution of jointz-scores for a gene set, it is necessary to obtain the covariance matrix ofz-scores among genes. Two genes should show correlation of their z-scores when they are physically close on the same chromo-some. The closer the two genes are located, the higher the expected correlation due to LD. Inspired by VEGAS, we esti-mated the correlation ofz-scores for a given gene pair based on the LD matrix of SNPs within the two genes. The LD in-formation can be obtained from ancestry-matched reference samples, such as those from HapMap, 1000 Genomes, or a custom set of individuals if genotype data are available. Figure S1shows the schematic diagram of our simulation process to estimate the correlation ofz-scores between two genes. In brief, for any given pair of genes, we simulta-neously generateKnumber of multivariate normal vectors Zfor both genes with mean 0 and covariance matrix equal to the pairwise LD matrix of SNPs within the two genes. Zis then transformed into a vector of chi-square variables Q with 1 d.f. For each simulation sample ofQ, we calculate a gene-based statistic by summing over the same proportion of top-rankedQ-values as done for the observed gene-based statistic in VEGAS. The null distribution of the gene-based statistic for each Q sample is approximated by the same gene-based statistics calculated from the remainingQ sam-ples. We then derive a gene-basedP-value for each simu-lated Q sample based on its gene-based statistic and null distribution. Therefore, we can getKnumber of gene-based P-values from each of theKsimulatedQsamples. After con-vertingKnumber of gene-basedP-values intoz-scores, we obtainKnumber ofz-scores for both genes simultaneously. The correlation of thez-score between two genes can then be calculated based on theKnumber of z-scores obtained for each gene.
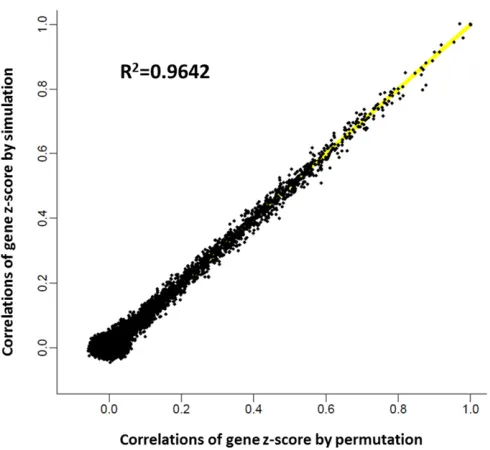
To investigate the validity of this LD-based simulation approach, we compared gene correlations estimated by the simulation approach to those obtained from a standard per-mutation-based approach. Specifically, we constructed null data sets with individual-level genotype data for thefirst 500 genes on chromosome 22, using real GWAS data from the
Atherosclerosis Risk in Communities (ARIC) study (Athero-sclerosis Risk in Communities Investigators 1989). We ran-domly selected 500 samples as cases and 500 samples as controls and permuted case–control status 10,000 times. For each permuted phenotype, we performed an association test for each SNP, using PLINK logistic regression (Purcell et al.2007). For each gene, we obtained 10,000P-value vec-tors with each vector recordingP-values of SNPs within that gene from one permuted phenotype. P-value vectors were then transformed into vectors of chi-square variablesQwith 1 d.f. We then calculated a gene-basedP-value for each gene in the same way as we did in the LD-based simulation ap-proach. We further converted gene-basedP-values toz-scores and obtained 10,000z-scores for each gene. The correlations of all pairwise genez-scores for these 500 genes were then calculated and compared with those obtained by the LD-based simulation approach.
A class of tests and an adaptive test
The proposed gene set method employs VEGAS to com-pute a gene-basedP-value for each gene, using SNP-level P-values and ancestry-matched LD information. The gene-based P-value is then converted to a z-score according to z¼F21ð12pÞ;whereF21denotes the inverse normal distribution function. FLAGS constructs a number of propor-tion test statistics that are the sum ofz-scores over various proportions of the most significant genes (for example, the top 10%, 20%, . . ., 100% of the most significant genes). Formally, we define the proportion test statistic as TðaÞ ¼Pi2aZi;wherearepresents the topa%of the most significant genes, andTðaÞrefers to the sum of the topa%of z-scores. With various values ofa;we obtain a class of pro-portion tests. Under the null hypothesis of no genetic effect, thez-scores of the gene set jointly follow a multivariate nor-mal distribution with mean 0 and covariance matrix PðGÞ; which can be estimated based on the simulation approach described above. To estimate the significance of each proportion test statistic, FLAGS simulates B number of multivariate normal vectors with mean 0 and covari-ance matrix PðGÞ: We calculated the null statistic TðbÞðaÞ ¼P
i2aZiðbÞ; b¼1; 2;. . .; B:The P-value for the TðaÞstatistic is
pðaÞ ¼ X
B
b¼1
h
ITðbÞðaÞ$TðaÞiþ1!.ðBþ1Þ; whereIðxÞis an indicator function.
