Hexaaquacopper(II) dinitrate: absence of
Jahn–Teller distortion. Corrigendum
Ramin Zibaseresht and Richard M. Hartshorn*
Department of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, New Zealand
Correspondence e-mail: richard.hartshorn@canterbury.ac.nz Received 28 November 2012; accepted 4 December 2012
The identity of the metal atom in the paper by Zibaseresht & Hartshorn [Acta Cryst.(2006), E62, i19–i22] is corrected.
The published crystal structure in the paper by Zibaseresht & Hartshorn (2006) is almost certainly that of the nickel complex and not the copper complex as originally identified. Refine-ment of the copper occupancy was consistent with the presence of a lighter transition metal ion in the structure, and residuals improved significantly when the complex was refined as the nickel or cobalt salts. Unfortunately the original samples are no longer available, so further analysis of the crystals is not possible, but based on the colour of the crystals (blue) we believe the metal to be nickel.
The authors are grateful to Frank R. Fronczek for assistance in identifying this error.
References
Zibaseresht, R. & Hartshorn, R. M. (2006).Acta Cryst.E62, i19–i22.
addenda and errata
Acta Cryst.(2013). E69, e1 doi:10.1107/S1600536812049732 Zibaseresht and Hartshorn
e1
Acta Crystallographica Section E
Structure Reports
Online
inorganic papers
Acta Cryst.(2006). E62, i19–i22 doi:10.1107/S1600536805041851 Zibaseresht and Hartshorn [Cu(H
2O)6](NO3)2
i19
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Hexaaquacopper(II) dinitrate: absence of
Jahn–Teller distortion
Ramin Zibaseresht and Richard M. Hartshorn*
Department of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, New Zealand
Correspondence e-mail:
richard.hartshorn@canterbury.ac.nz
Key indicators
Single-crystal X-ray study
T= 93 K
Mean(O–N) = 0.004 A˚
Rfactor = 0.034
wRfactor = 0.096 Data-to-parameter ratio = 9.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, [Cu(H2O)6](NO3)2, the geometry
around the CuIIion is approximately octahedral, formed by six O atoms from the coordinated water molecules. The Cu—O distances are rather similar [2.014 (2)–2.084 (2) A˚ ] and not related by symmetry. The Jahn–Teller effect is, at best, only weakly observed in this structure, in contrast to many other structures where the hexaaquacopper(II) ion has been characterized. An extensive mesh of hydrogen-bond inter-actions between the coordinated water molecules and nitrate ions is a feature of the structure and may limit the degree to which the Jahn–Teller effect can be observed.
Comment
During attempts to grow crystals of a copper complex of the ditopic ligand, 1-[40-p-tolyl-(2,20:60,200
-terpyridyl)]-1,4,8,11-tetraazacyclotetradecane, (I) (Padilla-Tostaet al., 2000), blue block-shaped crystals of [Cu(H2O)6](NO3)2, (II), formed
instead from the reaction mixture. Attempts to grow similar crystals in the absence of the ditopic ligand proved unsuc-cessful, which leads us to speculate that the ditopic ligand may be influencing the crystallization process. Unfortunately, the vagaries of nucleation and crystal growth make it difficult to test this hypothesis. We report here the structure of the hexaaquacopper(II) complex as its dinitrate salt.
The asymmetric unit of (II) consists of a [Cu(H2O)6] 2+
cation and two nitrate anions. The geometry around the Cu2+ can be best described as octahedral, with bonds to six water molecules (Fig. 1 and Table 1). The Cu—O bond lengths are rather similar, falling in the range 2.014 (2)–2.084 (2) A˚ , and there is an extended hydrogen-bonding network that links the coordinated water molecules and the nitrate anions throughout the crystal structure (Fig. 2 and Table 2). Bond lengths and angles in the nitrate anions [1.233 (4)–1.272 (4) A˚ and 118.5 (3)–121.1 (3), respectively] are unremarkable,
there being only small deviations from the ideal geometry. The similarity of the Cu—O bond lengths is rather unusual in that Jahn–Teller distortion often leads to two of the copper-ligand bonds that lie along one axis being much longer than
the remaining four copper–ligand bonds. A number of Jahn– Teller-distorted hexaaquacopper(II) complexes have been characterized by X-ray crystallography,viz. X–3(C2H10N22+)–
