MITOTIC REGULATION OF NUCLEAR ASSEMBLY, POSITIONING AND FUNCTION
Vincent Boudreau
A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department
of Biology in the College of Arts and Sciences.
Chapel Hill 2019
ABSTRACT
Vincent Boudreau: MITOTIC REGULATION OF NUCLEAR ASSEMBLY, POSITIONING AND FUNCTION
(Under the direction of Paul S. Maddox)
This chapter of my life is dedicated to the women who characterized it, punctuated it, supported it and made it the wonderful, sad and euphoric experience it was.
Elena Fanucchi, my mother Sofia Boudreau, my daughter
ACKNOWLEDGEMENTS
My time at UNC has made me feel part of a large scientific family and I have a lot to be thankful for. It can be challenging to fully thank a parent for all the intangibles they provide, especially through the tough love and constant challenge they supply, pushing you to better yourself. That best describes my relationship with Paul Maddox, who I am thankful to call a mentor, an advisor, a friend and a scientific father. Similarly, Amy Maddox represents a source of inspiration and positivity through science, family and my move to UNC. I am forever grateful to them both for providing me with the guidance, the environment, the scientific family and the lab roof above our heads.
To the faculty at UNC including, but not limited to, Kerry Bloom, Bob Goldstein, Amy Gladfelter, Ted Salmon, Steve Rogers, Scott Williams and Mark Peifer – thank you for making the community and my time at UNC so special.
To my thesis committee – thank you for keeping me focused and for all your support. To the UNC club hockey team – thank you for the fun, the pain, the hockey, the broken buses, the ACC championship, the ACC all-star tournament, the friendships, the fun, and the fun again.
To the Superfriends – you know what you did and I shall no repeat it. Another thank you to Bob Goldstein for the naming our collective.
To the past, present and future organizers of the Triangle Cytoskeleton Meeting – thank you for the incredible memories and the work in putting together an unbelievable event for the community that we should be proud of.
To the Marine Biological Laboratory and all involved in the Physiology course – thank you for providing a research environment that has not only informed the rest of my scientific career, but has provided me with an experience that has changed my life in several ways, several times over and that has allowed me to meet inspiring, lifelong colleagues.
To Carlos and Laura – thank you for sharing a home, being part of this communal life we made for ourselves, making dinner and sharing your wine, and simply being the amazing friends and humans that you are. I love you guys.
To my family – thank you for the support and for putting up with my inability to really convey what this experience and this career is all about.
And lastly,
PREFACE
Having the supportive dissertation mentor I had allowed me to explore the diversity in fields, demographics, backgrounds and thoughts that percolate cell biology today. To the young budding scientist, it is tempting to believe that science is the exclusively objective, somewhat cold, quantification of the world around us. In this is implied an optimal or unique approach to doing science. Undoubtedly, if this were the case the exploration of new mediums, new fields and new approaches would have run dry. It is so common to hear discoveries originate from intuition, guesses, something someone once said or flat out mistakes, it becomes difficult to suggest that we must be doing any given thing for any given reason. Diversity in persons and in thought is at the core of creative scientific work.
conventional thoughts surround the makings of a researcher, whether in dexterity, playfulness, a certain brilliance or some mechanical aptitude. It is truly difficult to ascertain if any one trait ensures a successful scientific career as the individuals in science are self-selected and most traits can be taught and learned. In the spirit of true diversity, the stories of the many must undo the stereotypical scientist and embrace as many who wish to play in nature’s sandbox.
The outcome of the work detailed in this thesis is very much the consequence of a diversity of people, of disciplines and of mediums. My work on nuclear expansion is the result of efforts to bring people from different fields together, and its outcome would have been drastically different without their contribution. The people I have met along my young scientific journey have directly informed what I pursued and how I chose to purse it. To assume choosing what one shall pursue is a solely objective process is to ignore the community in which the work has been done and the contributions of the many people having thought about those same questions before us.
A personal source of inspiration has always been the work of turn of the century artist-scientists including Walther Flemming and Santiago Ramón y Cajal. Their respective contributions to chromosome biology and neuroscience were marked by the stark contrast between the development of new technologies in tissue staining and the lack of cameras to record their observations. Their detailed illustrations leaves one wondering not only how technology has unlocked scientific progress over the past century, but how the context in which their work was done fostered creativity, craftsmanship and observational skill.
From the beauty of watching biology unfold, to learning to just observe, to assimilating the breadth of the personalities doing scientific research, the following quote captures these sentiments while describing much of how contemporary science is done today:
TABLE OF CONTENTS
LIST OF FIGURES ... xii
LIST OF ABBREVIATIONS ... xiv
CHAPTER 1: INTRODUCTION ... 1
1.1 Thesis overview and research philosophy ... 1
1.2 Cell mechanics, forces and sizes in development and disease... 1
1.3 Mitotic mechanotransduction ... 2
1.4 Biochemical cell cycle regulation ... 4
1.5 Mitotic exit regulation of nuclear assembly ... 7
1.6 Mitotic entry regulation of nucleo-cytoskeleton interactions ... 8
CHAPTER 2: BIOCHEMICAL REGULATORS, THE NUCLEAR LAMINA AND NUCLEO-CYTOPLASMIC TRAFFICKING COMPONENTS COLLABORATIVELY REGULATE CELL CYCLE PROGRESSION ... 11
2.1 Introduction ... 11
2.2 Methods ... 12
2.3 Results ... 13
2.4 Discussion ... 19
CHAPTER 3: NUCLEO-CYTOPLASMIC TRAFFICKING REGULATES NUCLEAR SURFACE AREA DURING MITOTIC EXIT AND NUCLEAR ASSEMBLY ... 21
3.1 Introduction ... 21
3.2 Methods ... 23
3.2.2 Mathematical model ... 24
3.2.3 Mammalian tissue culture cell maintenance, drug treatments, immunostaining and microscopy ... 24
3.2.4 C. elegans use, RNAi and microscopy ... 25
3.2.5 Drosophila husbandry and microscopy ... 25
3.2.6 Image analysis ... 26
3.3 Results ... 26
3.3.1 Neither nuclear surface area nor nuclear volume scale with cell size in multinucleate cytoplasm droplets ... 27
3.3.2 Modelling nuclear organogenesis captures the roles of nuclear volumetric and surface area factors in regulating nuclear expansion ... 28
3.3.3 Nuclear expansion is regulated by nucleo-cytoplasmic trafficking of nuclear volume and surface area factors ... 31
3.3.4 Surface area factor concentrations are regulated by nucleo-cytoplasmic trafficking ... 33
3.3.5 Disrupting nucleo-cytoplasmic trafficking rates in Drosophila embryos affects nuclear function following nuclear organogenesis ... 35
3.4 Discussion ... 38
3.5 Supplemental material ... 41
CHAPTER 4: MICROTUBULE-BASED PULLING FORCES AND CENTROSOME-NUCLEAR ENVELOPE TETHERING COORDINATE CENTROSOME SEPARATION DURING MITOTIC ENTRY ... 46
4.1 Introduction ... 46
4.2 Methods ... 48
4.2.1 Computational simulations... 48
4.2.2 C. elegans use, RNAi and microscopy ... 48
4.2.3 Image analysis ... 49
4.3.1 PP2A-B55/SUR-6 and LMN-1 play critical roles in centrosome separation
in the C. elegans zygote ... 49
4.3.2 Nuclear envelope-based Dynein motor activity is regulated by PP2A-B55/SUR-6 ... 51
4.3.3 PP2A-B55/SUR-6 regulates nuclear envelope-based Dynein density ... 54
4.3.4 PP2A-B55/SUR-6 regulates cortical Dynein motoring activity... 57
4.3.5 Computational simulations of centrosome migration partially recapitulate PP2A-B55/SUR-6’s role in regulating centrosome separation through pronuclear size and nuclear envelope Dynein density ... 59
4.3.6 PP2A-B55/SUR-6 collaborates with LMN-1 for proper centrosome separation ... 62
4.4 Discussion ... 64
4.5 Supplemental material ... 67
CHAPTER 5: DISCUSSION ... 73
5.1 Nuclear assembly during mitotic exit ... 73
5.2 Nuclear assembly following fertilization... 75
5.3 Centrosome pulling forces ... 77
LIST OF FIGURES
Figure 1.1. Biochemical phosphoregulation of mitotic entry and mitotic exit ... 5 Figure 1.2. NCT regulation of cytoplasm-nucleoplasm directionality ... 7 Figure 2.1. A second-site non-complementation screen identifies several nucleocytoplasmic
machinery components that collaborate with PP2A-B55/Tws for cell cycle progression ... 14
Figure 2.2. PP2A-B55/Tws collaborates with distinct genetic networks in regulating cell cycle
progression ... 16
Figure 2.3. CycB and Emb antagonize pp2a/cas and pp2a/lamB genetic interactions ... 18 Figure 3.1. Models of nuclear size scaling with cell size and nuclear surface area to volume ratios