Our empirical observation is that, depending on the pro-portion of causal genes in a gene set, some propro-portion tests may be more powerful than others. For example, if 10% of the genes in a set are causal, the test statistic summing over the top 10% of most significant genes tends to be the most powerful. Since we do not know the true proportion of causal genes in a gene set, it cannot be known in advance which proportion test statistic will be the most powerful. Nonetheless, we do know that one of these proportion tests should be more powerful than others. To adaptively aggregate information over genes and maintain high power across different association patterns, FLAGS scans a class of proportion test statistics and takes their minimumP-value as a data-adapted statistic. Formally, suppose that we have a class of values of a in G, e.g.,
G¼ f10; 20; 30; . . .; 100g;and suppose that theP-value ofTðaÞispðaÞ;then our adaptive statistic isT¼mina2GpðaÞ:
The null distribution of the adaptive statistic (minimum P-value) can be obtained from the same set of simulated z-score vectors. For each simulatedz-score vector, we calculate the corresponding test statisticTðbÞðaÞand theirP-values as
pðbÞðaÞ ¼ X
b16¼b
I
Tðb1ÞðaÞ$TðbÞðaÞ
þ1
!.
B:
Thus, we haveTðbÞ¼min
a2GpðbÞðaÞ;and theP-value of the
adaptive test statistic is
p¼ X
B
b¼1
I
TðbÞ#T
þ1!.ðBþ1Þ:
A brief review of some existing tests
We compared the power of FLAGS with that of several existing self-contained tests. Below, we briefly review a number of tests that have been widely applied to the analysis of GWAS data (SRT, PLINK-set, SKAT, and GRASS) or that have been re-cently reported (aSPUpath and MAGMA).
SRT:SRT evaluates the proportion of significant SNPs of all SNPs within genes of a gene set. It computes an empirical P-value, using a permutation-based approach. Specifically, the test statistic of SRT represents the ratio of the number of significant SNPs (P,0.05) to the total number of SNPs in a gene set. The null distribution of the test statistic is obtained by permutation. An empiricalP-value of the test statistic is the proportion of null statistics that are equal to or greater than the observed statistic.
PLINK-set: PLINK-set is a SNP set analysis implemented in PLINK and can be naturally extended for gene set analysis. For a given gene set, itfirst conducts a single SNP-level analysis. It then performs aP-value informed LD pruning to select the top independent SNPs with aP-value below a predefined thresh-old (e.g.,P,0.05). The test statistic is the mean of these single SNP statistics from the top independent SNPs. The P-value of the test statistic is estimated by permutation, which is the proportion of permuted statistics that are equal to or exceed the observed one.
SKAT: SKAT is a SNP set analysis approach that is con-ducted in a kernel-machine regression framework. Similar to PLINK-set, SKAT can also be applied to gene set analysis. It aggregates signals of variants through a kernel matrix. It is computationally efficient because it does not need permu-tation to deriveP-values. Itfirstfits the null model in which the phenotype is regressed on the covariates alone. A var-iance component score test is then applied to calculate the P-value analytically.
GRASS:GRASS investigates the association of a gene set with the phenotype, using principle components analysis (PCA) and regularized regression. It first derives principle compo-nents or “eigenSNPs”of each gene by PCA. It then uses a regularized regression technique, termed group ridge regres-sion, to select representative eigenSNPs for each gene and assess their joint association with disease risk. The test sta-tistic for a gene set is defined as Tl¼ ðb21þ. . .þb
2 GÞ
1=2
;
aSPUpath: The aSPUpath test is an adaptive test for gene sets extended from an adaptive gene-based test—the adap-tive sum of powered score test (SPU) (Pan et al. 2014, 2015). The essence of the aSPUpath test is that it adaptively aggregates signals at both the gene and the pathway level through a power-weighting scheme. At the power of one, aSPUpath treats each variant and gene equally and takes the average signals for both genes and pathway. If the power is raised to a very large number, it tends to use the maximum signal as the test statistic. By varying the value of the power, aSPUpath adaptively identifies a subset of variants and genes that are most likely to be causal. However, it needs individual-level data and permutations to deriveP-values for a gene set.
MAGMA: MAGMA is a novel tool for gene and gene set analysis of GWAS data (de Leeuwet al.2015). To perform gene set analysis, it converts the gene-basedP-value to a z-score based on the inverse normal distribution function. It then tests whether the mean ofz-scores of all genes in a gene set deviates from zero in a regression model while accounting for the correlations among genes. It does not require permu-tation to deriveP-values.
Simulations
We evaluated the performance of FLAGS, using genotype data with realistic LD patterns. Specifically, we conducted simula-tions using ARIC GWAS data from9000 samples (Athero-sclerosis Risk in Communities Investigators 1989). To reflect the complex configurations of gene sets, we simulated data
using gene sets in the MSigDB database (Subramanianet al. 2005). We performed standard quality control for the GWAS data before it was used to generate simulated data. Briefly, we excluded samples and SNPs based on predetermined quality control metrics, including sample call rate #95%, SNP call rate#95%, minor allele frequency (MAF)#0.01, and P-values of Hardy–Weinberg Equilibrium (HWE) tests
#1025. We computed principal components (PCs) for GWAS samples, using EIGENSOFT (Priceet al.2006). We removed outlier samples defined as subjects whose ancestry was at least three standard deviations from the mean of one of the two largest PCs. SNPs were mapped to genes based on NCBI 37.3 gene definitions.