2(O12P44) (Averbuch-Pouchot & Durif, 1989),X–2(ClO4)–
2(C6H10N2O2) (Benedetti et al., 1986), X–2(C6H4
Cl-O3S) (Bernardinelliet al., 1991),X–2(C7H7O3S) (Couldwell et al., 1978), X–2(C9H9O9S3
3
–1.3(H2O) (Dalrymple et al.,
2002),X–2(C2H10N2 2+
)–O18P6 6
(Durif & Averbuch-Pouchot, 1989),X–C6H8CuO10
2
(Filippova, 2000),X–2(C12H10O4P
)– 2(C2H5NO2) (Glowiak & Podgorska, 1986), X–C16H16
-CuO10 2
(Honghuiet al., 1988),X–C16H16CuO10 2
(Kennard & Smith, 1989),X–2(Cl4
),2(H2O) (Liet al., 2004),X–2 C l
– 2(C10H8N2O2)–2(H2O) (Ma et al., 2001), X–2(C7H5O6S
)– 2(H2O) (Maet al., 2003),X–2(NH4
+
)–2(SO4 2
) (Maslenet al., 1988), X–2(C24H44H16O4Pt4
4+
)–10(ClO4
)–9(H2O) (Navarro et al., 2000), X–(C6H8CuO10
2+
) (Rodriguez-Martin et al., 2002), X–2(C8H11N4O
+
)–2(SO4 2
)–2(H2O) (Shamuratov et al., 1993) and (X)n–2n(C5H8O4
)–4n(H2O) (Zviedre et al.,
1985), whereXis [Cu(H2O)6]2+. In these cases, the axial Cu—
O bond lengths fall in the range 2.202–2.423 A˚ , in comparison with the equatorial bond lengths (1.945–2.084 A˚ ). The mean axial bond length is between 8.7 and 24% longer than the mean equatorial bond length in these structures (the mean value of these percentage differences is 18.6% over 20 struc-tures). In our structure, the mean bond length along the longest axis (O2—Cu—O4) is only 1.6% longer than that along the remaining axes.
We are aware of only six crystallographic studies of copper(II) complexes where static Jahn–Teller distortions are not observed in complexes where all six donors are otherwise identical, viz. in X–(BrO3)2 (Blackburn et al., 1991),
Cu(en)3 2+
–SO4 2
(Cullen & Lingafelter, 1970), 2K+–Pb2+–
Cu(NO2)6 4
(Cullen & Lingafelter, 1971), Cu{[(CH3)2N]2
-P(O)OP(O)[N(CH3)2]2}3(Cl4)2 (Joesten et al., 1970), X–
(SiF6) 2
–6(H2O) (Rayet al., 1973) and 2Tl +
–Pb2+–Cu(NO2)6 2
(Takagiet al., 1976), whereXis [Cu(H2O)6] 2+
. The structure we report further stands out from these other six because, in this case, the Cu atom lies on a general position, with all Cu— O bond lengths being independently refined. In the other six cases, the Cu atoms are located on special positions in higher symmetry space groups (Pa3, P31c,Fm3,P3c1, R3 andFm3, respectively).
Jahn–Teller distortion may not be observed in a crystal-lographic study if either there is disorder in the structure (so that a defined long axis is randomly distributed over the three orientations relative to the unit cell axes), or there is sufficient thermal motion to allow the long and short bonds in a struc-ture to exchange over time (sometimes referred to as the dynamic Jahn–Teller effect). In these cases, the averaging inherent in the X-ray experiment (over spatial location in the crystal in the first case or time in the second) might be expected to manifest itself in the crystallographic modelling process as larger than expected anisotropic displacement parameters for the donor atoms along the direction of the copper–ligand bond. This effect has been discussed (Cullenet al., 1970) and may be significant in a number of the literature cases (Blackburnet al., 1991; Cullenet al., 1971; Takagiet al., 1976). Table 3 presents the anisotropic displacement para-meters of Cu and the water O atoms in the structure of (II). The largest principal axes of the ellipsoids are not directed along the Cu—O bonds (Fig. 1). Taken together, these data strongly suggest the lack of Jahn–Teller distortion (static or dynamic) in the structure of (II). Here, three marginally longer Cu—O bonds (Cu—O2, Cu—O3 and Cu—O4) are meridionally distributed around the Cu atom, as are the Cu— O shorter bonds. The variation in the Cu—O bond lengths of the structure, and the absence of any significant Jahn–Teller effect, may be explained by the influence of the hydrogen-bonding network in the crystal structure of the complex (Fig. 3
inorganic papers
i20
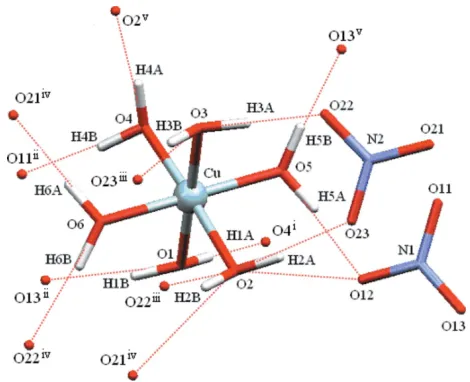
Zibaseresht and Hartshorn [Cu(H [image:3.610.329.538.72.239.2]2O)6](NO3)2 Acta Cryst.(2006). E62, i19–i22
Figure 1
A perspective view of the title CuIIcomplexes, (II), showing the atom-labelling scheme with 50% probability displacement ellipsoids. H atoms are drawn as small spheres of arbitrary radii.