within a defined cell size ... 26
Figure 3.2. in vitro encapsulation of nuclei in cytoplasmic droplets reveals nuclear surface area to volume regulation during nuclear expansion ... 27
Figure 3.3. A mechano-chemical mathematical model dependent on dynein-based transport supports an inverse scaling relationship between nuclear diameter and nuclear surface area ... 30
Figure 3.4. Nucleo-cytoplasmic trafficking regulates nuclear expansion ... 32 Figure 3.5. Inhibiting nuclear export during nuclear expansion reduces the nuclear concentration of
LMNB1, a proposed nuclear surface area factor ... 34
Figure 3.6. Depleting core nucleo-cytoplasmic trafficking components via RNAi affects the timing and rate of transcription activation in vivo... 37
Figure 3.S1. Nucleo-cytoplasmic trafficking inhibition at anaphase onset by IPZ and LMB ... 41 Figure 3.S2. Treating cells with IPZ or LMB at anaphase onset does not affect global mitotic exit
timing ... 43
Figure 3.S3. Targeting IMB-1 and XPO-1 via RNAi in the C. elegans embryo compromises nuclear expansion similarly to our mathematical model’s prediction... 44
Figure 3.S4. Depletion of Emb and Nup358 in the D. melanogaster embryo via RNAi leads to
embryonic lethality ... 45
Figure 4.1. sur-6 and lmn-1 RNAi affect centrosome separation through distinct mechanisms ... 50 Figure 4.2. PP2A-B55/SUR-6 regulates Dynein-dependent microtubule growth velocity acceleration
Figure 4.3. Quantification of endogenous DHC-1:mNeonGreen reveals nuclear envelope density regulation by PP2A-B55/SUR-6 ... 56
Figure 4.4. PP2A-B55/SUR-6 regulates Dynein-dependent microtubule growth velocity acceleration at the cortex ... 58
Figure 4.5. Cytosim-based computational simulations predict differences in pronuclear size and nuclear envelope-tethered Dynein density affect centrosome separation, while initial inner centrosome separation is dominant ... 61
Figure 4.6. Co-depleting sur-6 and lmn-1 or lis-1 and zyg-12 leads to centrosome attachment and centrosome separation defects ... 63
Figure 4.S1. Partial dhc-1/lis-1 and zyg-12/sun-1 RNAi affect centrosome separation through
separate mechanisms ... 67
Figure 4.S2. Cytoplasmic microtubule growth velocities prior to pronuclear migration ... 68 Figure 4.S3. Quantification of endogenous DHC-1:mNeonGreen mean amounts on female and
male pronuclear envelopes ... 70
Figure 4.S4. Pronuclear cross-sectional area measurements during pronuclear expansion ... 71 Figure 4.S5. Cytosim-based computational simulations predict 24 hour sur-6 RNAi effects on
LIST OF ABBREVIATIONS
APC-CDC20 Anaphase-promoting complex ATP Adenosine triphosphate C. elegans Caenorhabditis elegans
CDK1 Cyclin-dependent kinase 1 D. melanogaster Drosophila melanogaster
ECM Extracellular matrix GAP GTPase-activating protein
GEF Guanine nucleotide exchange factor
Gwl Greatwall
Impb Importin beta
Mts Microtubule star
NCT Nucleo-cytoplasmic trafficking NLS Nuclear localization signal NERF Nuclear envelope reformation PP2A Protein phosphatase 2A
Tws Twins
X. laevis Xenopus laevis
XPO1 Exportin-1
CHAPTER 1 INTRODUCTION
1.1Thesis overview and research philosophy
1.2Cell mechanics, forces and sizes in development and disease
Cell biology is executed at various size, length and contextual scales. The mitotic cell is in no way absolved from interpreting and responding to multiple mechanical cues. Mounting evidence points to an intimate functional relationship between cell mechanics and cell cycle progression that is undoubtedly essential to developmental and disease contexts (reviewed in Edens et al., 2013). How the mitotic apparatus integrates mechanical cues through cell cycle signaling pathways to ensure mitotic fidelity has emerged as a central question to developing wholistic models of cell cycle progression.
One critical question in cell mechanics is size regulation. For example, developing animals succeed in adjusting nuclear size as cells become smaller through multiple rounds of mitosis. Nuclear size scaling is achieved in part by regulating nuclear lamina stiffness (Levy et al., 2010). Beyond regulation of the molecular composition of the nucleus in size regulation, the nucleus and the cell must both be responsive to contextual cues including extracellular matrix (ECM) stiffness, osmolarity, pressure and temperature, to name a few. In a series of elegant experiments, Guo et al. (2017) demonstrate a relationship between ECM stiffness, osmolarity and cell volume. As ECM stiffness increases, adherent cells spread further thereby increasing their surface area while decreasing their volume. In line with previous reports, nuclear size scales with cell size in this context and hence with extracellular cues such as ECM stiffness. Critically, cell and nuclear volume as influenced either by ECM stiffness or osmolarity is sufficient to drive murine bone marrow (mMSC) stem cell fate. This work suggests that molecular crowding in the cytoplasm, nucleoplasm or both could play a critical role in mitotic function and illustrates the need to understand the mechanical properties being interpreted by the mitotic apparatus.
1.3Mitotic mechanotransduction
Currently, several aspects of cell mechanics have been found to be linked to mitosis. Cell size, for example, has been found to scale not only with nuclear size, but is also sufficient to scale chromosome size (Ladouceur et al., 2015) and spindle size (Hazel et al., 2013 and Good et al., 2013). Scaling mitotic components to cytoplasmic volume implies a limiting component model by which the amount and concentration of as of yet unidentified components directly influence mitotic function. This finding is also in line with the idea that molecular crowding as determined by several extrinsic factors plays critical roles in mitosis.
As introduced previously, ECM stiffness has a direct relationship with cell and nuclear size and is sufficient to drive stem cell fate. How external cues such as ECM stiffness are translated to mitotic progression remains an open question. Mechanistically, cells respond to biophysical cues such as ECM stiffness through cell adhesion regulation. For example, use of traction force microscopy has shown that cells exert a wide range of pulling forces on their substrates through the cell cycle, peaking during DNA replication (Vianay et al., 2018). This data suggests a relationship between the regulation of cell adhesion, ECM composition and cell cycle-based signaling. In fact, Jones et al. (2018) have recently shown that large cell adhesion areas in G1-S are promoted by the master kinase cyclin-dependent kinase 1 (CDK1) in conjunction with cyclin A2. For cells to enter mitosis and disassemble adhesions, CDK1-cyclin A2 is inactivated by inhibitory phosphorylation, allowing the mitotic CDK1-cyclin B1 complex to remodel the actin cytoskeleton and promote cell rounding. This work provides critical insight into the relationship between cell shape/size and cell cycle progression and how cell cycle signaling is coupled to mechanical cues.
Finally, inextricably linked to cell size, cell mechanics and cell cycle progression, metabolic requirements represent another underexplored area likely to be critical to our understanding of mitotic mechanisms. Recently, Rodenfels at al. (2019) repurposed an isothermal calorimeter to measure heatflow between developing embryos and their surrounding media. Critically, embryonic cell cycle progression was found to be responsible for heat flow oscillations. These heat flow oscillations are attributed to cell cycle-dependent phosphorylation and dephosphorylation events at mitotic entry and mitotic exit, respectively. These heat oscillations represent immense energetic costs to the cell to the tune of ~ 400 nM ATP/s compared, for example, to the estimated cost of DNA replication of ~ 1 nM ATP/s. How energetic burdens are budgeted during mitosis remains an open question that must be intrinsically linked to cell cycle signaling similarly to cell mechanics.
In exploring the relationship between molecular and physical mitotic processes, this thesis provides insight into two novel mechanisms. First, pathways critical to mitotic progression were identified using a genetic screen as described in section 1.4 and chapter 2. Second, the biophysical role of an uncovered pathway in regulating nuclear assembly during mitotic exit is introduced in section 1.5 and described in chapter 3. Lastly, the relationship between mitotic entry signaling and the microtubule cytoskeleton during centrosome separation is described in section 1.6 and chapter 4 of this thesis.
1.4Biochemical cell cycle regulation
CDC14, was found to be insufficient in higher eukaryotes (Berdougo et al., 2008 and Mocciaro and Schiebel, 2010). As CDK1 phosphorylates substrates critical for a plethora of mitotic events including chromosome condensation (Abe et al., 2011) and nuclear envelope breakdown (Hinchcliffe et al., 1999), PP2A has been thought to be equally important in dephosphorylating CDK1 substrates in order for cells to exit mitosis. However, despite our understanding of biochemical cell cycle regulation, the identity of PP2A substrates have largely remained elusive.
