Stable polymorph of morphine
1Thomas Gelbrich,* Doris E. Braun and Ulrich J. Griesser
Institute of Pharmacy, University of Innsbruck, Innrain 52c, 6020 Innsbruck, Austria Correspondence e-mail: [email protected]
Received 19 November 2012; accepted 28 November 2012
Key indicators: single-crystal X-ray study;T= 173 K; mean(C–C) = 0.009 A˚;
Rfactor = 0.068;wRfactor = 0.141; data-to-parameter ratio = 7.3.
In the stable polymorph of the title compound, C17H19
-NO3 [systematic name: (5,6
)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol], the molecular conformation is in agreement with the characteristics of previously reported morphine forms. The molecule displays the typical T-shape and its piperidine ring adopts a slightly distorted chair conformation. Intermolecular O—H O hydrogen bonds link the molecules into helical chains parallel to the b axis. Intramolecular O—H O hydrogen bonds are also observed.
Related literature
For related structures, see: Guguta et al. (2008); Gylbert (1973); Mackay & Hodgkin (1955); Bye (1976); Wongwei-chintana et al. (1984); Lutz & Spek (1998); Scheins et al. (2005); Gelbrich et al. (2012). For decriptions of morphine polymorphs, see: Kofler (1933); Kuhnert-Brandsta¨tter et al. (1975). For a description of the Cambridge Structural Data-base, see: Allen (2002). For the programXPac, see: Gelbrich & Hursthouse (2005).
Experimental
Crystal data
C17H19NO3
Mr= 285.33
Orthorhombic,P212121
a= 7.6989 (10) A˚
b= 12.737 (4) A˚
c= 13.740 (4) A˚
V= 1347.4 (6) A˚3
Z= 4
CuKradiation
= 0.78 mm1
T= 173 K
0.150.100.03 mm
Oxford Diffraction Xcalibur (Ruby, Gemini ultra) diffractometer Absorption correction: multi-scan
(CrysAlis PRO; Oxford Diffraction, 2003)
Tmin= 0.624,Tmax= 1.000
13009 measured reflections 1408 independent reflections 977 reflections withI> 2(I)
Rint= 0.118
Refinement
R[F2> 2(F2)] = 0.068
wR(F2) = 0.141
S= 1.01 1408 reflections
192 parameters
H-atom parameters constrained
max= 0.27 e A˚3
min=0.26 e A˚3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1 O3i
0.84 1.96 2.757 (6) 159
O3—H3 O2 0.84 2.17 2.629 (6) 114
Symmetry code: (i)x;yþ1 2;zþ
3 2.
Data collection: CrysAlis PRO(Oxford Diffraction, 2003); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure:SHELXL97(Sheldrick, 2008); molecular graphics:XPinSHELXTL(Bruker, 1998) andMercury
(Brunoet al., 2002); software used to prepare material for publica-tion:publCIF(Westrip, 2010).
We thank Volker Kahlenberg for access to the X-ray instrument used in this study. DEB acknowledges financial support from the Hertha Firnberg Programme of the Austrian Science Fund (FWF, project No. T593–N19).
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IM2413).
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Bruker (1998).XP. Bruker AXS Inc., Madison, Wisconsin, USA
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Bye, E. (1976).Acta Chem. Scand. Ser. B,30, 549–554.
Gelbrich, T., Braun, D. E. & Griesser, U. J. (2012).Acta Cryst.E68, o3358– o3359.
Gelbrich, T. & Hursthouse, M. B. (2005).CrystEngComm,7, 324–336. Guguta, C., Peters, T. P. J. & de Gelder, R. (2008).Cryst. Growth Des.8, 4150–
4158.
Gylbert, L. (1973).Acta Cryst.B29, 1630–1635. Kofler, L. (1933).Pharm. Monatsh.14, 220–222.
Kuhnert-Brandsta¨tter, M., Kofler, A. & Heindl, W. (1975).Pharm. Acta Helv.
50, 360–372.
Lutz, M. & Spek, A. L. (1998).Acta Cryst.C54, 1477–1479. Mackay, M. & Hodgkin, D. C. (1955).J. Chem. Soc.3261–3267.