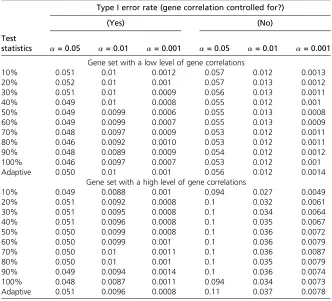
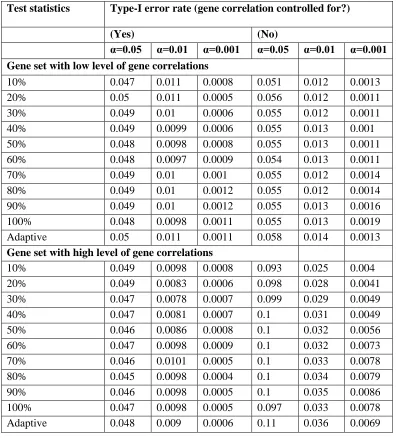
Type I error
We aimed to examine the type I error rates of FLAGS and to evaluate the extent to which the type I error would be influenced if gene correlations within a gene set were not considered. For this purpose, we generated null data sets for two pathways selected from the MSigDB database, rep-resenting gene sets with a low or a high level of gene cor-relations. Specifically, the gene set of low-level gene correlations has 19 genes and contains only one gene pair with az-score correlation$0.2. The gene set of high-level gene correlations includes 13 genes, among which eight gene pairs have a z-score correlation $0.2. To generate null data sets, 1000 random samples were drawn without replacement from the ARIC GWAS data. We randomly assigned half the samples as cases and the other half as controls. We then permuted case–control labels 10,000 times to generate 10,000 null phenotypes and hence 10,000 null data sets for each gene set. For each null data set, we ran PLINK logistic regression to get SNP-based P-values, followed by applying FLAGS to get the gene set-basedP-values with and without considering gene correla-tions. The empirical type I error rate was calculated as the proportion of 10,000P-values from the null data that were equal to or smaller than a given alevel (0.05, 0.01, and 0.001). To evaluate the effect of sample size on type I error, we repeated the simulation for a sample size of 2000 (1000 cases and 1000 controls).
Power
To compare the power of FLAGS and other existing methods, we simulated data under alternative hypotheses, using cu-rated gene sets from the MSigDB database, including KEGG, BioCarta, and Reactome. We reasoned that a simulation based on a variety of gene sets across the genome would ensure that the power estimation was not dependent on the choice of a specific set, and hence it would more realistically reflect the performance of various methods. We selected 1000 gene sets with 10–200 genes as“causal gene sets.”The phenotype was generated based on the ARIC GWAS data for each causal set. Specifically, for each gene set, we randomly selectedK num-ber of genes as causal genes, and one causal SNP (0.05, MAF ,0.25) was randomly taken from each causal gene, Figure 2 Comparison of gene correlations inz-scores estimated by an
yieldingKcausal genotypesðg1;g2;. . .;gKÞ. Then, we gener-ated a quantitative trait by a simple linear regression model,
y¼b1g1þb2g2þ. . .þbKgKþe;
wherebiði¼1;2;⋯;KÞis the additive effect for the causal variants, and e is a random term that follows a standard normal distributionNð0;1Þ:A dichotomous trait was created by taking samples with the highest ($75th percentile) and lowest (#25th percentile) quartiles of the simulated quanti-tative trait as cases and controls, respectively. The genetic effect of all causal variants was set as the same logð1:1Þ: We considered four different settings for the proportion of causal genes contained in each causal gene set (10%, 20%, 30%, or 40%). We therefore created 1000 causal data sets under each disease model. For each causal data set, FLAGS and other competing methods were run to get a gene set-basedP-value. The power was defined as the proportion of P-values from 1000 causal data sets that were smaller than a given a¼0:01 level. We considered two sample sizes of 1000 (500 cases and 500 controls) and 2000 (1000 cases and 1000 controls).
Real data analysis
To evaluate the performance of FLAGS on real data sets and compare it with existing methods, we applied FLAGS and other appropriate methods to two data sets: (1) Wellcome Trust Case Control Consortium (WTCCC) GWAS data for
Crohn’s disease (CD) (Wellcome Trust Case Control Consor-tium 2007) and (2) GWAS meta-analysis results for bipolar disorder (BD) from the Psychiatric Genomic Consortium (PGC) (Psychiatric GWAS Consortium Bipolar Disorder Work-ing Group 2011). The CD data include the raw data and therefore enabled us to compare FLAGS with four other methods that need raw data as input (aSPUpath, GRASS, SKAT, and MAGMA). We did not include STR and PLINK-set for CD, because these two methods are too computation-ally demanding. In the comparative analysis of BD, we applied only MAGMA because it is the sole method that can take summary statistics as input. We focused our analysis on autosomal SNPs. We assigned a SNP to a gene if it was lo-cated within the gene, based on NCBI 37.3 gene annotation, or within 20 kb upstream and downstream of the gene, to capture regulatory variants. To reduce multiple testing and choose gene sets that are relatively well defined, we used only gene sets from the KEGG database (Kanehisa et al. 2010). Following previous authors, to facilitate interpreta-tion of the results, we excluded gene sets that were either unusually small (,10 genes) or unusually large (.500 genes). In total, there were 64,557 and 178,873 SNPs mapped to 4572 and 4904 genes for CD and BD, respectively, which resulted in 197 gene sets for CD and 186 gene sets for BD. To reduce systematic bias and minimize the chance of false positive findings, we used genomic control-corrected SNP P-values for both disorders. For gene set analyses that require gene-based P-values, we computed these with Table 1 Empirical type I error rates for two gene sets at three nominal error rates
(a= 0.05,a= 0.01, anda= 0.001) based on 500 cases and 500 controls
Type I error rate (gene correlation controlled for?)