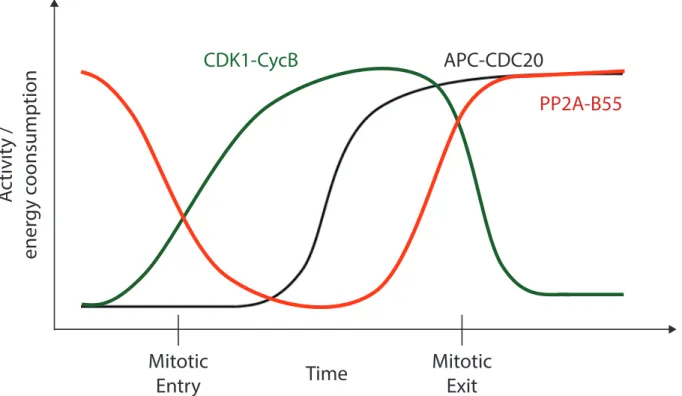
Figure 1.1 | Biochemical phosphoregulation of mitotic entry and mitotic exit. Mitotic entry corresponds to an energy intensive process driven by CDK1-CycB kinase activity. As cells exit mitosis, CDK1-CycB is inactivated through the directed degradation of CycB. CycB becomes ubiquitinated by the mitotic E3 ligase APC-CDC20 and is degraded by the proteasome at the metaphase-anaphase transition. During mitotic exit (anaphase onwards) PP2A-B55 is the main protein phosphatase responsible for CDK1 substrate dephosphorylation in metazoans.
To shed light on potential PP2A-related pathways and substrates, we used an unbiased genetic screen. Taking advantage of the genetic prowess of D. melanogaster, over 70% of the fly genome was screened using a second-site non-complementation screen to identify PP2A-specific genetic collaborators (Mehsen et al., 2018). PP2A functions as a heterotrimeric holoenzyme composed of a scaffolding subunit, a catalytic subunit (Microtubule star or Mts in flies) and a regulatory subunit conferring substrate recognition specificity to the phosphatase. During mitotic
A
ctivit
y /
ener
gy c
oonsumption
Mitotic
Entry
Mitotic
Exit
CDK1-CycB
APC-CDC20
PP2A-B55
exit, the B55 regulatory subunit is responsible for directing the phosphatase towards CDK1 substrates. The mammalian B55 subunit has homologs both in flies (Twins or Tws) and worms (SUR-6). To identify PP2A-B55 genetic collaborators, we screened both tws and mts hypomorphic alleles against a library of deficiencies in a trans-heterozygous fashion. This approach and its product are further discussed in Chapter 2 of this thesis.
One key finding from this work was the identification of several components of the nucleo-cytoplasmic trafficking machinery that function either as PP2A-B55 protagonists or antagonists. This finding is further supported by (Schmitz et al., 2010) who identified a functional interaction between a key nuclear import factor (importin beta or Impb) and PP2A-B55 in regulating chromosome decondensation and nuclear envelope reformation (NERF). Another product of our approach was the identification of a genetic interaction between PP2A-B55 and the nuclear lamina. This genetic interaction became the focus of work published in Mehsen et al. (2018), and raised important questions regarding the regulation of nuclear assembly during mitotic exit and the regulatory role the nuclear envelope plays in assembling a functional nucleus of the appropriate biophysical composition.
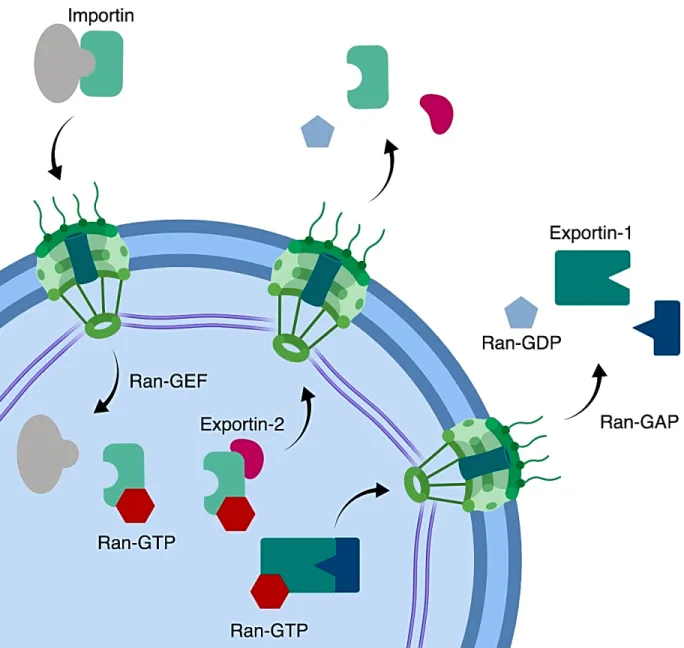
Figure 1.2 | NCT regulation of cytoplasm-nucleoplasm directionality. In ensuring cytoplasm-nucleoplasm directionality, the Ran GTPase functions with a nuclear GEF to promote nuclear GTP-bound Ran while Ran GAPs promote GTP hydrolysis in the cytoplasm. Ran association with GTP in the nucleus is required for import cargo dissociation, export cargo binding and importin recognition by Exportin-2 in promoting importin recycling to the cytoplasm. Similarly, GTP hydrolysis in the cytoplasm is required for export cargo dissociation and importin binding to import cargo. The nuclear lamina (purple) forms a network of intermediate filaments required for nuclear mechanics.
1.5Mitotic exit regulation of nuclear assembly
and Heald, 2010). The implication of the nuclear lamina in nuclear size scaling implies a potential role for the nuclear lamina in regulating nuclear size and nuclear assembly.
Nuclei must be assembled into functional organelles in at least two biological contexts: assembly of the sperm nucleus following fertilization in sexually reproducing animals and assembly of daughter nuclei during mitotic exit. How nuclei are assembled into functional organelles remains a poorly understood question in cell biology. During nuclear assembly, chromatin must be decondensed, the nuclear envelope must be re-assembled and directionality between the nucleoplasm and the cytoplasm must be re-established. How these events are coupled in space and time remains unknown. The product of these molecular pathways is a highly dynamic organelle whose structural integrity is critical in maintaining a barrier between the nucleoplasm and cytoplasm and in regulating genome organization and gene expression (reviewed in Van de Vosse et al., 2011). In fact, in addition to behaving as a viscoelastic material, the shape and size of the nucleus have been shown to be directly influenced by external cues such as the extracellular environment (Kim et al., 2016 and Guo et al., 2017).
As introduced previously, NCT has also been reported to regulate nuclear size in interphase nuclei (Matsuyama et al., 2006, Neumann and Nurse, 2007 and Levy and Heald, 2010). In the case of nuclear size scaling, nuclear import regulation of the nuclear lamina component lamin B3 allows for scaling in the developing Xenopus embryo. Conversely, nuclear export inhibition in fission yeast has been shown to lead to larger nuclei. Taken together, nuclear size is likely set by the combination of bulk protein localization in addition to nuclear size factors such as lamin B3 in Xenopus.
During nuclear assembly, similar nuclear size mechanisms are likely to be at work in allowing chromatin to expand to a given size. In chapter three of this thesis, we use a combination of in vitro cytoplasm encapsulation, mathematical modeling and in vivo approaches to determining the critical factors in nuclear assembly and propose a novel function for nuclear export in regulating nuclear expansion through the regulation of nuclear surface tension.
1.6Mitotic entry regulation of nucleo-cytoskeleton interactions
elegans embryo as a cell biological model conducive to the visualization of the earliest mitotic events, we examined the role of the genetic interaction between PP2A-B55 and the nuclear lamina in regulating mitotic progression. Following fertilization in the embryo, the paternal pronucleus must be assembled from sperm chromatin. Initially condensed, sperm chromatin expands to form a functional nucleus capable of NCT, while centrosomes, which are provided through fertilization and tethered to the paternal nuclear envelope, must separate in order to form a mitotic spindle (Hyman and White, 1987). Compromising PP2A-B55 or nuclear lamina function in the embryo led to distinct yet complimentary centrosome separation phenotypes. These experiments, discussed further in chapter four of this thesis, raised several questions including how PP2A-B55 could regulate centrosome separation and whether PP2A-B55-regulated centrosome separation and nuclear lamina-driven centrosome-nuclear envelope tethering collaborate in positioning centrosomes correctly prior to mitosis.