Oxford Diffraction (2003). CrysAlis CCD and CrysAlis RED. Oxford Diffraction Ltd., Abingdon, Oxfordshire, England.
Scheins, S., Messerschmidt, M. & Luger, P. (2005).Acta Cryst. B61, 443– 448.
Sheldrick, G. M. (2008).Acta Cryst.A64, 112–122. Westrip, S. P. (2010).J. Appl. Cryst.43, 920–925.
Wongweichintana, C., Holt, E. M. & Purdie, N. (1984).Acta Cryst.C40, 1486– 1490.
Structure Reports
Online
supporting information
Acta Cryst. (2013). E69, o2 [https://doi.org/10.1107/S1600536812048945]
Stable polymorph of morphine
Thomas Gelbrich, Doris E. Braun and Ulrich J. Griesser
S1. Comment
Morphine is the main alkaloid of opium, the dried latex of the opium poppy (Papaver somniferum). The Cambridge
Structural Database (CSD; version 5.33 and updates; Allen, 2002) contains a number of free base and salt structures of
morphine: a monohydrate (Bye, 1976), a hydrochloride trihydrate (Gylbert, 1973), a hydroiodide dihydrate (Mackay &
Hodgkin, 1955), a complex with β-phenylhydracrylic acid (Lutz & Spek, 1998) and a bis(morphinium) dihydrogensulfate
pentahydrate (Wongweichintana et al., 1984). A hydrochloride anhydrate structure was recently reported by us (Gelbrich
et al., 2012). The title structure was previously solved from powder data by Guguta et al. (2008), however the
corresponding atomic coordinates are not available from the CSD or from supplementary materials accompanying this
report.
According to Kofler (1933), morphine can exist in two distinct polymorphic modifications, and the characteristics of the
crystals investigated by us match Kofler's description of the stable form. Our thermomicroscopy experiments have shown
that the investigated crystals melt under decomposition at 254 °C (the applied heating rate was 5 °C per minute). This
behaviour is in agreement with reports given by Kofler (1933) and Kuhnert-Brandstätter et al. (1975).
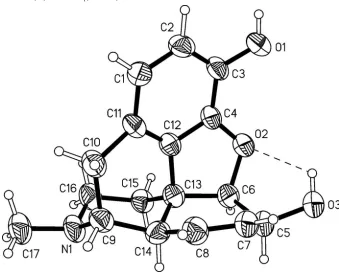
The geometry of the molecular morphine scaffold (Figure 1) with its five rings agrees with the characteristics of the
related salt and free base structures mentioned above. The title structure displays two sets of O—H···O bonds, one of
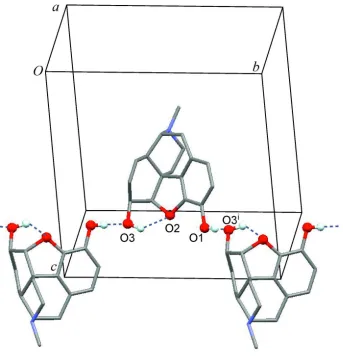
which is intramolecular and the other is intermolecular (Table 1). Intermolecular hydrogen bonds link the morphine
molecules into an infinite helical chain that propagates parallel to the b-axis (Figure 2).
The packing of the geometrically inflexible morphine moieties in the title structure was compared with corresponding
packing arrangements present in the six morphine forms mentioned above (Bye, 1976; Gelbrich et al., 2012; Gylbert,
1973; Mackay & Hodgkin, 1955; Lutz & Spek, 1998; Wongweichintana et al., 1984), using the program XPac (Gelbrich
& Hursthouse, 2005). These comparisons have shown that the packing mode of morphine molecules in the stable form is
unique and has no supramolecular constructs in common with any of the other structures in this group.
S2. Experimental
Morphine was obtained from Heilmittelwerke Wien, Austria. Very thin, plate-shaped crystals of the stable polymorph
were yielded from a sublimation experiment carried out on a Kofler hot bench at approximately 150 °C, using two glass
slides separated by a spacer ring of 1 cm height.