Figure 2
[image:3.610.84.258.75.311.2]and Table 2). All of the coordinated water molecules are involved in several hydrogen bonds, which means that, while the copper centre may not be in its lowest energy Jahn–Teller distorted state, this could be made up for by the large number of weak interactions that may each be marginally stronger in the less distorted structure.
Experimental
A solution of Cu(NO3)23H2O (50 mg) in ethanol (5 ml) was added to
a cooled filtered solution of ligandL, (I) (0.15 g), in ethanol (5 ml). The reaction mixture was heated at reflux for 1 h, and, upon cooling to room temperature, afforded a blue–green insoluble precipitate (0.11 g). The precipitate was suspended in ethanol–water (1:1, 5 ml), then the mixture was filtered after it was heated to reflux for 1 h. The solution was allowed to cool to room temperature overnight. The solution was kept in a refrigerator for about two months during which time blue crystals of (II) suitable for X-ray analysis were produced. No crystals of (I) or its copper complex were produced in this way.
Crystal data
[Cu(H2O)6](NO3)2 Mr= 295.67
Triclinic,P1
a= 5.7404 (8) A˚
b= 7.6452 (10) A˚
c= 11.4655 (15) A˚ = 106.428 (2)
= 98.399 (2)
= 101.504 (2)
V= 461.84 (11) A˚3
Z= 2
Dx= 2.126 Mg m
3 MoKradiation Cell parameters from 2722
reflections = 2.9–26.4
= 2.43 mm1 T= 93 (2) K Block, blue
0.550.340.12 mm
Data collection
Bruker SMART CCD diffractometer ’and!scans
Absorption correction: multi-scan (SADABS; Bruker, 1999)
Tmin= 0.341,Tmax= 0.744 2917 measured reflections
1556 independent reflections 1494 reflections withI> 2(I)
Rint= 0.020 max= 25.1
h=6!6
k=8!8
l=13!13
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.034 wR(F2) = 0.096
S= 0.91 1556 reflections 172 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0546P)2 + 2.7318P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.60 e A˚
3
min=1.14 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
Cu—O5 2.014 (2)
Cu—O1 2.034 (2)
Cu—O6 2.041 (2)
Cu—O4 2.064 (2)
Cu—O3 2.074 (2)
Cu—O2 2.084 (2)
N1—O11 1.241 (4)
N1—O12 1.245 (4)
N1—O13 1.268 (4)
N2—O21 1.233 (4)
N2—O23 1.252 (4)
N2—O22 1.272 (4)
O5—Cu—O1 89.49 (10)
O5—Cu—O6 175.94 (10)
O1—Cu—O6 93.58 (10)
O5—Cu—O4 91.38 (10)
O1—Cu—O4 88.81 (10)
O6—Cu—O4 91.34 (10)
O5—Cu—O3 91.72 (10)
O1—Cu—O3 178.31 (10)
O6—Cu—O3 85.17 (10)
O4—Cu—O3 92.35 (10)
O5—Cu—O2 89.50 (10)
O1—Cu—O2 87.93 (10)
O6—Cu—O2 87.96 (10)
O4—Cu—O2 176.61 (9)
[image:4.610.52.287.71.262.2]O3—Cu—O2 90.89 (10)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1A O4i
0.98 (2) 1.91 (2) 2.894 (3) 179 (3) O1—H1B O13ii
0.97 (2) 1.79 (2) 2.741 (4) 168 (4) O2—H2B O22iii
0.98 (2) 2.12 (2) 3.038 (4) 156 (3) O2—H2A O23 0.98 (2) 2.00 (2) 2.940 (4) 162 (3) O2—H2B O21iv 0.98 (2) 2.38 (3) 2.912 (4) 113 (3) O3—H3A O22 0.98 (2) 1.83 (2) 2.779 (4) 162 (3) O3—H3B O23iii
0.97 (2) 1.88 (2) 2.827 (4) 167 (3) O4—H4A O2v 0.97 (2) 1.99 (2) 2.942 (4) 167 (4) O4—H4A O1v
0.97 (2) 2.60 (4) 3.070 (3) 110 (3) O4—H4B O11ii
0.97 (2) 1.79 (2) 2.763 (4) 175 (3) O5—H5A O12 0.97 (2) 1.78 (2) 2.735 (3) 166 (4) O5—H5A N1 0.97 (2) 2.50 (3) 3.417 (4) 156 (3) O5—H5A O11 0.97 (2) 2.58 (3) 3.285 (3) 130 (3) O5—H5B O13v
0.96 (2) 1.78 (2) 2.740 (4) 172 (4) O5—H5B N1v 0.96 (2) 2.47 (2) 3.365 (4) 155 (3) O5—H5B O12v
0.96 (2) 2.45 (3) 3.123 (3) 126 (3) O6—H6B O21iv
0.98 (2) 2.44 (3) 3.154 (3) 130 (3) O6—H6B O22iv 0.98 (2) 1.91 (2) 2.860 (4) 162 (4) O6—H6B N2iv
0.98 (2) 2.51 (2) 3.436 (4) 157 (3)
Symmetry codes: (i)x;y;z; (ii)x1;y1;z; (iii)xþ1;yþ1;zþ1; (iv)
[image:4.610.313.564.387.570.2]x;y1;z; (v)x1;y;z.