As microtubule organizing centers, centrosomes are subject to force being exerted on microtubules. In the embryo, the principal source of microtubule force generation is produced by the molecular motor Dynein. As a microtubule minus-end directed motor, Dynein exerts force on centrosomes and on the nucleus through two principal mechanisms: cortex-associated Dynein and nuclear envelope-associated Dynein. Tethered to a given organelle, Dynein exerts force on the microtubule cytoskeleton by walking towards the minus end of the microtubule, effectively generating a pulling force towards the plus end.
centrosome separation and downstream spindle assembly remains an open question. At the nuclear envelope, Malone et al., (2003) identified the requirement for the hook protein ZYG-12 in tethering Dynein. Given ZYG-12 dependent Dynein localization to the nuclear envelope, probing nuclear envelope Dynein function has relied on the depletion of its anchor. Interestingly, zyg-12 depletions led to centrosome detachment from the male pronucleus and excess separation in a cortical Dynein-dependent manner (De Simone et al., 2016). Whether these phenotypes represent a loss of Dynein function at the nuclear envelope or a loss of centrosome-nuclear envelope cohesion remains a confounding issue.
CHAPTER 2
BIOCHEMICAL REGULATORS, THE NUCLEAR LAMINA AND NUCLEO-CYTOPLASMIC TRAFFICKING COMPONENTS COLLABORATIVELY REGULATE
CELL CYCLE PROGRESSION1
2.1 Introduction
Mitotic fidelity is ensured by the strict regulation of dynamic, morphological changes affecting every cellular component from chromatin to the cell cortex. A distinctive feature of most metazoan mitoses is the disassembly of the nuclear envelope – the defining feature of open mitoses. This cellular barrier is crucial for the spatio-temporal regulation of several cellular processes including mitosis (Gavet and Pines, 2010; Wang et al., 2014). As chromosomes condense and kinetochores are built at the onset of mitosis, the nuclear envelope begins disassembling and the nucleoplasm is dissolved in the cytoplasm. Following the metaphase-anaphase transition and as chromosomes are segregated, chromosomes must decondense, the nuclear envelope must reform (NERF) and the newly formed nucleus must re-establish its capacity to act as a cellular compartment. Although mitotic entry progression has been extensively studied, the timing and regulation of its processes is poorly understood.
Biochemically, the entry into mitosis is characterized by high levels of kinase activity driven mainly by Cyclin-Dependent Kinase 1 (CDK1) in complex with Cyclin B (CycB) (Santamaria et al., 2006). Proper entry into mitosis requires the spatiotemporal regulation of CycB and other mitotic regulators prior to nuclear envelope breakdown (NEBD) (Gavet and Pines, 2010; Wang et al., 2011). Protein Phosphatase 2 A (PP2A), bound to its regulatory B55 subunits (Twins, or Tws, in flies) was identified as the main phosphatase responsible for CDK1-CycB substrate dephosphorylation during mitotic exit, which coincides with the kinase’s inactivation (Mochida et
1This chapter previously appeared as part of a peer reviewed article in the Journal of Cell Biology.
al., 2009). In addition to PP2A-B55/Tws, PP2A-B56 and PP1 phosphatases also play key roles in mitotic exit processes (Bastos et al., 2014; Chen et al., 2007; Vagnarelli et al., 2011). How these phosphatases collaborate to ensure mitotic exit progression is not understood.
More recently, a biochemical anaphase midzone gradient was identified as a key regulatory component of the anaphase-telophase transition (ATT) (Afonso et al., 2014). The chromosomal passenger complex (CPC) plays several signalling roles in mitotic exit from regulating kinetochore-microtubule attachments to furrow ingression in cytokinesis (Kitagawa et al., 2013; Wang et al., 2011). The transition of the CPC from the centromere to the spindle midzone in anaphase has been shown to provide a phosphorylation gradient that spatially restricts chromosome decondensation and NERF (Afonso et al., 2014). Aurora B, the CPC’s catalytic unit, provides a spatial framework for decondensation and NERF, CDK1 and PP1/PP2A seem to regulate these processes temporally. Aurora B likely plays central roles in regulating mitotic exit progression, potentially in collaboration with mitotic exit phosphatases and other kinases in yet to be uncovered pathways.
nucleocytoplasmic trafficking collaborates with mitotic exit phosphatases in regulating mitotic exit.
2.2 Methods
For the genetic screen, a collection of 417 lines containing most Drosdel deficiencies and a few additional ones (obtained from Martin Lefrançois and Marc Therrien, Université de Montréal, Montréal, Canada) covering most of chromosomes II and III was used. Each line was crossed to the yw; twsP/TM6B or mtsXE-2258/CyO lines. Flies heterozygous for both the deletion and the twsP or mtsXE-2258 mutation were selected based on the absence of balancer chromosomes, and their fertility was tested. Mutant fly lines were obtained from the Bloomington Drosophila Stock Center (Bloomington, IN), Aldelaide Carpenter and David Glover (University of Cambridge, Cambridge, England), Hiroyuki Ohkura (Wellcome Trust Centre for Cell Biology, Edinburgh, Scotland), or Marc Therrien (Université de Montréal, Montréal, QC, Canada).
For fertility tests, three to five virgin females 1 to 4 days old were crossed with three to five Oregon R males per tube and kept at 25°C for 1 day. Flies were then transferred to tubes containing grape juice agar with yeast paste. After 1 day, flies were transferred again to new tubes. 24 hours later, the percentage of hatched embryos was counted. Approximately 100 embryos were counted each time, and this scoring was repeated at least three times. For the genetic screen, the fertility of a group of three to five females crossed to three to five Oregon R males was scored on three consecutive days, and numbers were pooled.
2.3 Results
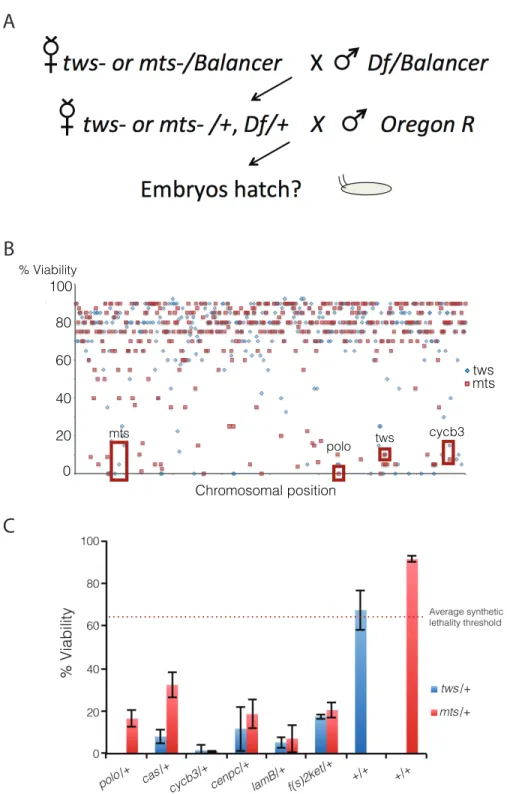
Figure 2.1 | A second-site non-complementation screen identifies several nucleocytoplasmic machinery components that collaborate with PP2A-B55/Tws for cell cycle progression.a, Schematic of the genetic screen approach. Over 400 deficiencies were screened against mts and tws loss of function alleles. b, Several deficiencies led to synthetic embryonic lethality when combined with Mts and Tws loss of function alleles. Data points represent individual deficiencies plotted along their relative chromosomal position. c, Five novel genes were mapped to PP2A-B55/Tws interacting deficiencies. Loss of function alleles of the five identified genes genetically interact with both mts and tws loss of function alleles. Error bars represent SD.
!" #!" $!" %!" &!" '!" (!" )!" *!" +!" #!!"
tws
mts
0 20 40 60 80 100
Chromosomal position
tws mts
mts
polo
cycb3 tws
% Viability
A
B
A library of over 400 deficiencies was screened against heterozygous loss of functions alleles of either the regulatory Tws subunit or the catalytic Mts of the phosphatase (Fig 2.1A). Several deficiencies led to synthetic lethality when combined with either tws, mts or with both alleles (Fig 2.1B). Several deficiencies expected to interact with tws or mts were identified, such as Df(3L)ED4858 that removes the polo gene, a previously identified genetic interaction (Wang et al., 2011b). Several genes were mapped to deficiencies interacting with both the tws and the mts alleles (Fig 2.1C). To determine if the uncovered genes collaborate with one another, alleles of identified genes were assayed as described above against one another for synthetic embryonic lethality. The summary of the presence and absence of genetic interactions (Fig 2.2A) was plotted as an interactome, which highlights the presence of distinct genetic networks each contain the Polo and PP2A-B55/Tws master regulators (Fig 2.2B).