(Yes) (No)
Test
statistics a= 0.05 a= 0.01 a= 0.001 a= 0.05 a= 0.01 a= 0.001
Gene set with a low level of gene correlations
10% 0.051 0.01 0.0012 0.057 0.012 0.0013
20% 0.052 0.01 0.001 0.057 0.013 0.0012
30% 0.051 0.01 0.0009 0.056 0.013 0.0011
40% 0.049 0.01 0.0008 0.055 0.012 0.001
50% 0.049 0.0099 0.0006 0.055 0.013 0.0008
60% 0.049 0.0099 0.0007 0.055 0.013 0.0009
70% 0.048 0.0097 0.0009 0.053 0.012 0.0011
80% 0.046 0.0092 0.0010 0.053 0.012 0.0011
90% 0.048 0.0089 0.0009 0.054 0.012 0.0012
100% 0.046 0.0097 0.0007 0.053 0.012 0.001
Adaptive 0.050 0.01 0.001 0.056 0.012 0.0014
Gene set with a high level of gene correlations
10% 0.049 0.0088 0.001 0.094 0.027 0.0049
20% 0.051 0.0092 0.0008 0.1 0.032 0.0061
30% 0.051 0.0095 0.0008 0.1 0.034 0.0064
40% 0.051 0.0096 0.0008 0.1 0.035 0.0067
50% 0.050 0.0099 0.0008 0.1 0.036 0.0072
60% 0.050 0.0099 0.001 0.1 0.036 0.0079
70% 0.050 0.01 0.0011 0.1 0.036 0.0087
80% 0.050 0.01 0.001 0.1 0.035 0.0079
90% 0.049 0.0094 0.0014 0.1 0.036 0.0074
100% 0.048 0.0087 0.0011 0.094 0.034 0.0073
VEGAS, using a statistic summarizing the top 5% of most significant SNPs. For CD, we used genotype information from healthy controls in the CD GWAS as reference samples to compute the gene-basedP-values and estimate gene correla-tions, whereas for BP, we did so by using the unrelated CEU (Utah residents with ancestry from northern and western Europe) samples from the 1000 Genomes Project. Because the gene correlation in z-scores is close to zero when two genes were .2 Mb away (Figure S2), we calculated only correlations for gene pairs within 2 Mb.
Data availability
R code and data examples for implementing FLAGS are posted at http://www.medicine.uiowa.edu/psychiatry/ han/software.
Results
Estimation of gene correlations
two genes were .2 Mb apart (Figure S3). Based on this empirical observation, we recommend that the estimation of gene correlation is necessary only for closely located gene pairs, for example, within 2 Mb.
Type I error
We evaluated the type I error of FLAGS and to what extent the type I error would be influenced when gene correlations are not considered within a gene set. Table 1 shows the empirical type I error rates at three nominal error rates (a= 0.05,a= 0.01, anda= 0.001) for two gene sets, which represent a low and a high level of gene correlation, respectively. Overall, all FLAGS test statistics maintained an appropriate type I error rate when gene correlation was accounted for. However, the type I error rates tended to be inflated if gene correlations were ignored. The extent of inflation of type I error was de-pendent on the level of gene correlation within the gene set. For example, the type I error rates were only slightly higher than the nominal levels for the gene set in the presence of a low level of gene correlation. However, the type I error rates were considerably inflated when gene correlation was ig-nored for the gene set with a high level of gene correlation. We found a similar pattern of type I error rates based on simulations with a larger sample size (1000 cases and 1000 controls;Table S1).