Table 3
Selected anisotropic displacement parameters (A˚2).
U11 U22 U33 U12 U13 U23
Cu 0.0058 (3) 0.0106 (3) 0.0126 (3) 0.00344 (18) 0.00374 (17) 0.00079 (17) O1 0.0078 (12) 0.0084 (13) 0.0119 (12) 0.0020 (10) 0.0045 (10) 0.0012 (9) O2 0.0050 (11) 0.0102 (13) 0.0130 (12) 0.0047 (10) 0.0024 (9) 0.0003 (9) O3 0.0091 (12) 0.0099 (13) 0.0123 (12) 0.0032 (10) 0.0053 (10) 0.0027 (10) O4 0.0052 (11) 0.0079 (13) 0.0134 (12) 0.0031 (10) 0.0035 (9) 0.0013 (9) O5 0.0067 (12) 0.0120 (13) 0.0160 (13) 0.0076 (10) 0.0064 (10) 0.0021 (10) O6 0.0058 (12) 0.0114 (13) 0.0137 (12) 0.0063 (10) 0.0046 (10) 0.0014 (9)
inorganic papers
Acta Cryst.(2006). E62, i19–i22 Zibaseresht and Hartshorn [Cu(H
2O)6](NO3)2
i21
Figure 3
A perspective view of the CuIIcomplex, (II), showing the hydrogen-bonding interactions (dashed lines) involving the dication. [Symmetry codes: (i)x,y,z; (ii)x1,y1,z; (iii)x+ 1,y+ 1,z+ 1; (iv)
[image:4.610.314.566.645.727.2]H atoms were located in a difference Fourier map. The O—H distances were restrained to 1.00 (2) A˚ , withUiso(H) = 1.2Ueq(O). In
the final difference map the deepest hole is located 0.89 A˚ from the Cu atom.
Data collection:SMART(Bruker, 1999); cell refinement: SAINT-Plus(Bruker, 1999); data reduction:SAINT-Plus; program(s) used to solve structure: SHELXTL (Sheldrick, 2001); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL and
MERCURY (Version 1.4; Bruno et al., 2002); software used to prepare material for publication:SHELXTL.
The authors thank Professor W. T. Robinson and Dr J. Wikaira for their help with X-ray structural investigations associated with this project. A University of Canterbury Doctoral Scholarship (to RZ) is gratefully acknowledged.
References
Averbuch-Pouchot, M. T. & Durif, A. (1989).Acta Cryst.C45, 46–49. Benedetti, E., Bavoso, A., Di Blasio, B., Pavone, V. & Pedone, C. (1986).Inorg.
Chim. Acta,123, 155–159.
Bernardinelli, G., Lucken, E. A. C. & Costines, M. (1991).Z. Kristallogr.195, 141–1422.
Blackburn, A. C., Gallucci, J. C. & Gerkin, R. E. (1991).Acta Cryst.C47, 1786– 1789.
Bruker (1999).SADABS,SAINT-Plus(Version 6.22) andSMART(Version 5.045), Bruker AXS Inc., Madison, Wisconsin, USA.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Couldwell, C., Prout, K., Robey, D., Taylor, R. & Rossotti, F. J. C. (1978).Acta Cryst.B34, 1491–1499.
Cullen, D. L. & Lingafelter, E. C. (1970).Inorg. Chem.9, 1858–1864. Cullen, D. L. & Lingafelter, E. C. (1971).Inorg. Chem.10, 1264–1268. Dalrymple, S. A., Parvez, M. & Shimizu, G. K. H. (2002).Inorg. Chem.41,
6986–6996.
Durif, A. & Averbuch-Pouchot, M. T. (1989).Acta Cryst.C45, 1884–1887. Filippova, I. G. (2000).Russ. J. Coord. Chem.26, 276–280; translation of
Koord. Khim.26, 295.
Glowiak, T. & Podgorska, I. (1986).Inorg. Chim. Acta,125, 83–88. Honghui, W., Naijue Z, Heng F.,Ronncchang L. & Kui W. (1988).Sci. Sin. Ser.