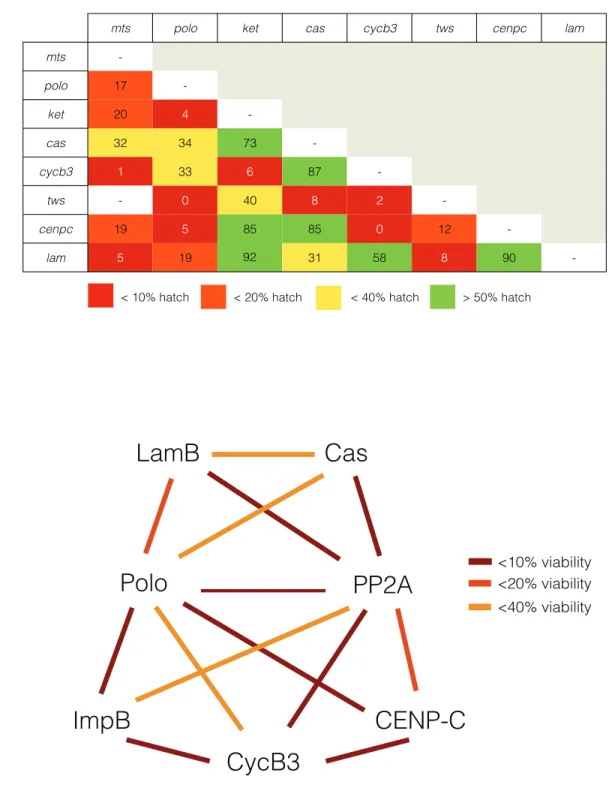
This genetic approach to identifying mitotic exit molecules highlighted three nucleocytoplasmic trafficking components: LamB, Fs(2)Ket and Cas (Fig 2.1C). LamB is a key structural component of the nuclear envelope and is a known CDK1 substrate during NEBD (Muhlhausser and Kutay, 2007; Peter et al., 1990). It is therefore likely that LamB is dephosphorylated by a mitotic exit phosphatase during NERF. Fs(2)Ket is the fly homologue of Importinb, the main component of protein import to the nucleus (Chook and Blobel, 2001). Impb
Figure 2.2 | PP2A-B55/Tws collaborates with distinct genetic networks in regulating cell cycle progression.a, All combinations of loss of function alleles of identified genes were tested for synthetic embryonic lethality. b, Interactome representation of uncovered genetic networks. The absence of a line between genes indicates no genetic interaction, yellow lines indicate embryonic viability of under 40%, orange lines indicate embryonic viability of under 20% and red lines indicate embryonic viability of under 10%.
LamB
PP2A
Polo
CENP-C
ImpB
CycB3
Cas
<10% viability <20% viability <40% viability
92
mts polo ket cas cycb3 tws cenpc lam
mts
-polo 17
-ket 20 4
-cas 32 34 73
-cycb3 1 33 6 87
-tws - 0 40 8 2
-cenpc 19 5 85 85 0 12
-lam 5 19 31 58 8 90
-< 10% hatch < 20% hatch < 40% hatch > 50% hatch
A
Figure 2.3 | CycB and Emb antagonize pp2a/cas and pp2a/lamB genetic interactions. a, Trans-heterozygous embryonic lethality between cse1k03902 and twsP is rescued by embk16715. b, Trans-heterozygous
embryonic lethality between lamk2 and twsP is rescued by embk16715. c, Trans-heterozygous embryonic
lethality between cse1k03902 and twsP is rescued by cycb2. d, Trans-heterozygous embryonic lethality
between lamk2 and twsP is rescued by cycb2. e, Interactome representation of uncovered genetic networks
including antagonistic genetic interactions with cycb and emb. Error bars represent SD.
WT emb k16715 /+;tws/+ emb k16715 /cse1 k03902 ;TM6B/+ cse1 k03902 /+;tws/+ emb k16715 /cse1 k03902 ;tws/+ 0 20 40 60 80 100
Hatched Embryos (%
) WT emb k16715 /+;tws/+ emb k16715 /lam K2;TM6B/+ lam K2/+;tws P/+ emb k16715 /lam K2;tws P/+ 0 20 40 60 80 100
Hatched Embryos (%
) WT cycb 2/+;tws P/TM6B cycb 2/cse1 k03902 ;TM6B/+ cse1 k03902 /+;tws P/+ cycb 2/cse1 k03902 ;tws P/+ 0 20 40 60 80 100
Hatched Embryos (%
) WT cycb 2/+;tws P/TM6B cycb 2/lam K2;TM6B/+ lam K2/+;tws/+ cycb 2/lam K2;tws P/+ 0 20 40 60 80 100
Hatched Embryos (%
2.4 Discussion
Despite our considerable understanding of mitotic entry mechanisms, key mitotic exit regulatory mechanisms have yet to be uncovered. The relatively recent identification of PP2A-B55 as the metazoan CDK1-CycB counteracting phosphatase has raised important mitotic exit questions. For example, key PP2A-B55 substrates have remained elusive. Here, we used an unbiased classical genetics approach to identify genes that collaborate with PP2A-B55 for cell cycle progression in the fly embryo.
Several genomic deficiencies were found to interact genetically with hypomorphic alleles of the PP2A-B55 catalytic subunit mts, the regulatory B55 subunit tws or both. Although our follow up of genomic deficiencies was restricted to deficiencies interacting with both mts and tws alleles, several deficiencies interacting with either allele individually were uncovered and could contain valuable insight into PP2A function broadly. From deficiencies identified as mts and tws collaborators, individual genes responsible for genetic interactions were identified using previously developed loss of function alleles. Although several genes were identified through this approach, several strong collaborators remain unknown given the relatively scarce availability of mutants.
Synthetic lethal interactions within the PP2A-B55/Lam/Cas axis could be rescued with a third hypomorphic allele for an antagonistic component (Fig 2.3). Given the relevance of these interactions to NCT or nuclear import broadly, we hypothesized that partially compromising nuclear export could rescue embryonic lethality in these conditions. Generating trans-heterozygous conditions with loss of function alleles between pp2a-b55/cas and pp2a-b55/lam with emb (XPO-1 or Exportin-1), embryonic viability could be significantly rescued (Fig 2.3A and B). This suggests that the uncovered genetic interactions are relevant to NCT and nuclear import during embryonic development as they are antagonized by the principal nuclear export component in flies, Embargoed (XPO-1).
Similarly, to test the relevance of the PP2A-B55/Lam/Cas genetic axis in cell cycle progression, we reasoned the PP2A-B55 counteracting kinase CDK1-CycB would be antagonistic to the uncovered genetic interactions. Embryonic viability between loss of function alleles between pp2a-b55/cas and pp2a-b55/lam could be rescued by incorporating a loss of function allele for cycb (Fig 2.3C and D). As a central mitotic progression kinase, we conclude that CDK1-CycB antagonism suggests that the identified genetic interactions between PP2A-B55/Lam/Cas are relevant to mitotic entry and/or exit.
CHAPTER 3
NUCLEO-CYTOPLASMIC TRAFFICKING REGULATES NUCLEAR SURFACE AREA
DURING MITOTIC EXIT AND NUCLEAR ASSEMBLY22
3.1 Introduction
Assembling the nucleus into a functional organelle is a regulated, stepwise process essential for nuclear function. Most of our understanding of nuclear organogenesis stems from the examination of nuclear envelope reformation (NERF) at the end of mitosis in organisms that undergo nuclear envelope breakdown (NEBD) at mitotic entry (reviewed in Güttinger et al., 2009). Although the temporal recruitment of many nuclear assembly components during mitotic exit and their regulation has become clearer, how the nucleus expands into a functional organelle within a cell of a limited volume remains elusive.
NERF is a stepwise process implicating several biochemical cues that sequentially trigger chromosome arm compaction (Mora-Bermudez et al., 2007), the recruitment of nuclear pore (Mansfeld et al., 2006; Rasala et al., 2006; Stavru et al., 2006) and membrane precursors (Hallberg et al., 1993), and nuclear lamina assembly (Tsenga and Chena, 2011). PP2A-B55, the principal CyclinB-CDK1 substrate dephosphorylating component during mitotic exit (Mochida et al., 2009; Gharbi-Ayachi et al., 2010), is one of the core biochemical factors driving nuclear expansion and NERF, and is known to collaborate with Importin β (Impβ, Schmitz et al., 2010). The involvement of Impβ, and of nucleo-cytoplasmic trafficking (NCT) generally, in regulating nuclear assembly has long been suggested by several lines of evidence, including the requirement of Ran’s GTPase activity in NERF (Hetzer et al., 2000). Additionally, the nuclear export factor Crm1 has been shown to be important for chromosome segregation (Arnaoutov et al., 2005) and the translocation of the chromosomal passenger complex from the centromere to the midzone in anaphase (Knauer
2This chapter previously appeared as a preprint on the bioRxiv. The original citation is as follows:
et al., 2006). Therefore substantial evidence suggests that both protein import and export pathways are important in mitotic exit models of nuclear organogenesis. Despite the highlighted role for NCT, there is little mechanistic information to suggest how NCT regulates nuclear organogenesis or what molecules must be trafficked. It also remains unclear how disrupting NCT during nuclear assembly affects function in the subsequent interphase.