Power
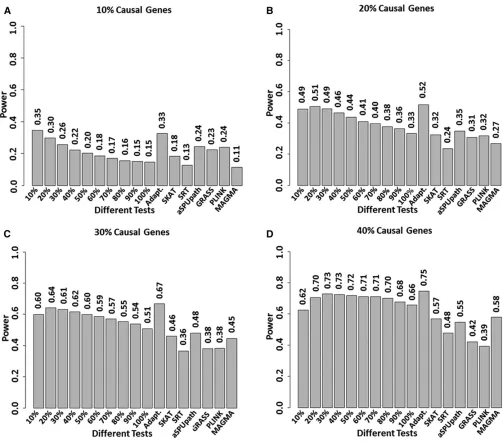
Using a number of simulated disease models, we investigated the power of FLAGS and compared its performance with that of several other self-contained tests (Figure 3). We considered four disease models with various proportions of causal genes (10%, 20%, 30%, and 40%). As expected, among all FLAGS
proportion tests, we observed the highest power for tests that combined the top significant genes with a proportion that was equal or close to the proportion of causal genes. For example, for disease models with 10% and 20% causal genes, the highest power was observed for the test that combined the top 10% and 20% of significant genes, respectively. The power of the adaptive test was close to that of the most pow-erful proportion test. It was noted that, for disease models with a low proportion of causal genes (10% or 20%), the performance of FLAGS proportion tests deteriorated dramat-ically when an increasing proportion of genes were included in the tests. On the other hand, for the disease model with a high proportion of causal genes (40%), FLAGS proportion tests did not suffer from substantial power loss when more genes were added into the tests. In comparison with other self-contained tests, the FLAGS adaptive test was clearly the winner in all simulated models. The power of the FLAGS adaptive test is10–30% higher than that of other methods across various disease models. In the scenario of a low pro-portion of causal genes (10% and 20%), SRT and MAGMA have the lowest power, possibly due to their low performance in the situation of low signal-to-noise ratio. However, aSPU-path performs slightly better than other methods, perhaps because of its adaptive nature. In the case of a high pro-portion of causal genes (40%), MAGMA performs the best among all other compared methods. Power simulations using a larger sample size (1000 cases and 1000 controls) yielded the same conclusions (Figure S4).
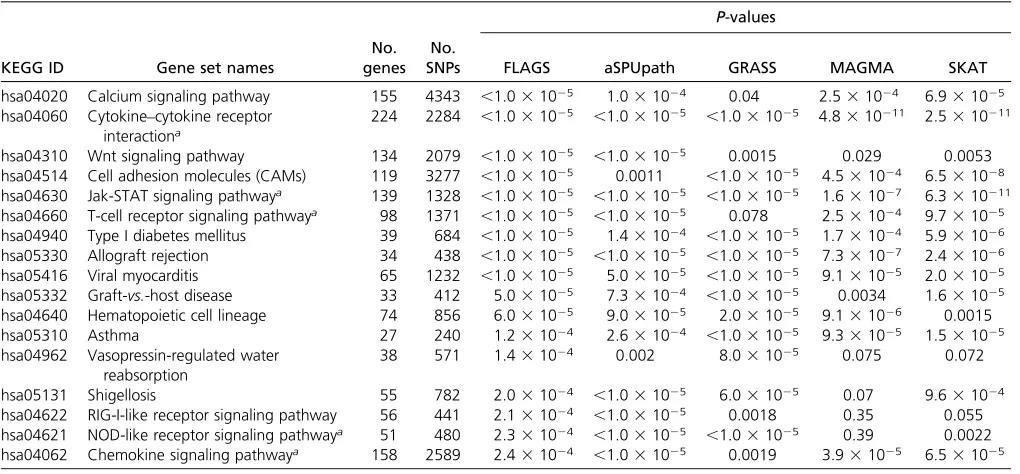
Real data
CD:We applied the FLAGS adaptive test, as well as four other methods, to the GWAS data for CD. Table 2 shows 17 KEGG Table 2 Results of the WTCCC CD GWAS data application: KEGG gene sets withP-values<2.531024by FLAGS and corresponding results
by aSPUpath, GRASS, MAGMA, and SKAT
P-values
KEGG ID Gene set names
No. genes
No.