S3. Refinement
All H atoms were identified in a difference map. Methyl H atoms were idealized and included as rigid groups allowed to
rotate but not tip (C—H = 0.98 Å) and H atoms bonded to oxygen atoms (O—H = 0.84 Å), tertiary CH (C—H = 0.99 Å),
secondary CH2 (C—H = 0.99 Å) and aromatic carbon atoms (C—H = 0.95 Å) were positioned geometrically. The
Figure 1
Molcular structure of morphine with displacement ellipsoids are drawn at the 50% probability level and hydrogen atoms
Figure 2
Hydrogen bonded helical chain. Intramolecular and intermolecular O—H···O bonds are drawn as broken lines; H and O
atoms involved in hydrogen bonding are drawn as balls [symmetry code: (i) -x, y + 1/2, -z + 3/2.].
(5α,6α)-7,8-Didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol
Crystal data
C17H19NO3 Mr = 285.33
Orthorhombic, P212121
Hall symbol: P 2ac 2ab
a = 7.6989 (10) Å
b = 12.737 (4) Å
c = 13.740 (4) Å
V = 1347.4 (6) Å3 Z = 4
F(000) = 608
Dx = 1.407 Mg m−3
Cu Kα radiation, λ = 1.54180 Å Cell parameters from 1360 reflections
θ = 3.2–68.2°
µ = 0.78 mm−1 T = 173 K Plate, colourless 0.15 × 0.10 × 0.03 mm
Data collection
Oxford Diffraction Xcalibur (Ruby, Gemini ultra)
diffractometer
Radiation source: Enhance (Mo) X-ray Source Graphite monochromator
Detector resolution: 10.3575 pixels mm-1 ω scans
Absorption correction: multi-scan
(CrysAlis PRO; Oxford Diffraction, 2003)
1408 independent reflections 977 reflections with I > 2σ(I)
Rint = 0.118
h = −9→8
k = −14→15
l = −16→16
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.068 wR(F2) = 0.141 S = 1.01 1408 reflections 192 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0112P)2 + 2.P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.27 e Å−3
Δρmin = −0.26 e Å−3
Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0043 (4)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
The Flack x parameter (Flack, 1983) and the Hooft y parameter (Hooft et al., 2008) were both indeterminate due to a lack of significant resonant scattering. Accordingly, Friedel opposites were merged prior to the final refinement. [Flack, H. D. (1983). Acta Cryst. A39, 876–881; Hooft, R. W. W., Straver, L. H. & Spek, A. L. (2008). J. Appl. Cryst.41, 96–103.]
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.0998 (5) 0.6564 (3) 0.7856 (3) 0.0520 (12)
H1 0.0431 0.7103 0.7705 0.062*
O2 0.3165 (5) 0.4742 (3) 0.7958 (3) 0.0466 (11)
O3 0.1429 (6) 0.2969 (3) 0.7838 (3) 0.0546 (12)
H3 0.1096 0.3587 0.7941 0.066*
N1 0.8422 (6) 0.4378 (4) 0.5555 (4) 0.0492 (14)