Another important question in understanding nuclear organogenesis is how nuclei expand to a given nuclear size in a given cellular volume. It has been well established that cell size (or cytoplasmic volume) scales with nuclear size in different metabolic and genetic conditions (Jorgensen et al., 2007; Neumann and Nurse, 2007) and in different developmental situations (Levy et al., 2010). The mechanisms by which nuclear size is regulated, particularly in the context of genomic content, are less clear as genetic perturbations modifying ploidy by an order of magnitude have been reported to have minimal effects on nuclear size (Neumann and Nurse, 2007). In contrast, the increase in genomic content following DNA replication has been reported to cause a 20% to 100% increase in nuclear volume (Jorgensen et al., 2007), whereas inhibiting replication has been shown to result in a significant decrease in nuclear size (Levy et al., 2010). In a model in which nuclear size is dependent on genomic content, genomic content and nuclear volume should increase linearly with one another whereas nuclear volume should be unaffected by changes in cytoplasmic volume (Fig 3.1A). An alternative nuclear scaling model in which nuclear size is determined by nucleoplasmic or nuclear envelope components (Fig 3.1B) would be independent of genomic content. This model would be in line with current evidence of nuclear to cell size scaling (Jorgensen et al., 2007, Levy et al., 2010, Neumann and Nurse, 2007), yet it remains unclear whether nucleoplasmic or nuclear envelope components set nuclear size.
Taking advantage of this model trained solely on in vitro data from Xenopus egg extracts, we predicted in silico and tested in cells and in vivo how NCT regulates nuclear organogenesis. These results uncover a role for nuclear import and nuclear export in regulating nuclear expansion. Nuclear export was predicted and found to regulate nuclear expansion through the localization of a proposed surface area factor (Lamin B). In developing D. melanogaster embryos, compromising NCT affects nuclear expansion in a manner consistent with our model. To test how NCT’s role in nuclear organogenesis affects downstream nuclear function, we show that disrupting NCT impairs transcription activation in interphase following nuclear assembly using an in vivo live-cell transcription reporter (Garcia et al., 2013). Overall, we demonstrate NCT regulates nuclear expansion through proposed nuclear surface area factors during nuclear organogenesis - a process required for downstream nuclear function.
3.2 Methods
3.2.1 Xenopus extract use, microfluidics and microscopy
Cytostatic factor (CSF)-arrested egg extracts were prepared as described previously (Desai et al. 1999; Hannak and Heald, 2006). Interphase extract was maintained by the addition of calcium and cycloheximide. De-membranated sperm nuclei and fluorophores were subsequently added to the extract, which was immediately loaded into microfluidic devices. m-Cherry HURP or GFP-NLS were added to the extract at 2 uM to visualize nuclear import. The standard nuclear assembly reaction was 100 µl fresh extract, 0.4 mM CaCl2, 100 µg/ml cycloheximide and 1000 Xenopus
sperm per µl. Reactions were incubated at 16-18°C and spherical, import-competent nuclei generally formed within 30-45 min.
channel network was exposed to oxygen plasma (Harrick Plasma, Ithaca, NY) and placed in contact with a cover glass slip (#1.5, Thomas Scientific) to form an irreversible bond.
3.2.2 Mathematical model
The mathematical model is derived from a combination of mechanical force-balance determining the size of the nucleus, and transport of surface and volume factors throughout the cytoplasm and into the nucleus. The almost-spherical shape of in vitro nuclei allows us to impose spherical symmetry on the nucleus and describe its size with a single variable, the radius r(t). Mechanical force-balance thus becomes a single ordinary differential equation (ODE) for dr/dt. To describe transport in the cytoplasm, we make the simplifying assumption that it can be divided into two compartments, “proximal” to the nucleus and “distal” to the nucleus. This leads to 6 ODEs: for proximal, distal and nuclear amounts of surface and volume factors. The full model is thus a coupled system of 7 nonlinear ODEs. These equations are solved numerically in Matlab (Mathworks). There are 15 biophysical parameters in the model. Of these, 9 are taken from independent experiments or estimated from previous work. The remaining 6 (nuclear import and export rates and initial amounts of both factors) are fit to the time series for growth, as described in the Supplemental Math (Boudreau et al., 2018).
3.2.3 Mammalian tissue culture cell maintenance, drug treatments, immunostaining and microscopy
HeLa cells stably expressing H2B:GFP were cultured in DMEM (containing 1g/L glucose) supplemented with 10% fetal bovine serum and penicillin-streptomycin (Sigma) at 37oC in a
humidified 95% air, 5% CO2 incubator. To target nuclear import, the small molecule inhibitor
Importazole (IPZ) that targets the interaction between Ran-GTP and Importin-ß was used at concentrations consistent with previously published results (Soderholm et al., 2011). To target nuclear export, Leptomycin-B (LMB), an irreversible inhibitor of the main nuclear protein export component Crm1, was used (Kudo et al., 1999). Immunostaining was performed using a 5 minute 0.2% TritonX-100 permeabilization, followed by a 20-minute fixation with 4% EM-grade paraformaldehyde (Ted Pella) in PHEM. All steps are performed with coverslips in tissue culture dishes placed on top of a hot plate heated to 37oC. LMNB1 was stained with a rabbit polyclonal
Live-cell microscopy of tissue-culture cells was performed at 37oC in a humidified
chamber on a DeltaVision microscope using Softworx software (Applied Precision) with a CoolSnap HQ2 camera (Photometrics) and a x60 planApo objective. Cells were incubated in CO2
-independent media (ThermoFisher) in 6-channel µ-Slides (Ibidi) during image acquisition. Fixed-cell microscopy was performed on the same DeltaVision microscope as mentioned above at room temperature.
To measure nuclear import or nuclear export, an NLS or an NES respectively was cloned into pOD69 with an mCherry tag as to obtain fusion proteins containing either the NLS or the NES with two fluorophores to prevent diffusion across nuclear pore complexes. The SV40 NLS (PKKKRKV) was used to follow Impb-directed nuclear import (Kalderon et al., 1984). HIV-1’s viral Rev protein’s NES (LPPLERLTL) was used to follow nuclear export (Fischer et al., 1995; Neville et al., 1997).
3.2.4 C. elegans use, RNAi and microscopy
The worm strain TH32 (pie-1::bg-1::GFP + unc-119(+), pie-1::GFP::H2B + unc-119(+)), was grown and maintained at 20°C using standard procedures. Bacterial strains containing a vector expressing dsRNA under the IPTG promoter were obtained from the Ahringer library (from Bob Goldstein’s laboratory, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA). Targets were confirmed by sequencing.
Worm embryos were mounted in Egg buffer (118 mmol/L NaCl, 48 mmol/L KCl, 2 mmol/L CaCl2, 2 mmol/L MgCl2, 25 mmol/L HEPES, pH 7.3) between a 1.5 coverslip and a
microscope slide spaced by 22.81µm glass beads (Whitehouse Scientific) and sealed with Valap (1:1:1 lanolin, petroleum jelly, and parafilm wax). Embryos were then imaged on a Nikon A1r resonant scanning confocal microscope using a x60 Apo water immersion objective (Nikon), GaASP PMT detectors and NIS-Elements (Nikon) at 22oC.
3.2.5 Drosophila husbandry and microscopy
either UAS-emb or UAS-nup358 were obtained and subsequently crossed with males of the reporter line (hb P2 enhancer and promoter). Flies expressing single copies of MCP:GFP and H2A:RFP were used as controls. Embryos were dechorionated, mounted between gas permeable lumox® dish 50 dishes and a coverslip and imaged on a Nikon A1r resonant scanning confocal microscope. Images were acquired using a x60 Apo TIRF objective. All fly husbandry, crosses and imaging were performed at 22oC.
3.2.6 Image analysis
Nuclear expansion, transcription activation and DIC morphology assay ImageJ plugins are available on VB’s GitHub page (https://github.com/viboud12). Other image analysis plugins used in nuclear import/export, pronuclear size and droplet nuclear size analysis are available upon request.