SNPs FLAGS aSPUpath GRASS MAGMA SKAT
hsa04020 Calcium signaling pathway 155 4343 ,1.031025 1.031024 0.04 2.531024 6.931025
hsa04060 Cytokine–cytokine receptor interactiona
224 2284 ,1.031025 ,1.031025 ,1.031025 4.8310211 2.5310211
hsa04310 Wnt signaling pathway 134 2079 ,1.031025 ,1.031025 0.0015 0.029 0.0053
hsa04514 Cell adhesion molecules (CAMs) 119 3277 ,1.031025 0.0011 ,1.031025 4.531024 6.531028
hsa04630 Jak-STAT signaling pathwaya 139 1328 ,1.031025 ,1.031025 ,1.031025 1.631027 6.3310211
hsa04660 T-cell receptor signaling pathwaya 98 1371 ,1.031025 ,1.031025 0.078 2.531024 9.731025
hsa04940 Type I diabetes mellitus 39 684 ,1.031025 1.431024 ,1.031025 1.731024 5.931026
hsa05330 Allograft rejection 34 438 ,1.031025 ,1.031025 ,1.031025 7.331027 2.431026
hsa05416 Viral myocarditis 65 1232 ,1.031025 5.031025 ,1.031025 9.131025 2.031025
hsa05332 Graft-vs.-host disease 33 412 5.031025 7.331024 ,1.031025 0.0034 1.631025
hsa04640 Hematopoietic cell lineage 74 856 6.031025 9.031025 2.031025 9.131026 0.0015
hsa05310 Asthma 27 240 1.231024 2.631024 ,1.031025 9.331025 1.531025
hsa04962 Vasopressin-regulated water reabsorption
38 571 1.431024 0.002 8.031025 0.075 0.072
hsa05131 Shigellosis 55 782 2.031024 ,1.031025 6.031025 0.07 9.631024
hsa04622 RIG-I-like receptor signaling pathway 56 441 2.131024 ,1.031025 0.0018 0.35 0.055
hsa04621 NOD-like receptor signaling pathwaya 51 480 2.331024 ,1.031025 ,1.031025 0.39 0.0022
gene sets that were identified by FLAGS and remained sig-nificant after multiple-testing correction (P,2.531024). Interestingly, 5 positive control gene sets that have been
con-firmed to be associated with CD are all among the 17 gene sets. These gene sets are all related to the immune system and represent the best understood pathways underlying CD. It is noteworthy that only FLAGS and aSPUpath identified all pos-itive controls atP-values,2.531024. All methods detected the JAK-STAT signaling pathway (hsa04630) and the cyto-kine–cytokine receptor interaction pathway (hsa04060). However, GRASS did not identify the T-cell receptor signal-ing pathway (hsa04660,P= 0.078) and the chemokine sig-naling pathway (hsa04062, P = 0.0019). The NOD-like receptor signaling pathway (hsa04621) was missed by MAGMA (P= 0.39) and SKAT (P= 0.002). FLAGS also identified three additional gene sets—Graft-vs.-host dis-ease (hsa05332,P= 531025), cell adhesion molecules (hsa04514,P,131025), and vasopressin-regulated water reabsorption (hsa04962,P= 1.431024)—which were not significant by aSPUpath, but were significant by GRASS or SKAT.
BD:FLAGS and MAGMA were applied to the BD GWAS meta-analysis results. We applied only MAGMA for comparison with FLAGS because it is the sole method that can take summary statistics as input. The significance threshold isP= 2.73 1024after Bonferroni correction for the number of gene sets tested for the BD data set. To evaluate both approaches, we included one positive control gene set from Gene Ontology, histone H3-K4 methylation (term 51568). This gene set was the most strongly associated one for BD in a recent study (Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium 2015). Interestingly, FLAGS detected the positive control gene set with a P-value that remained significant after multiple-testing correction (P = 2.3 3 1025), but it showed only nominal significance by MAGMA (P= 0.003). Besides the positive control gene set, there were eight other gene sets that showedP-values,0.001 by either method (Table 3). Of note, none of these survived multiple-testing correction in the MAGMA analysis. However, FLAGS detected five additional gene sets that were significant after such correction, including type II diabetes mellitus
(P= 2.2 31025), MAPK signaling pathway (P= 1.1 3 1024), glycosphingolipid biosynthesis-globo series (P = 1.631024), GnRH signaling pathway (P= 2.231024), and gap junction (P= 2.431024).
Discussion
Motivation for the proposed FLAGS method is based on the consideration that only a fraction of genes are causal within a gene set that underlies disease susceptibility. Moreover, the proportion of risk genes may also vary across different gene sets. A robust test statistic should take into account the unique association patterns of gene sets. Accordingly, FLAGS was designed to adaptively aggregate information over the most likely causal genes while minimizing the noise of noncausal genes. FLAGS showed consistent high performance over a wide range of disease models. The robustness of FLAGS is consistent with a recent study that found the adaptive rank truncated product (ARTP) to perform best in a comparison of a number of set-based association tests (Suet al.2015). The ARTP adaptively seeks a subset of SNPs that yields the best evidence for genetic association. The ARTP method is analo-gous to FLAGS in that both methods form a model selection that aims to maximize the statistical evidence. However, the ARTP method needs raw data and permutations to derive P-values, which is not only computationally intensive, but also not possible or very difficult in many situations. In contrast, FLAGS needs only SNP-levelP-values and ancestry-matched LD information. Moreover, FLAGS does not use permutation to estimateP-values; instead it uses a simulation-based ap-proach to deriveP-values, and it is therefore much faster and more computationally efficient.