B: Chem. Biol. Agri. Med. Earth Sci. (Engl. Ed.),31, 20–27.
Joesten, M. D., Hussain, M. S. & Lenhert, P. G. (1970).Inorg. Chem.9, 151– 161.
Kennard, C. H. L. & Smith, G. (1989).Z. Kristallogr.188, 63–68.
Li, X. H., Xia, F. Y., Xiao, H. P. & Hu, M. L. (2004).Acta Cryst.E60, i31–i32. Ma, B.-Q., Sun, H.-L., Gao, S. & Xu, G.-X. (2001).Inorg. Chem.40, 6247–6253. Ma, J. F., Yang, J. & Liu, J. F. (2003).Acta Cryst.E59, m485–m486. Maslen, E. N., Watson, K. J. & Moore, F. H. (1988).Acta Cryst.B44, 102–107. Navarro, J. A., Freisinger, E. & Lippert, B. (2000).Inorg. Chem.39, 2301–2305. Padilla-Tosta, M. E., Lloris, J. M., Martinez-Manez, R., Benito, A., Soto, J., Pardo, T., Miranda, M. A. & Marcos, M. D. (2000).Eur. J. Inorg. Chem.741– 748.
Ray, S., Zalkin, A. & Templeton, D. H. (1973).Acta Cryst.B29, 2748–2751. Rodriguez-Martin, Y., Sanchiz, J., Ruiz-Perez, C., Lloret, F. & Julve, M. (2002).
Cryst. Eng. Commun.pp. 631–637.
Shamuratov, E. B., Sharipov, K. T., Batsanov, A. S., Struchkov, Yu. T., Khudoyarov, A. B. & Mirdzhalalov, F. F. (1993).Koord. Khim.19, 155–159. (In Russian.)
Sheldrick, G. M. (2001).SHELXTL. Version 6.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Takagi, S., Joesten, M. D. & Lenhert, P. G. (1976).Acta Cryst.B32, 326–328. Zviedre, I., Bel’skii, V. K. & Shvarts, E. M. (1985).Latv. PSR Zinat. Akad.
Vestis Kim. Ser.pp. 672–677. (In Russian.)
inorganic papers
i22
Zibaseresht and Hartshorn [Cu(Hsupporting information
sup-1
Acta Cryst. (2006). E62, i19–i22
supporting information
Acta Cryst. (2006). E62, i19–i22 [doi:10.1107/S1600536805041851]
Hexaaquacopper(II) dinitrate: absence of Jahn
–
Teller distortion
Ramin Zibaseresht and Richard M. Hartshorn
S1. Comment
During attempts to grow crystals of the copper complex of the ditopic ligand, 1-[4′-p
-tolyl-(2,2′:6′,2′′-terpyridyl)]-1,4,8,11-tetraazacyclotetradecane, (I) (Padilla-Tosta et al., 2000), blue block-shaped crystals of [Cu(H2O)6]
(NO3)2, (II), formed instead from the reaction mixture. Attempts to grow similar crystals in the absence of the ditopic
ligand proved unsuccessful, which leads us to speculate that the ditopic ligand may be influencing the crystallization process. Unfortunately, the vagaries of nucleation and crystal growth make it difficult to test this hypothesis. We report here the structure of hexaaquacopper complex as its dinitrate salt.
The asymmetric unit of (II) consists of a [Cu(H2O)6]2+ cation and two nitrate anions. The geometry around the Cu2+ can
be best described as an octahedron, with bonds to six water molecules (Fig. 1 and Table 1). The Cu—O bond lengths are rather similar, falling in the range 2.014 (2)–2.084 (2) Å, and there is an extended hydrogen-bonding network that links the coordinated water molecules and the nitrate anions throughout the crystal lattice (Fig. 2 and Table 2). Bond lengths and angles in the nitrate anions [1.233 (4)–1.272 (4) Å and 118.5 (3)–121.1 (3)°, respectively] are unremarkable, there being only small deviations from the ideal geometry.