3.3 Results
Figure 3.1 | Models of nuclear size scaling with cell size and nuclear surface area to volume ratios within a defined cell size. Simple models of nuclear size in which either the number of nuclei/genome size or cell size/cytoplasmic size are fixed predict nuclear size scaling relationships and nuclear surface area:volume ratios under different regimes. a, In Hypothesis 1, nuclear size is driven by genome size. As cell size/cytoplasmic size increases, nuclear size is constant as genome size is constant. By keeping cell size constant and varying the number of genomes in this regime, nuclear volume and nuclear surface area increase. b, In Hypothesis 2, nuclear size to cell size scaling is independent of genome size. Considering
Envelope
Volume-filling network Chromatin
Hypothesis 1
Hypothesis 2
0 5 10
Cell/cytoplasmic size
1 2 3 1 2 3
Number of Nuclei
1 2 3
0 5 10 1 2 3 1 2 3 1 2 3
0 5 10 1 2 3 1 2 3 1 2 3
Number of Nuclei Number of Nuclei
A
B
Fig. 1
FIXED GENOME SIZE FIXED CELL/CYTOPLASMIC SIZE
Nuclear size Total nuclear
volume
the nucleus as a viscoelastic material composed of a nuclear envelope and a nucleoplasmic volumetric network, nuclear size is hypothesized to be regulated in a nuclear envelope-limited regime (upper panel) or a volumetric network-limited regime (lower panel), where either nuclear surface or nuclear volume set nuclear size respectively. * = p < 0.05.
3.3.1 Neither nuclear surface area nor nuclear volume scale with cell size in multinucleate cytoplasm droplets
Nuclear, organelle, chromosome, and spindle size have all been shown to scale with cell size in either developmental or in vitro contexts (Hazel et al., 2013; Ladouceur et al., 2015; Good et al., 2013; Wilbur et al., 2013). How the contribution of genomic content and cytoplasmic volume define a given nuclear size have not been examined with controlled biophysical variables.
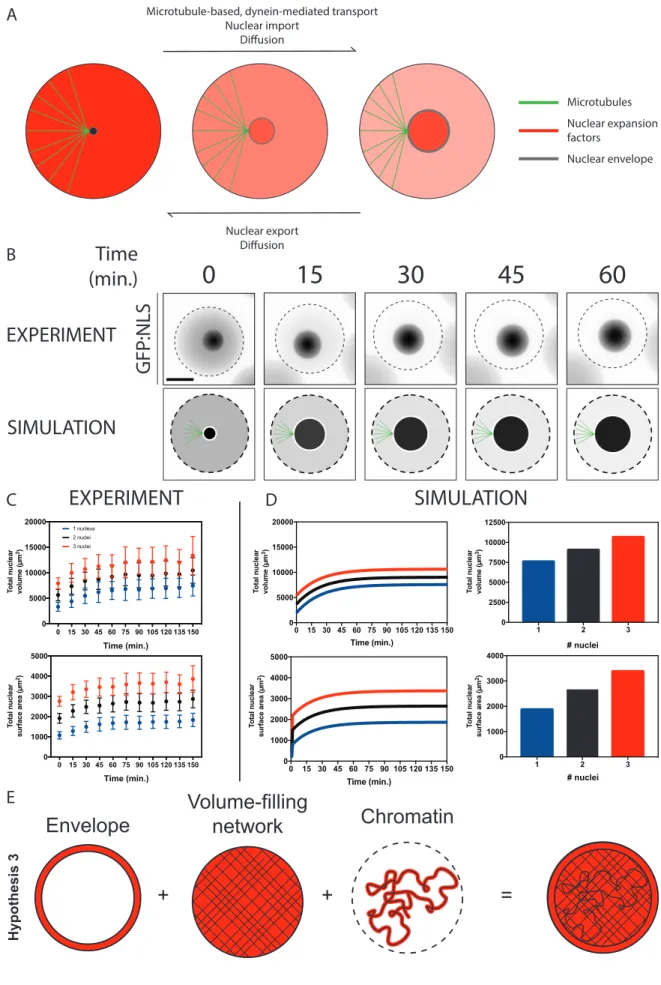
Figure 3.2 | in vitro encapsulation of nuclei in cytoplasmic droplets reveals nuclear surface area to volume regulation during nuclear expansion. a, Representative images of 1, 2, or 3 nuclei assembled in droplets with a diameter of 50 µm. Nuclei were visualized by import of an GFP:NLS reporter to assess nuclear morphology. Scale bar, 20 µm. b, Total nuclear volume plotted as a function of the number of encapsulated nuclei at 90 minutes post encapsulation in droplets (n > 1000). c, Total nuclear surface area plotted as a function of the number of encapsulated nuclei at 90 minutes post encapsulation (n > 1000). d,
Surface area to volume ratios of mean surface areas and volumes of nuclei 90 minutes post encapsulation in droplets across four experimental replicates. Error bars represent SD.
Here, a previously described microfluidics approach (Hazel et al., 2013) was used to encapsulate fully functional cytoplasm (derived from X. laevis eggs; Desai et al., 1999) of defined
0.225 0.250 0.275 0.300 0.325 0.350 Total nuclear
surface area to volume rati
o
A
B
C
Fig. 2
D
# nuclei: 1 2 3 # nuclei: 1 2 3
*
# nuclei: 1 2 3
GFP :NLS 0 4000 8000 12000 16000 20000
Total nuclear volume
volumes to probe how physical parameters affect nuclear assembly processes. Using de-membranated sperm nuclei as a source of chromatin in addition to 2 µM of GFP:NLS, droplets containing mainly spherical nuclei were observed, which expand and reach steady state within the duration of the experiment.
To determine how genomic content, nuclear volume, and nuclear surface area influence nuclear assembly, we measured nuclear size in cytoplasmic droplets of fixed volumes (50µm in diameter) containing a single or several genomes (Fig 3.2A). Whereas total nuclear volume increased slightly in droplets containing two or more nuclei (Fig 3.2B), total nuclear surface area increased faster (Fig 3.2C). This observation is inconsistent with models in which nuclear size is driven solely by chromatin amount (Fig 3.1A). Additionally, total nuclear volume and surface area both increased in droplets with multiple nuclei, indicating that nuclear size is likely not dictated by nucleoplasmic or nuclear envelope components exclusively, as hypothesized in Figure 3.1B. Indeed, these data suggest nuclear size is regulated by a combination of nuclear envelope and nucleoplasmic components, in addition to chromatin amount.
3.3.2 Modelling nuclear organogenesis captures the roles of nuclear volumetric and surface area factors in regulating nuclear expansion
To explore conditions under which nuclear scaling would be expected to occur, we developed a mathematical model of the mechanochemistry of nuclear assembly. In the model, the nucleus is comprised of a viscoelastic body (containing chromatin and internal architectural networks such as NuMA that provide mechanical rigidity) surrounded by a viscoelastic envelope (the lamina and membranes). The fundamental assumption of the model is that two molecular components must be imported into the nucleus for its growth: a surface factor that assembles into the envelope and a volume factor that assembles into the interior. We do not a priori specify the molecular identity of these components.
Fig. 3
A 0 1000 2000 3000 4000 50000 15 30 45 60 75 90 105 120 135 150 Time (min.) To tal nuclear surface area ( m 2)
Microtubule-based, dynein-mediated transport Nuclear import
Diffusion
Nuclear export
Diffusion
Microtubules Nuclear expansion factors
Nuclear envelope Nuclear import
Diffusion
0 15 30 45 60 75 90 105 120 135 150 Time (min.) 0 5000 10000 15000 20000 To tal nuclear volume ( m 3) 1 nucleus 2 nuclei 3 nuclei
0 15 30 45 60 75 90 105 120 135 150
0 1000 2000 3000 4000 5000 Time (min.) To tal nuclear surface area ( m 2)
0 15 30 45 60 75 90 105 120 135 150
0 5000 10000 15000 20000 Time (min.) To tal nuclear volume ( m 3)
1 2 3
0 1000 2000 3000 4000 # nuclei To tal nuclear surface area ( m 2)
1 2 3
0 2500 5000 7500 10000 12500 # nuclei To tal nuclear volume ( m 3)
SIMULATION
Time
(min.)