Although the FLAGS method has been developed for GWAS data, the same adaptive framework can be applied to gene set analysis of rare variants, such as those generated from whole-genome or -exome sequencing studies. However, to calculate gene correlations based on rare variants, we recommend using a standard permutation-based approach, which requires that the raw data be available. In the current application of FLAGS we used VEGAS to compute gene-basedP-values, although other gene-based tests may also be applied that can take SNP-level summary statistics as input. However, we point out that Table 3 Results of the PGC BD GWAS meta-analysis application: KEGG gene sets withP-values<0.001 by either FLAGS or MAGMA
P-values
KEGG ID Gene set names No. genes No. SNPs FLAGS MAGMA
GO51568a Histone H3-K4 methylation 13 303 2.331025 0.003
hsa04930 Type II diabetes mellitus 45 2,576 2.231025 0.1
hsa04010 MAPK signaling pathway 254 12,118 1.131024 0.12
hsa00603 Glycosphingolipid biosynthesis-globo series 13 528 1.631024 0.0062
hsa04912 GnRH signaling pathway 97 5,699 2.231024 4.931024
hsa04540 Gap junction 85 5,969 2.431024 0.024
hsa04720 Long-term potentiation 65 5,486 5.531024 0.11
hsa04722 Neurotrophin signaling pathway 120 5,084 9.631024 0.0028
hsa05010 Alzheimer’s disease 151 6,574 0.0016 6.031024
the current approach to estimating gene correlations was based on gene-level test statistics from VEGAS that use LD information from ancestry-matched reference samples. To estimate gene correlations based on other gene test statis-tics, we recommend that a permutation-based approach be employed using genotype data from reference samples. In addition, our method introduces a natural way to incorporate weights on genes that are more likely to be causal. Specifi -cally, the weights can be incorporated in the null distributions of jointz-scores of genes within a gene set. The weights may come from prior information, such as gene expression differ-ences, linkage signals, and other biological knowledge.
In the analysis of two real data sets for CD and BD, FLAGS not only detected all positive control gene sets for both diseases, but also identified novel gene sets that may underlie the pathophysiology. For example, one top hit gene set for CD is the“calcium signaling pathway.”Calcium signals are cru-cial for the proper activation of lymphocytes, regulation of cell differentiation, and effector functions (Vig and Kinet 2009). Dysregulated calcium responses have been implicated in several autoimmune and inflammatory diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis (Feske 2007). Detection of the calcium sig-naling pathway may reveal new insights into CD pathogene-sis and provide potential targets for treatment. In the test of BD meta-analysis results, the most significant pathway was identified for“type II diabetes mellitus,”which is in line with prior evidence supporting a shared genetic component be-tween diabetes and BD (Torkamaniet al.2008). The second top gene set was the “MAPK signaling pathway.” Interest-ingly, a number of studies have shown that the two most commonly used antimanic agents, lithium and valproate, ac-tivate the MAPK signaling cascade, which may in part be re-sponsible for their therapeutic effects (Boeckeleret al.2006). The third-ranked gene set “glycosphingolipid biosynthesis-globo series” produces glycosphingolipids (GSLs) that are especially abundant in the nervous system. Mounting evi-dence supports regulatory roles of GSLs in neurogenesis and signal transduction in the developing human brain (Furukawaet al.2014). FLAGS also identified another sig-nificant gene set,“gap junction,”which is consistent with its important roles in the nervous system.
In large-scale data analysis, not only statistical power but also computational demand is a major consideration. We compared the running time offive different methods applied to CD data based on a 16-core Xeon Phi (2.6 GHz) with 256 GB of RAM (Table S2). In the analysis of 197 KEGG gene sets using only a single core, it took 5 and 57 min for MAGMA and SKAT, respectively. The running times for FLAGS and aSPU-path were 42 and 68 hr, respectively. GRASS is the most computationally demanding, as it can take.10 days. How-ever, in our real practice where we have submitted jobs in parallel, the computational time was greatly reduced for FLAGS, aSPUpath, and GRASS. For example, it took11 hr for FLAGS, 13 hr for aSPUpath, and 5 days for GRASS, through running 80 parallel jobs on the Linux clusters.
In summary, we have developed aflexible and powerful adaptive test for gene sets, using summary statistics. Although our method was simulated and applied based on binary traits, it can be utilized for other types of traits, such as quantitative and ordinal ones. Given the fact that GWAS summary results are increasingly publicly available, and given the difficulties that can arise in accessing raw data, our method will allow for the broader application of gene set analysis to GWAS of complex diseases.
Acknowledgments
We are grateful for the insightful comments and sug-gestions by the reviewers. This study was supported by National Institutes of Health grant R01AA022994. This study was also supported by the National Alliance for Research in Schizophrenia and Affective Disorders Young Investigator Award (to S.H.). This study made use of data generated by the Wellcome Trust Case Control Consortium (WTCCC). A full list of the investigators who contributed to the gener-ation of the WTCCC data is available from http://www. wtccc.org.uk. Funding for the WTCCC project was provided by the Wellcome Trust under award 076113.
Literature Cited
Atherosclerosis Risk in Communities Investigators, 1989 The Ath-erosclerosis Risk in Communities (ARIC) Study: design and ob-jectives. Am. J. Epidemiol. 129: 687–702.
Atias, N., S. Istrail, and R. Sharan, 2013 Pathway-based analysis of genomic variation data. Curr. Opin. Genet. Dev. 23: 622–626. Boeckeler, K., K. Adley, X. Xu, A. Jenkins, T. Jinet al., 2006 The neuroprotective agent, valproic acid, regulates the mitogen-ac-tivated protein kinase pathway through modulation of protein kinase A signalling in Dictyostelium discoideum. Eur. J. Cell Biol. 85: 1047–1057.
Chen, D., and U. Gyllensten, 2014 Lessons and implications from association studies and post-GWAS analyses of cervical cancer. Trends Genet. 31: 41–54.