The similarity of the Cu—O bond lengths is relatively unusual in that Jahn–Teller distortion often leads to two of the copper-ligand bonds that lie along one axis being much longer than the remaining four copper–ligand bonds. A number of Jahn–Teller-distorted hexaaquacopper complexes have been characterized by X-ray crystallography, viz. X–3(C2H10N22+)–
2(O12P44−) (Averbuch-Pouchot & Durif, 1989), X–2(ClO4−)–2(C6H10N2O2) (Benedetti et al., 1979, 1986), X–
2(C6H4ClO3S−) (Bernardinelli et al., 1991), X–2(C7H7O3S−) (Couldwell et al., 1978), X–2(C9H9O9S33−–1.3(H2O)
(Dalrymple et al., 2002), X–2(C2H10N22+)–O18P66− (Durif & Averbuch-Pouchot, 1989), X–C6H8CuO102− (Filippova, 2000),
X–2(C12H10O4P−)–2(C2H5NO2) (Glowiak & Podgorska, 1986), X–C16H16CuO102− (Honghui et al., 1988), X–C16H16CuO102−
(Kennard & Smith, 1989), X–2(Cl4−),2(H2O) (Li et al., 2004), X–2 C l−–2(C10H8N2O2)–2(H2O) (Ma et al., 2001), X–
2(C7H5O6S−)–2(H2O) (Ma et al., 2003), X–2(NH4+)–2(SO42−) (Maslen et al., 1988), X–2(C24H44H16O4Pt44+)–10(ClO4−)–
9(H2O) (Navarro et al., 2000), X–(C6H8CuO102+) (Rodriguez-Martin or Rodriquez-Martin et al., 2002), X–2(C8H11N4O+)–
2(SO42−)–2(H2O) (Shamuratov et al., 1993), X–(C16H16CuO102−) (Wang et al., 1988) and (X)n–2n(C5H8O4−)–4n(H2O)
(Zviedre et al., 1985), where X is [Cu(H2O)6]2+. In these cases, the axial Cu—O bond lengths fall in the range 2.202–
2.423 Å, in comparison with the equatorial bond lengths (1.945–2.084 Å). The mean axial bond length is between 8.7 and 24% longer than the mean equatorial bond length in these structures (the mean value of these percentage differences is 18.6% over 20 structures). In our structure, the mean bond length along the longest axis (O2—Cu—O4) is only 1.6% longer than that along the remaining axes.
We are aware of only six crystallographic studies of copper(II) complexes where static Jahn–Teller distortions are not observed in complexes where all six donors are otherwise identical, viz. in X–(BrO3)2 (Blackburn et al., 1991), Cu(en)32+–
SO42− (Cullen & Lingafelter, 1970), 2 K+–Pb2+–Cu(NO2)64− (Cullen & Lingafelter, 1971), Cu{[(CH3)2N]2P(O)OP(O)
supporting information
sup-2
Acta Cryst. (2006). E62, i19–i22
1976), where X is [Cu(H2O)6]2+. The structure we report further stands out from these other six because, in this case, the
Cu atom lies on a general position, with all Cu—O bonds lengths being independently refined. In the remaining six cases, the Cu atoms are located on the special positions in higher symmetry space groups (Pa3, P31c, Fm3, P3c1, R3 and Fm3, respectively).
Jahn–Teller distortion may not be observed in a crystallographic study if either there is disorder in the structure (so that a defined long axis is randomly distributed over the three orientations relation to the unit-cell axes), or there is sufficient thermal motion to allow the long and short bonds in a structure to exchange over time (sometimes referred to as the dynamic Jahn–Teller effect). In these cases, the averaging inherent in the X-ray experiment (over spatial location in the crystal in the first case or time in the second) might be expected to manifest itself in the crystallographic modelling process as larger than expected anisotropic displacement parameters for the donor atoms along the direction of the copper–ligand bond. This effect has been discussed (Cullen et al., 1970) and may be significant in a number of the literature cases (Blackburn et al., 1991; Cullen et al., 1971; Takagi et al., 1976). Table 3 presents the anisotropic displacement parameters of Cu and the water O atoms in the structure of (II). The largest principal axes of the ellipsoids are not directed along the Cu—O bonds (Fig. 1). Taken together, these data strongly suggest the lack of Jahn–Teller distortion (static or dynamic) in the structure of (II). Here, three marginally longer Cu—O bonds (Cu—O2, Cu—O3 and Cu—O4) are meridionally distributed around the Cu atom, as are the Cu—O shorter bonds. The variation in the Cu—O bond lengths of the structure, and the absence of any significant Jahn–Teller effect may be explained by the influence of the hydrogen-bonding network in the lattice of the complex (Fig. 3 and Table 2). All of the coordinated water molecules are involved in several hydrogen bonds, which means that, while the copper centre may not be in its lowest energy Jahn– Teller distorted state, this could be made up for by the large number of weak interactions that may each be marginally stronger in the less distorted structure.
S2. Experimental
A solution of Cu(NO3)2·3H2O (50 mg) in ethanol (5 ml) was added to a cooled filtered solution of ligand L, or (I) (0.15 g)
in ethanol (5 ml). The reaction mixture was heated at reflux for 1 h, and upon cooling to room temperature afforded a blue–green insoluble precipitate (0.11 g). The precipitate was suspended in ethanol–water (1:1, 5 ml), then the mixture was filtered after it was heated to reflux for 1 h. The solution was allowed to cool to room temperature overnight. The solution was kept in the refrigerator for about two months during which time blue crystals of (II) suitable for X-ray analysis were produced. No crystals of (I) were produced in this way.