0
15
30
45
60
Figure 3.3 | A mechano-chemical mathematical model dependent on dynein-based transport supports an inverse scaling relationship between nuclear diameter and nuclear surface area. a, Model assumptions of regulatory mechanisms governing nuclear surface area and volumetric factor concentration. The model assumes factors can become proximal to the expanding nucleus based on diffusion and dynein-based transport and are regulated at the nuclear envelope by nucleo-cytoplasmic trafficking. b,
Experimental and simulated expansion of single nuclei in a defined cytoplasmic volume. Experimental nuclear expansion was visualized using a GFP:NLS reporter in encapsulated cytoplasmic droplets of ~50
µm in diameter. Dashed lines represent cytoplasm edges. c, Quantification of sum nuclear volume and nuclear surface area in experimental droplets containing one or several nuclei. Error bars represent SD. d,
Total nuclear surface area and nuclear volume during simulated nuclear expansion and at nuclear expansion steady state. e, Quantification of nuclear expansion in defined cytoplasmic volumes suggests a model in which nuclear organogenesis occurs in a regime in which genomic size, nucleoplasmic factors and nuclear envelope factors collaborate.
We use this model to simulate nuclear expansion following the anaphase-telophase transition. For a given set of biophysical input parameters (nuclear rigidity, import and export rates), the simulation produces time trajectories of nuclear size (Fig 3.3B) and the concentrations of volume and surface factors. The model captures experimental nuclear expansion measurements of expanding nuclei in cytoplasmic droplets (Fig 3.3C and 3.3D), in which nuclear size reaches steady-state over the course of the experiment (Fig 3.3C).
Under Hypothesis 2 (Fig 3.1B), the model demonstrates that, depending on the ratio of nuclear surface rigidity to nuclear volume factor rigidity, multi-nucleated droplets could scale with either constant total volume or constant total surface area (SuppMath Fig S3/Boudreau et al., 2018), or combinations of these. Notably, all Hypothesis 2 simulations give non-increasing total nuclear volumes. Our experimental data (Fig 3.2 and Fig 3.3) place the model parameters in a regime in which nuclear volume rigidity is dominant, or, equivalently, in the regime in which the surface factor is not limited, and that chromatin confinement contributes significantly to nuclear size (SuppMath Fig S4/Boudreau et al., 2018), which we refer to as Hypothesis 3 (Fig 3.3E). By changing biophysical parameters, the model can simulate perturbations including to import/export or microtubules. This is discussed below (Fig 3.4A, 3.4B and 3.5B).
3.3.3 Nuclear expansion is regulated by nucleo-cytoplasmic trafficking of nuclear volume and surface area factors
in proximity to the developing nucleus, these factors must be subject to NCT to enable their nuclear functions. Modifying NCT rates in the mathematical model detailed above and simulating nuclear expansion suggested that by inhibiting nuclear export by a factor of two the nucleus would expand further than its wild-type counterpart (Fig 3.4A and 3.4B). The inverse effect is predicted by inhibiting nuclear import.
Figure 3.4 | Nucleo-cytoplasmic trafficking regulates nuclear expansion. a, Images of simulated nuclei undergoing nuclear expansion in a defined cytoplasmic volume. Nuclear export is compromised by 50% in this simulation. Dashed lines in all images represent nuclear size at t = 5 minutes in each condition. Shades of red represent the predicted nuclear concentrations of nuclear surface area factor. b, Predicted nuclear cross-sectional area of simulated nuclei through nuclear expansion in control or 50% nuclear export compromised conditions. c, Representative still images of time-lapse movies of HeLa cells stably expressing H2B:GFP treated with either DMSO or 20 nM LMB at anaphase onset specifically. Scale bar, 10 µm. d, Chromatin-occupied area of H2B:GFP expressing HeLa cells treated with DMSO or 20 nM LMB at anaphase onset. Areas are normalized to nuclear cross-sectional area of DMSO treated cells at the anaphase to telophase transition (corresponding to ~ 10 minutes post anaphase onset) to compare nuclear expansion dynamics. Error bars represent SD.
To test this prediction, HeLa cells stably expressing H2B:GFP were imaged as of anaphase-onset through mitotic exit – a transition in the cell cycle during which condensed mitotic chromatin must decondense to form an interphase-like nucleus. Cells were treated with inhibitors targeting nuclear import or export at anaphase onset specifically (Fig 3.4C). A previously described area-based live-cell image analysis assay was adapted to measure the area occupied by chromatin as a
A
B
D
Time (min.)5
10
15
20
25
DMSO (20nM)LMB
Fig. 4
0 5 10 15 20 25 30 50 60 70 80 90 100 110 120 130 140 0 Time (min.) Chromatin-occupied area ( m 2) DMSO LMB (20nM)
C
SIMULATION EXPERIMENT0 5 10 15 20 25 30 50 60 70 80 90 100 110 120 130 140 0 Time (min.) Nuclear area ( m
2) CTRLExport
-Time (min.)
5
10
15
20
25
CTRL Export
measure of nuclear expansion (Maddox et al., 2006). To improve temporal resolution computationally, a temporal super-resolution method (Berro et al., 2014) was implemented. Inhibiting nuclear export with Leptomycin B (LMB) (Kudo et al., 1999) at anaphase onset (Fig 3.S1) induced further nuclear expansion as predicted (Fig 3.4B), with nuclei reaching volumes approximately 50% larger than control nuclei (Fig 3.4D). Inhibiting nuclear import with Importazole (Soderholm, et al., 2011) IPZ failed to display significant effects on nuclear expansion, which can be attributed to the compound’s subtle effects on nuclear import inhibition (Fig 3.S1). To exclude a role for NCT in regulating progression through mitotic exit globally, we developed an edge-based quantitative time-lapse microscopy assay to measure cell shape changes during cytokinesis in DIC images. Treating cells at anaphase with LMB did not significantly affect cell shape changes from metaphase through mitotic exit (Fig 3.S2).
In many animals, following fertilization of the oocyte the highly condensed sperm chromatin must expand to form an interphase-like pronucleus prior to pronuclear meeting (Matsumoto et al., 1999) - a process akin to nuclear expansion following mitotic exit in fully differentiated somatic cells. To test whether NCT regulates nuclear expansion of sperm chromatin following fertilization, we measured male pronuclear size following fertilization in the 1-cell C. elegans embryo. Compromising nuclear import or nuclear export by partially depleting IMB-1 (Importin β, 12 h treatment) and XPO-1 (Exportin-1, 24 h treatment) respectively, pronuclear expansion rates recapitulated our model’s prediction (Fig 3.S3).
Taken together, experimental inhibition of nuclear export during nuclear expansion yielded nuclei expanding further than their control counterparts as predicted by halving nuclear export rates of nuclear volumetric and surface area factors in silico.
when slowing nuclear import or export rates, the nuclear concentration of surface area factors was perturbed (Fig 3.5B).
Figure 3.5 | Inhibiting nuclear export during nuclear expansion reduces the nuclear concentration of LMNB1, a proposed nuclear surface area factor. a, Predicted nuclear concentrations of a proposed nuclear volumetric factor, NuMa. b, Predicted nuclear concentrations of a proposed nuclear surface area factor, LMNB1, through nuclear expansion in control and nuclear export compromised conditions. Nuclear export activity is compromised by 50% in the simulation. c, Representative still images of fixed HeLa cells synchronized in G2/M, released into mitosis, treated with small molecule inhibitors at approximately anaphase onset and fixed and stained for DAPI and LMNB1. Scale bar, 10 µm. d, Normalized LMNB1 fluorescence intensity in 3D maximum intensity projections proximal to DNA as highlighted by DAPI. Extrapolated times were determined for individual nuclei based on chromatin-occupied area (measured in Fig 3.4D).
To test the prediction that a nuclear surface area factor must be trafficked between the cytoplasm and the nucleus during nuclear expansion, we measured the amount of endogenous Lamin B1 (LMNB1) during nuclear organogenesis using a fixed cell assay that allows temporal resolution using nuclear cross-sectional area as a predictor (Kafri et al., 2013). HeLa cells were synchronized at the G2/M transition using the CDK1 inhibitor RO-3306 (Vassilev et al., 2006) (Fig 3.5C), released in mitosis and treated with small molecule inhibitors 80 minutes following
Fig. 5
0 5 10 15 20 25 30
0.00 0.25 0.50 0.75 1.00 1.25
Extrapolated Time (min.)
Normalized Lamin B Fluorescence
Intensity per Unit
Area (a.u. ) LMB (20nM) DMSO
A
C
B
Thymidine Block Release RO-3306 Block Release Small Molecule Treatments Fix Stain Image24h 5h 8h 80min 20min
G1/S G2/M Anaphase
Onset
LMNB1
Extrapolated Time (min.):
DAPI
5 10 15 20 25
SIMULATION EXPERIMENT
0 5 10 15 20 25 30 0.7 0.8 0.9 1.0 1.1 Time (min.)
Nuclear volumetric factor
concentration (a.u.
)
CTRL Export
-0 5 10 15 20 25 30 0.7 0.8 0.9 1.0 1.1 Time (min.)
Nuclear surface area factor
concentration (a.u.
)