Chen, L. S., C. M. Hutter, J. D. Potter, Y. Liu, R. L. Prenticeet al., 2010 Insights into colon cancer etiology via a regularized ap-proach to gene set analysis of GWAS data. Am. J. Hum. Genet. 86: 860–871.
de Leeuw, C. A., J. M. Mooij, T. Heskes, and D. Posthuma,
2015 MAGMA: generalized gene-set analysis of GWAS data.
PLoS Comput. Biol. 11: e1004219.
Feske, S., 2007 Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7: 690–702.
Furukawa, K., Y. Ohmi, Y. Ohkawa, O. Tajima, and K. Furukawa, 2014 Glycosphingolipids in the regulation of the nervous sys-tem, pp. 307–320 inGlycobiology of the Nervous System, edited by R. K. Yu and C.-L. Schengrund. Springer-Verlag, New York. Goeman, J. J., and P. Bühlmann, 2007 Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23: 980–987.
Holmans, P., E. K. Green, J. S. Pahwa, M. A. R. Ferreira, S. M. Purcellet al., 2009 Gene Ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 85: 13–24.
Kanehisa, M., S. Goto, M. Furumichi, M. Tanabe, and M. Hirakawa,
networks involving diseases and drugs. Nucleic Acids Res. 38: D355–D360.
Liu, J. Z., A. F. McRae, D. R. Nyholt, S. E. Medland, N. R. Wray
et al., 2010 A versatile gene-based test for genome-wide asso-ciation studies. Am. J. Hum. Genet. 87: 139–145.
Manolio, T. A., F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorffet al., 2009 Finding the missing heritability of com-plex diseases. Nature 461: 747–753.
McCarthy, M. I., G. R. Abecasis, L. R. Cardon, D. B. Goldstein, J. Little et al., 2008 Genome-wide association studies for com-plex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9: 356–369.
Mooney, M. A., J. T. Nigg, S. K. McWeeney, and B. Wilmot, 2014 Functional and genomic context in pathway analysis of GWAS data. Trends Genet. 30: 390–400.
Network and Pathway Analysis Subgroup of the Psychiatric Geno-mics Consortium, 2015 Psychiatric genome-wide association study analyses implicate neuronal, immune and histone path-ways. Nat. Neurosci.18:199–209.
O’Dushlaine, C., E. Kenny, E. A. Heron, R. Segurado, M. Gillet al.,
2009 The SNP ratio test: pathway analysis of genome-wide
association datasets. Bioinformatics 25: 2762–2763.
Pan, W., J. Kim, Y. Zhang, X. Shen, and P. Wei, 2014 A powerful and adaptive association test for rare variants. Genetics 197: 1081–1095.
Pan, W., I. Y. Kwak, and P. Wei, 2015 A powerful pathway-based adaptive test for genetic association with common or rare vari-ants. Am. J. Hum. Genet. 97: 86–98.
Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadicket al., 2006 Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38: 904–909.
Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011 Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43: 977–983.
Purcell, S., B. Neale, K. Todd-Brown, L. Thomas, M. A. R. Ferreira
et al., 2007 PLINK: a tool set for whole-genome association
and population-based linkage analyses. Am. J. Hum. Genet. 81: 559–575.
Su, Y. C., W. J. Gauderman, K. Berhane, and J. P. Lewinger,
2015 Adaptive set-based methods for association testing.
Genet. Epidemiol. 2015 Dec 28. DOI: 10.1002/gepi.21950. [Epub ahead of print].
Subramanian, A., P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert et al., 2005 Gene set enrichment analysis: a knowl-edge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102: 15545–15550. Torkamani, A., E. J. Topol, and N. J. Schork, 2008 Pathway
anal-ysis of seven common diseases assessed by genome-wide asso-ciation. Genomics 92: 265–272.
Vig, M., and J.-P. Kinet, 2009 Calcium signaling in immune cells. Nat. Immunol. 10: 21–27.
Wang, K., M. Li, and M. Bucan, 2007 Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 81: 1278–1283.
Wang, K., M. Li, and H. Hakonarson, 2010 Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 11: 843–854.
Wang, K., H. Zhang, S. Kugathasan, V. Annese, J. P. Bradfieldet al., 2009 Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn disease. Am. J. Hum. Genet. 84: 399–405.
Wang, L., P. Jia, R. D. Wolfinger, X. Chen, and Z. Zhao,
2011 Gene set analysis of genome-wide association studies: methodological issues and perspectives. Genomics 98: 1–8.
Wellcome Trust Case Control Consortium, 2007 Genome-wide
association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678.
Wu, M. C., P. Kraft, M. P. Epstein, D. M. Taylor, S. J. Chanock
et al., 2010 Powerful SNP-set analysis for case-control ge-nome-wide association studies. Am. J. Hum. Genet. 86: 929– 942.
GENETICS
Supporting Information www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.185009/-/DC1
FLAGS: A Flexible and Adaptive Association Test for
Gene Sets Using Summary Statistics
Jianfei Huang, Kai Wang, Peng Wei, Xiangtao Liu, Xiaoming Liu, Kai Tan, Eric Boerwinkle, James B. Potash, and Shizhong Han