C1 0.3549 (8) 0.6662 (5) 0.5563 (5) 0.0458 (15)
H1A 0.3611 0.7068 0.4984 0.055*
C2 0.2382 (8) 0.6935 (5) 0.6290 (5) 0.0466 (16)
H2 0.1665 0.7534 0.6198 0.056*
C3 0.2223 (7) 0.6362 (5) 0.7147 (5) 0.0429 (15)
C4 0.3281 (7) 0.5499 (4) 0.7238 (5) 0.0408 (14)
C5 0.2959 (8) 0.2985 (5) 0.7233 (5) 0.0494 (17)
H5 0.3554 0.2293 0.7320 0.059*
C6 0.4208 (7) 0.3836 (5) 0.7615 (5) 0.0462 (16)
H6 0.4902 0.3548 0.8168 0.055*
H7 0.1398 0.2980 0.5961 0.060*
C8 0.3781 (9) 0.3338 (5) 0.5537 (5) 0.0506 (17)
H8 0.3524 0.3340 0.4861 0.061*
C9 0.6652 (8) 0.4267 (5) 0.5160 (5) 0.0450 (15)
H9 0.6748 0.3841 0.4551 0.054*
C10 0.5732 (8) 0.5321 (5) 0.4878 (5) 0.0491 (17)
H10A 0.6630 0.5838 0.4686 0.059*
H10B 0.4984 0.5194 0.4304 0.059*
C11 0.4634 (7) 0.5792 (5) 0.5682 (4) 0.0410 (14)
C12 0.4530 (7) 0.5257 (5) 0.6546 (4) 0.0386 (14)
C13 0.5465 (8) 0.4258 (5) 0.6810 (4) 0.0415 (15)
C14 0.5584 (8) 0.3617 (5) 0.5888 (5) 0.0466 (16)
H14 0.6227 0.2953 0.6033 0.056*
C15 0.7290 (7) 0.4477 (5) 0.7212 (5) 0.0443 (15)
H15A 0.7199 0.4948 0.7783 0.053*
H15B 0.7818 0.3809 0.7432 0.053*
C16 0.8453 (8) 0.4978 (5) 0.6457 (5) 0.0521 (17)
H16A 0.8056 0.5704 0.6329 0.062*
H16B 0.9657 0.5012 0.6708 0.062*
C17 0.9652 (8) 0.4788 (6) 0.4841 (5) 0.060 (2)
H17A 0.9335 0.5510 0.4672 0.090*
H17B 0.9621 0.4350 0.4254 0.090*
H17C 1.0826 0.4778 0.5117 0.090*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.040 (2) 0.053 (2) 0.063 (3) 0.012 (2) 0.008 (2) 0.002 (2)
O2 0.031 (2) 0.048 (2) 0.060 (3) 0.0019 (19) 0.004 (2) 0.000 (2)
O3 0.040 (2) 0.050 (2) 0.074 (3) −0.006 (2) 0.014 (3) −0.002 (2)
N1 0.026 (2) 0.063 (3) 0.059 (3) 0.001 (3) 0.005 (2) 0.003 (3)
C1 0.040 (3) 0.043 (3) 0.055 (4) 0.003 (3) 0.002 (3) 0.006 (3)
C2 0.035 (3) 0.044 (3) 0.061 (5) 0.008 (3) −0.004 (3) 0.000 (3)
C3 0.027 (3) 0.047 (3) 0.055 (4) 0.003 (3) 0.003 (3) −0.003 (3)
C4 0.025 (3) 0.046 (3) 0.051 (4) 0.000 (3) 0.001 (3) 0.000 (3)
C5 0.034 (3) 0.048 (4) 0.066 (5) −0.006 (3) 0.009 (3) −0.001 (3)
C6 0.030 (3) 0.048 (4) 0.061 (4) 0.000 (3) −0.001 (3) 0.003 (3)
C7 0.039 (3) 0.047 (4) 0.064 (5) −0.005 (3) 0.006 (3) −0.005 (3)
C8 0.044 (3) 0.052 (4) 0.056 (4) −0.005 (3) 0.003 (3) −0.008 (3)
C9 0.037 (3) 0.048 (4) 0.050 (4) 0.004 (3) 0.003 (3) −0.003 (3)
C10 0.040 (3) 0.058 (4) 0.050 (4) 0.008 (3) 0.007 (3) 0.006 (3)
C11 0.030 (3) 0.047 (3) 0.045 (4) 0.004 (3) 0.003 (3) −0.006 (3)
C12 0.024 (3) 0.044 (3) 0.048 (4) 0.002 (3) −0.003 (3) 0.001 (3)
C13 0.030 (3) 0.046 (3) 0.048 (4) 0.000 (3) −0.002 (3) 0.000 (3)
C14 0.038 (3) 0.038 (3) 0.065 (5) −0.001 (3) −0.001 (3) 0.002 (3)