S3. Refinement
The H atoms were located in a difference Fourier map. The O—H distances were constrained to 1.0 Å, with Uiso(H) =
1.2Ueq(O). The highest peak in the final difference map is located 0.99 Å from Cu and the deepest hole 0.89 Å from the
supporting information
sup-3
[image:8.610.137.464.76.514.2]Acta Cryst. (2006). E62, i19–i22 Figure 1
A perspective view of diagram of the title CuII complex, (II), showing the atom-labelling scheme with 50% probability
supporting information
sup-4
[image:9.610.129.486.72.352.2]Acta Cryst. (2006). E62, i19–i22 Figure 2
supporting information
sup-5
[image:10.610.127.484.75.368.2]Acta Cryst. (2006). E62, i19–i22 Figure 3
A perspective view of diagram of the CuII complex, (II), showing the hydrogen-bonding interactions (dashed lines)
involving the dication. [Symmetry codes: (i) −x, −y, −z; (ii) x − 1, y − 1, z; (iii) −x + 1, −y + 1, −z + 1; (iv) x, y − 1, z; (v) x
− 1, y, z.]
Hexaaquacopper(II) dinitrate
Crystal data
[Cu(H2O)6](NO3)2
Mr = 295.67 Triclinic, P1 Hall symbol: -P 1
a = 5.7404 (8) Å
b = 7.6452 (10) Å
c = 11.4655 (15) Å
α = 106.428 (2)°
β = 98.399 (2)°
γ = 101.504 (2)°
V = 461.84 (11) Å3
Z = 2
F(000) = 302
Dx = 2.126 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2722 reflections
θ = 2.9–26.4°
µ = 2.43 mm−1
T = 93 K Block, blue
0.55 × 0.34 × 0.12 mm
Data collection
Bruker SMART CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.192 pixels mm-1
φ and ω scans
Absorption correction: multi-scan (SADABS; Bruker, 1999)
Tmin = 0.341, Tmax = 0.744
2917 measured reflections 1556 independent reflections 1494 reflections with I > 2σ(I)
supporting information
sup-6
Acta Cryst. (2006). E62, i19–i22 θmax = 25.1°, θmin = 3.7°
h = −6→6
k = −8→8
l = −13→13
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.034
wR(F2) = 0.096
S = 0.91 1556 reflections 172 parameters 18 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0546P)2 + 2.7318P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.60 e Å−3
Δρmin = −1.14 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-7
Acta Cryst. (2006). E62, i19–i22
N2 0.5813 (5) 0.7634 (4) 0.3903 (3) 0.0102 (6) O21 0.6533 (5) 0.9126 (4) 0.3697 (2) 0.0124 (5) O22 0.3683 (4) 0.7234 (4) 0.4107 (2) 0.0145 (6) O23 0.7148 (4) 0.6530 (4) 0.3925 (2) 0.0125 (5)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Cu 0.0058 (3) 0.0106 (3) 0.0126 (3) 0.00079 (17) 0.00374 (17) 0.00344 (18) O1 0.0078 (12) 0.0084 (13) 0.0119 (12) 0.0012 (9) 0.0045 (10) 0.0020 (10) O2 0.0050 (11) 0.0102 (13) 0.0130 (12) 0.0003 (9) 0.0024 (9) 0.0047 (10) O3 0.0091 (12) 0.0099 (13) 0.0123 (12) 0.0027 (10) 0.0053 (10) 0.0032 (10) O4 0.0052 (11) 0.0079 (13) 0.0134 (12) 0.0013 (9) 0.0035 (9) 0.0031 (10) O5 0.0067 (12) 0.0120 (13) 0.0160 (13) 0.0021 (10) 0.0064 (10) 0.0076 (10) O6 0.0058 (12) 0.0114 (13) 0.0137 (12) 0.0014 (9) 0.0046 (10) 0.0063 (10) N1 0.0081 (14) 0.0111 (17) 0.0077 (14) 0.0011 (12) 0.0033 (11) 0.0015 (11) O11 0.0095 (12) 0.0124 (14) 0.0152 (13) 0.0043 (10) 0.0052 (10) 0.0043 (10) O12 0.0095 (12) 0.0079 (14) 0.0216 (14) 0.0007 (10) 0.0067 (10) 0.0068 (10) O13 0.0051 (12) 0.0120 (14) 0.0223 (14) −0.0007 (10) 0.0054 (10) 0.0058 (11) N2 0.0080 (14) 0.0121 (16) 0.0088 (14) 0.0016 (12) 0.0032 (11) 0.0009 (12) O21 0.0119 (12) 0.0108 (14) 0.0180 (13) 0.0023 (10) 0.0081 (10) 0.0078 (10) O22 0.0065 (12) 0.0137 (15) 0.0227 (14) −0.0003 (10) 0.0065 (10) 0.0049 (11) O23 0.0106 (12) 0.0135 (14) 0.0143 (13) 0.0054 (10) 0.0043 (10) 0.0037 (10)
Geometric parameters (Å, º)
Cu—O5 2.014 (2) O4—H4A 0.970 (18)
Cu—O1 2.034 (2) O4—H4B 0.974 (18)
Cu—O6 2.041 (2) O5—H5A 0.974 (18)
Cu—O4 2.064 (2) O5—H5B 0.963 (18)
Cu—O3 2.074 (2) O6—H6A 0.973 (18)
Cu—O2 2.084 (2) O6—H6B 0.978 (18)
O1—H1A 0.981 (18) N1—O11 1.241 (4)
O1—H1B 0.966 (18) N1—O12 1.245 (4)
O2—H2A 0.976 (18) N1—O13 1.268 (4)
O2—H2B 0.981 (18) N2—O21 1.233 (4)
O3—H3A 0.977 (18) N2—O23 1.252 (4)
O3—H3B 0.966 (18) N2—O22 1.272 (4)
O5—Cu—O1 89.49 (10) H2A—O2—H2B 112 (2)
O5—Cu—O6 175.94 (10) Cu—O3—H3A 108 (2)
O1—Cu—O6 93.58 (10) Cu—O3—H3B 110 (2)
O5—Cu—O4 91.38 (10) H3A—O3—H3B 113 (2)
O1—Cu—O4 88.81 (10) Cu—O4—H4A 111 (2)
O6—Cu—O4 91.34 (10) Cu—O4—H4B 108 (2)
O5—Cu—O3 91.72 (10) H4A—O4—H4B 113 (2)
O1—Cu—O3 178.31 (10) Cu—O5—H5A 114 (2)
supporting information
sup-8
Acta Cryst. (2006). E62, i19–i22
O4—Cu—O3 92.35 (10) H5A—O5—H5B 114 (3)
O5—Cu—O2 89.50 (10) Cu—O6—H6A 110 (2)
O1—Cu—O2 87.93 (10) Cu—O6—H6B 122 (2)
O6—Cu—O2 87.96 (10) H6A—O6—H6B 112 (2)
O4—Cu—O2 176.61 (9) O11—N1—O12 120.8 (3)
O3—Cu—O2 90.89 (10) O11—N1—O13 120.7 (3)
Cu—O1—H1A 116 (2) O12—N1—O13 118.5 (3)
Cu—O1—H1B 113 (2) O21—N2—O23 121.1 (3)
H1A—O1—H1B 113 (2) O21—N2—O22 118.9 (3)
Cu—O2—H2A 112 (2) O23—N2—O22 120.0 (3)
Cu—O2—H2B 113 (2)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H1A···O4i 0.98 (2) 1.91 (2) 2.894 (3) 179 (3)
O1—H1B···O13ii 0.97 (2) 1.79 (2) 2.741 (4) 168 (4)
O2—H2B···O22iii 0.98 (2) 2.12 (2) 3.038 (4) 156 (3)
O2—H2A···O23 0.98 (2) 2.00 (2) 2.940 (4) 162 (3) O2—H2B···O21iv 0.98 (2) 2.38 (3) 2.912 (4) 113 (3)
O3—H3A···O22 0.98 (2) 1.83 (2) 2.779 (4) 162 (3) O3—H3B···O23iii 0.97 (2) 1.88 (2) 2.827 (4) 167 (3)
O4—H4A···O2v 0.97 (2) 1.99 (2) 2.942 (4) 167 (4)
O4—H4A···O1v 0.97 (2) 2.60 (4) 3.070 (3) 110 (3)
O4—H4B···O11ii 0.97 (2) 1.79 (2) 2.763 (4) 175 (3)
O5—H5A···O12 0.97 (2) 1.78 (2) 2.735 (3) 166 (4) O5—H5A···N1 0.97 (2) 2.50 (3) 3.417 (4) 156 (3) O5—H5A···O11 0.97 (2) 2.58 (3) 3.285 (3) 130 (3) O5—H5B···O13v 0.96 (2) 1.78 (2) 2.740 (4) 172 (4)
O5—H5B···N1v 0.96 (2) 2.47 (2) 3.365 (4) 155 (3)
O5—H5B···O12v 0.96 (2) 2.45 (3) 3.123 (3) 126 (3)
O6—H6B···O21iv 0.98 (2) 2.44 (3) 3.154 (3) 130 (3)
O6—H6B···O22iv 0.98 (2) 1.91 (2) 2.860 (4) 162 (4)
O6—H6B···N2iv 0.98 (2) 2.51 (2) 3.436 (4) 157 (3)