C15 0.032 (3) 0.051 (4) 0.050 (4) 0.001 (3) −0.005 (3) 0.000 (3)
C16 0.028 (3) 0.064 (4) 0.064 (4) −0.003 (3) 0.000 (3) 0.000 (4)
O1—C3 1.380 (7) C7—H7 0.9500
O1—H1 0.8400 C8—C14 1.512 (9)
O2—C4 1.385 (7) C8—H8 0.9500
O2—C6 1.483 (7) C9—C14 1.538 (9)
O3—C5 1.442 (8) C9—C10 1.566 (9)
O3—H3 0.8400 C9—H9 1.0000
N1—C16 1.456 (8) C10—C11 1.516 (8)
N1—C17 1.460 (8) C10—H10A 0.9900
N1—C9 1.474 (8) C10—H10B 0.9900
C1—C2 1.387 (9) C11—C12 1.371 (8)
C1—C11 1.397 (8) C12—C13 1.507 (8)
C1—H1A 0.9500 C13—C14 1.511 (9)
C2—C3 1.390 (9) C13—C15 1.535 (8)
C2—H2 0.9500 C14—H14 1.0000
C3—C4 1.374 (8) C15—C16 1.512 (8)
C4—C12 1.387 (8) C15—H15A 0.9900
C5—C7 1.486 (9) C15—H15B 0.9900
C5—C6 1.542 (8) C16—H16A 0.9900
C5—H5 1.0000 C16—H16B 0.9900
C6—C13 1.563 (9) C17—H17A 0.9800
C6—H6 1.0000 C17—H17B 0.9800
C7—C8 1.331 (9) C17—H17C 0.9800
C3—O1—H1 109.5 C11—C10—C9 114.3 (5)
C4—O2—C6 106.2 (4) C11—C10—H10A 108.7
C5—O3—H3 109.5 C9—C10—H10A 108.7
C16—N1—C17 112.0 (5) C11—C10—H10B 108.7
C16—N1—C9 112.2 (5) C9—C10—H10B 108.7
C17—N1—C9 112.7 (5) H10A—C10—H10B 107.6
C2—C1—C11 120.1 (6) C12—C11—C1 117.4 (6)
C2—C1—H1A 119.9 C12—C11—C10 117.9 (5)
C11—C1—H1A 119.9 C1—C11—C10 124.2 (6)
C1—C2—C3 122.3 (6) C11—C12—C4 121.6 (5)
C1—C2—H2 118.8 C11—C12—C13 126.9 (5)
C3—C2—H2 118.8 C4—C12—C13 110.7 (5)
C4—C3—O1 119.3 (6) C12—C13—C14 106.5 (5)
C4—C3—C2 116.4 (6) C12—C13—C15 111.7 (5)
O1—C3—C2 124.1 (5) C14—C13—C15 110.1 (5)
C3—C4—O2 125.7 (5) C12—C13—C6 99.5 (5)
C3—C4—C12 121.7 (6) C14—C13—C6 116.4 (5)
O2—C4—C12 112.4 (5) C15—C13—C6 112.0 (5)
O3—C5—C7 113.0 (5) C13—C14—C8 109.8 (5)
O3—C5—C6 108.9 (5) C13—C14—C9 106.7 (5)
C7—C5—C6 113.5 (5) C8—C14—C9 114.2 (6)
O3—C5—H5 107.0 C13—C14—H14 108.7
C6—C5—H5 107.0 C9—C14—H14 108.7
O2—C6—C5 108.5 (5) C16—C15—C13 111.9 (5)
O2—C6—C13 107.1 (5) C16—C15—H15A 109.2
C5—C6—C13 112.8 (5) C13—C15—H15A 109.2
O2—C6—H6 109.5 C16—C15—H15B 109.2
C5—C6—H6 109.5 C13—C15—H15B 109.2
C13—C6—H6 109.5 H15A—C15—H15B 107.9
C8—C7—C5 121.3 (6) N1—C16—C15 110.7 (5)
C8—C7—H7 119.4 N1—C16—H16A 109.5
C5—C7—H7 119.4 C15—C16—H16A 109.5
C7—C8—C14 119.7 (6) N1—C16—H16B 109.5
C7—C8—H8 120.1 C15—C16—H16B 109.5
C14—C8—H8 120.1 H16A—C16—H16B 108.1
N1—C9—C14 107.8 (5) N1—C17—H17A 109.5
N1—C9—C10 115.3 (5) N1—C17—H17B 109.5
C14—C9—C10 112.4 (5) H17A—C17—H17B 109.5
N1—C9—H9 107.0 N1—C17—H17C 109.5
C14—C9—H9 107.0 H17A—C17—H17C 109.5
C10—C9—H9 107.0 H17B—C17—H17C 109.5
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H1···O3i 0.84 1.96 2.757 (6) 159
O3—H3···O2 0.84 2.17 2.629 (6) 114